
A 2.8 Å Structure of Zoliflodacin in a DNA Cleavage Complex with Staphylococcus aureus DNA Gyrase

 ,
, 
Abstract
:1. Introduction
2. Results
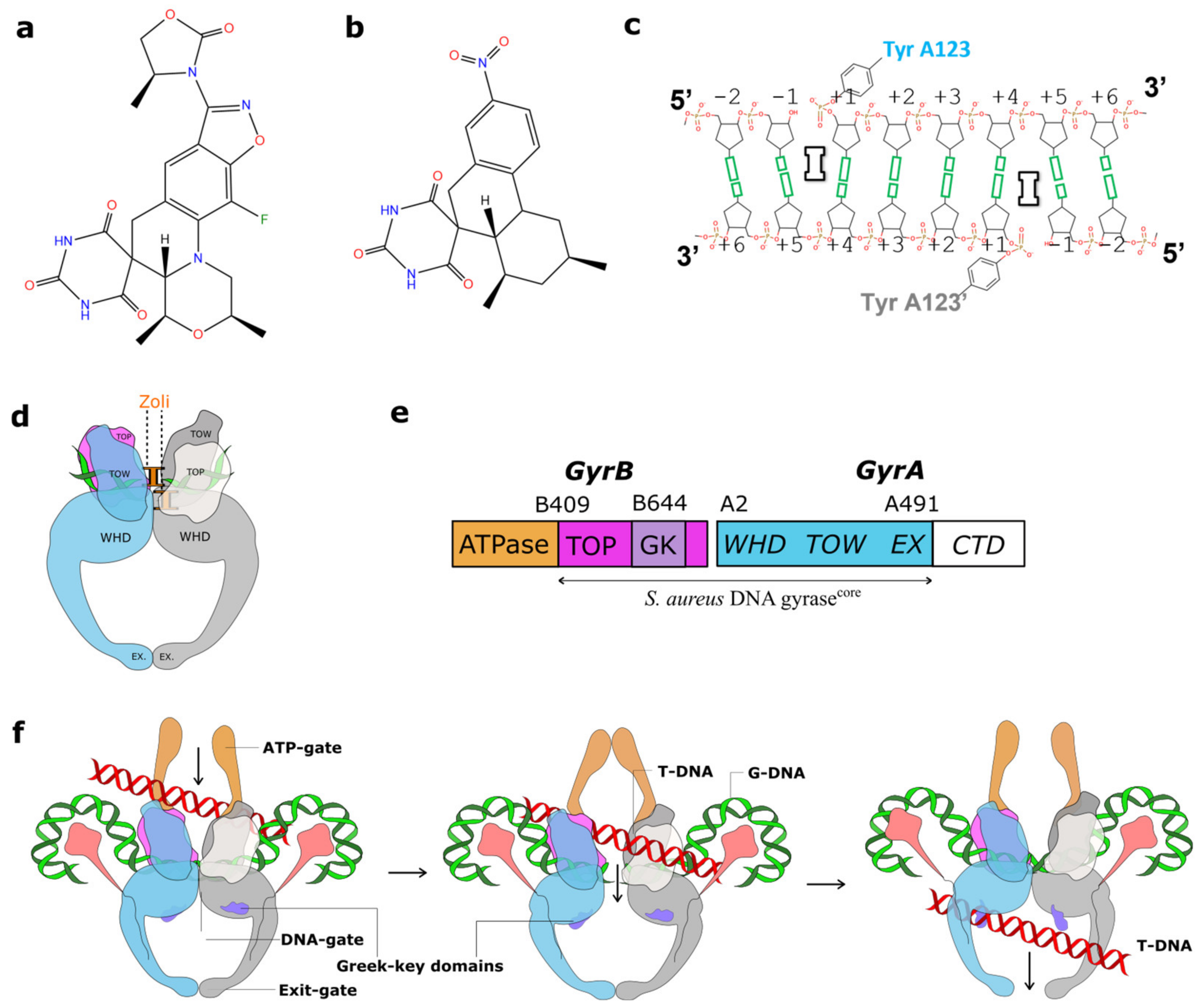
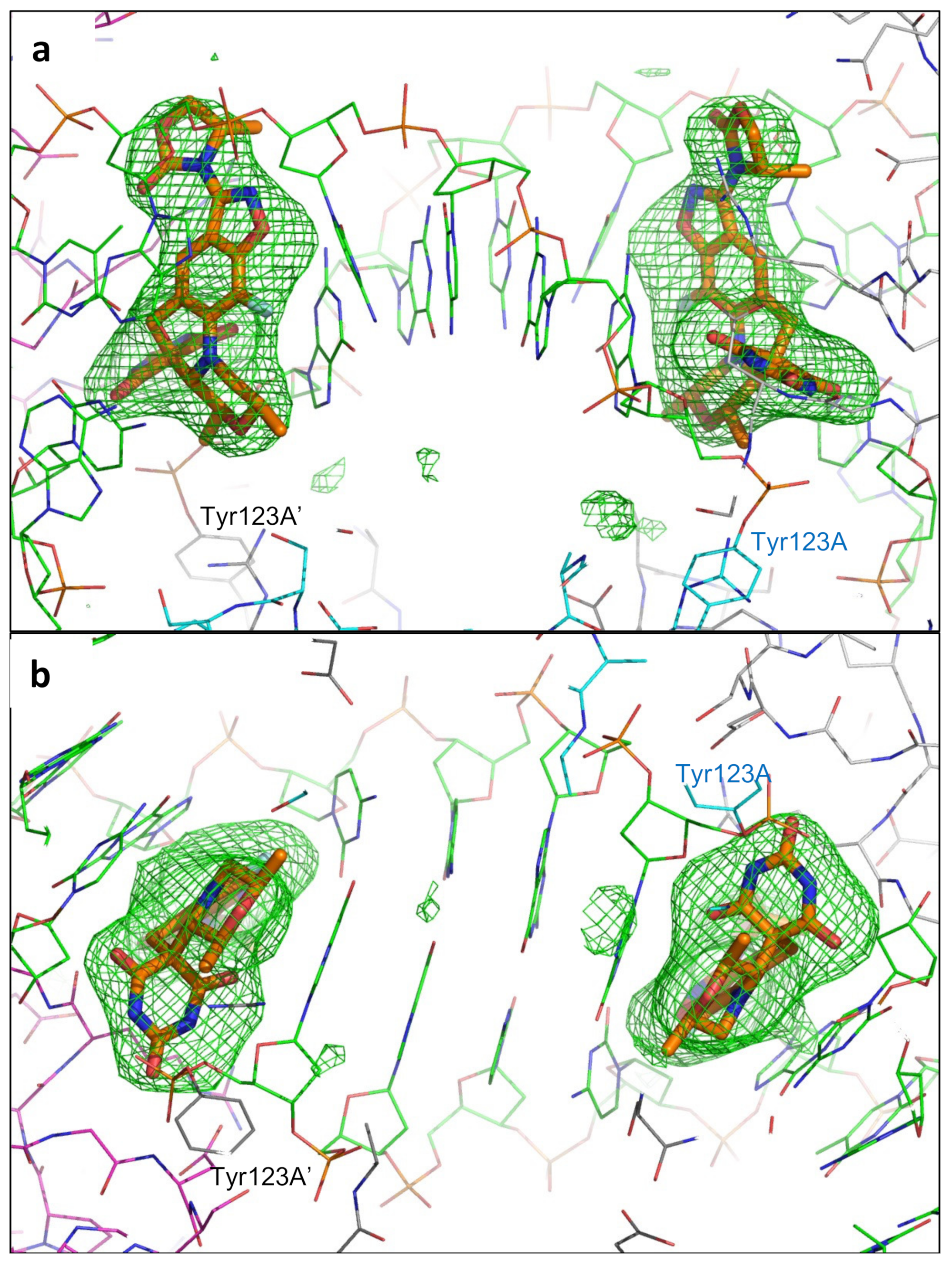
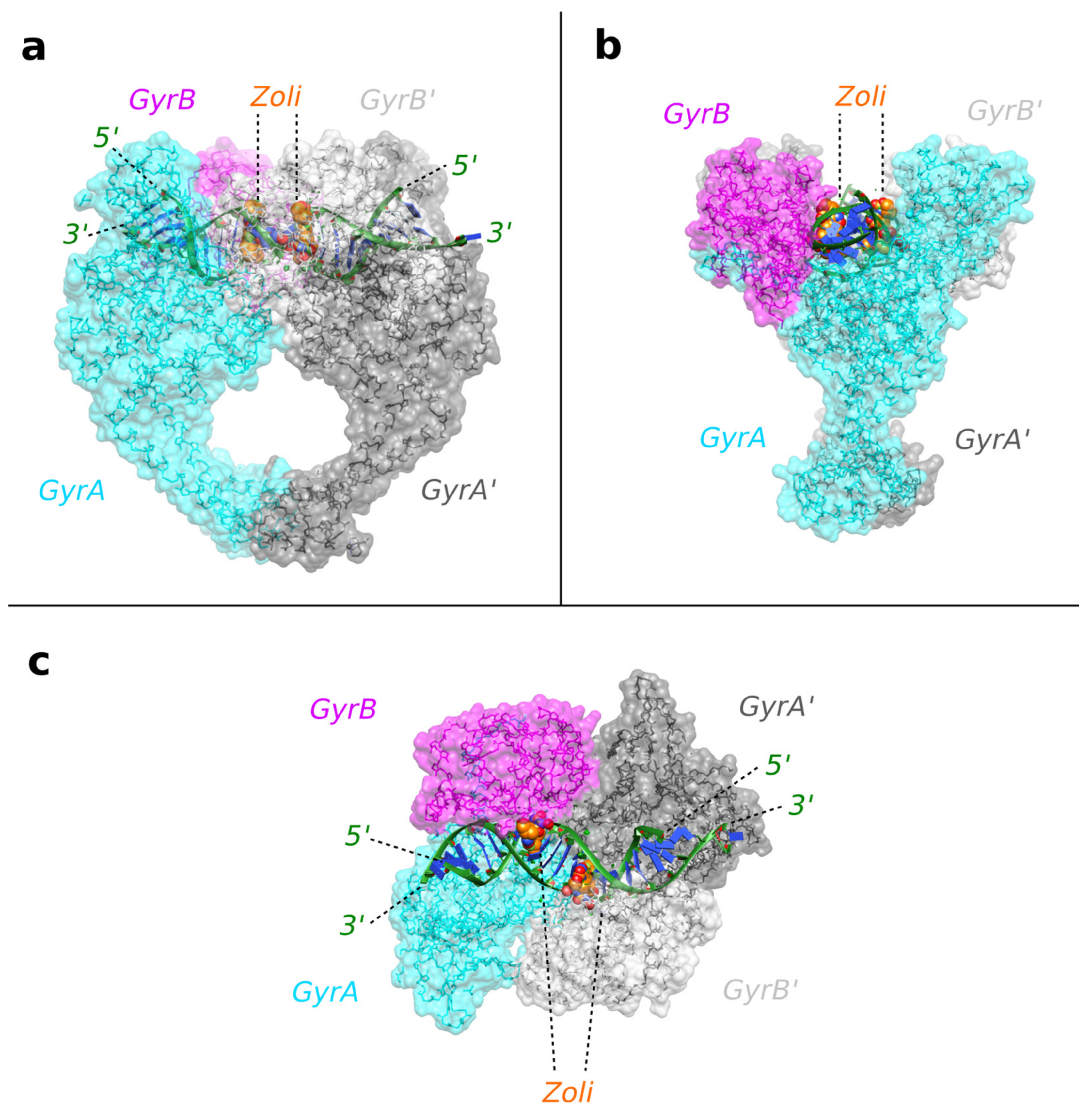
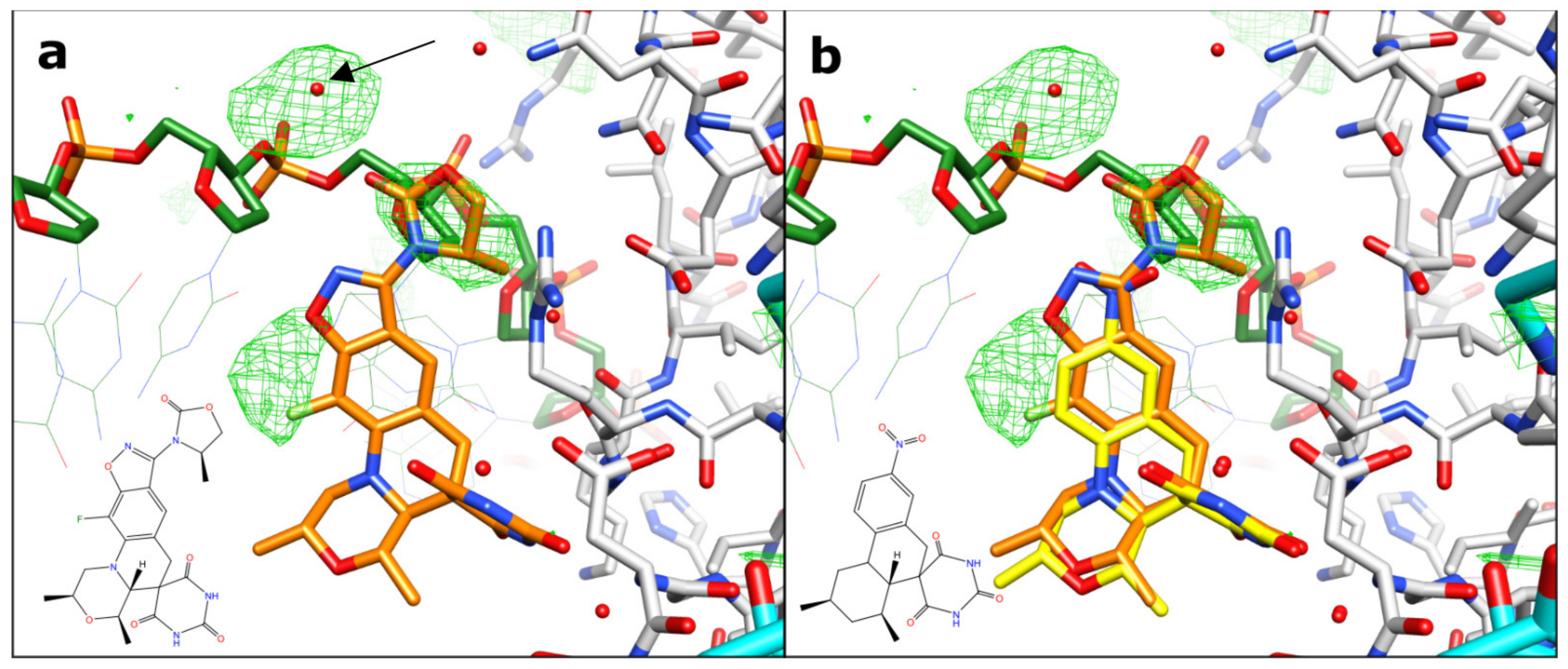
2.1. A 2.8 Å Zoliflodacin DNA Cleavage Complex with S. aureus DNA Gyrase
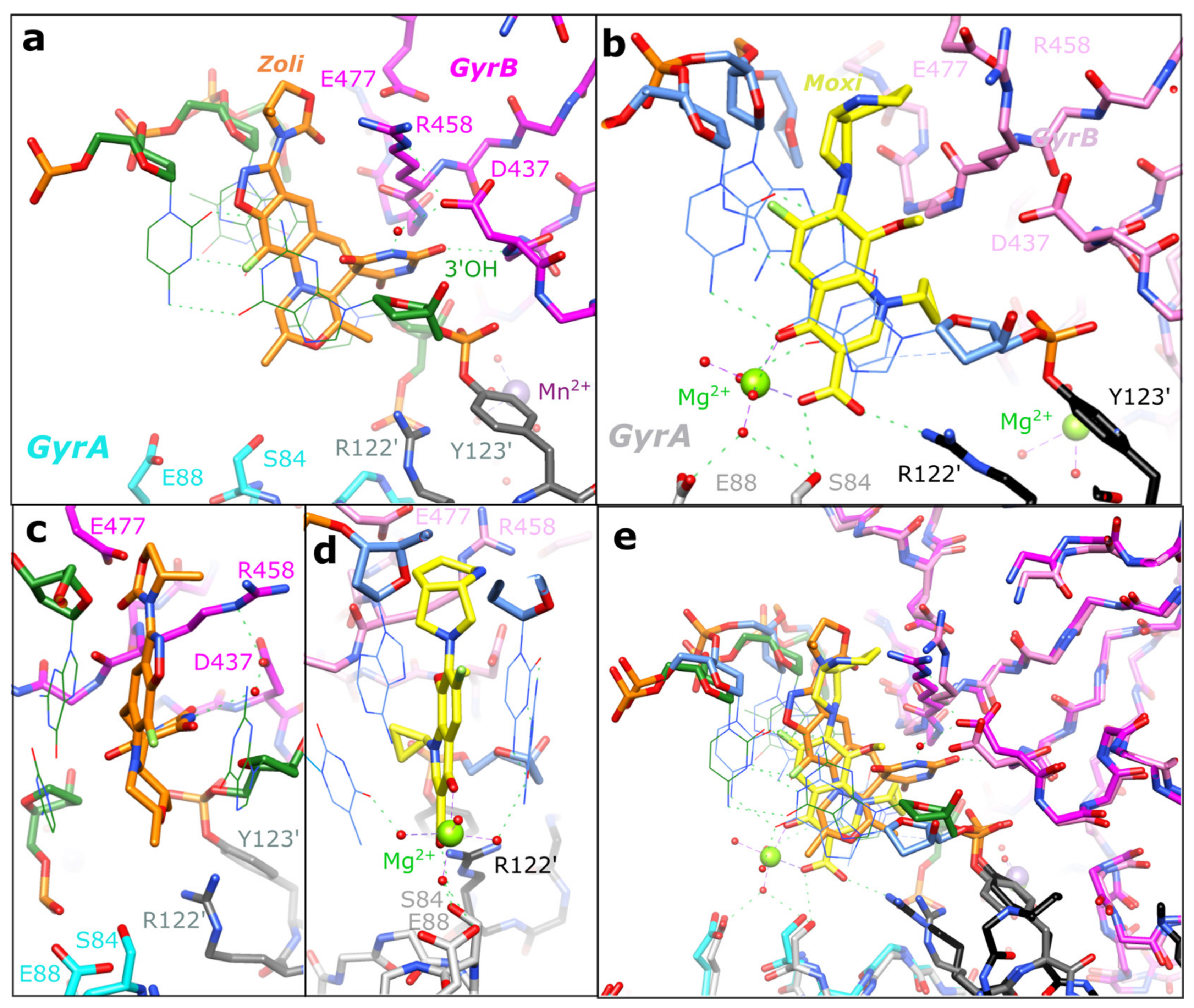
2.2. Zoliflodacin Interacts with GyrB, Whereas Moxifloxacin Interacts with GyrA
2.3. Target Mediated Resistance to Zoliflodacin in N. Gonorrhoeae
2.4. Improved Activity of Zoliflodacin against A. baumannii Compared to QPT-1
3. Discussion
4. Materials and Methods
4.1. Protein Purification and Crystallization of a Zoliflodacin DNA Cleavage Complex
4.2. Data Collection, Structural Determination, and Refinement
4.3. Structural Analysis
4.4. Minimum Inhibitory Concentration Assay
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jacobsson, S.; Golparian, D.; Oxelbark, J.; Alirol, E.; Franceschi, F.; Gustafsson, T.N.; Brown, D.; Louie, A.; Drusano, G.; Unemo, M. Pharmacodynamic Evaluation of Dosing, Bacterial Kill, and Resistance Suppression for Zoliflodacin Against Neisseria gonorrhoeae in a Dynamic Hollow Fiber Infection Model. Front. Pharm. 2021, 12, 682135. [Google Scholar] [CrossRef]
- Unemo, M.; Ahlstrand, J.; Sanchez-Buso, L.; Day, M.; Aanensen, D.; Golparian, D.; Jacobsson, S.; Cole, M.J.; European Collaborative, G. High susceptibility to zoliflodacin and conserved target (GyrB) for zoliflodacin among 1209 consecutive clinical Neisseria gonorrhoeae isolates from 25 European countries, 2018. J. Antimicrob. Chemother. 2021, 76, 1221–1228. [Google Scholar] [CrossRef]
- Bradford, P.A.; Miller, A.A.; O’Donnell, J.; Mueller, J.P. Zoliflodacin: An Oral Spiropyrimidinetrione Antibiotic for the Treatment of Neisseria gonorrheae, Including Multi-Drug-Resistant Isolates. ACS Infect. Dis. 2020, 6, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Jacobsson, S.; Kularatne, R.; Kittiyaowamarn, R.; Maseko, V.; Paopang, P.; Sangprasert, P.; Sirivongrangson, P.; Piddock, L.; Wi, T.; Alirol, E.; et al. High in vitro susceptibility to the first-in-class spiropyrimidinetrione zoliflodacin among consecutive clinical Neisseria gonorrhoeae isolates from Thailand (2018) and South Africa (2015–2017). Antimicrob. Agents Chemother. 2019, 63, e01479-19. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.A.; Bundy, G.L.; Mott, J.E.; Skepner, J.E.; Boyle, T.P.; Harris, D.W.; Hromockyj, A.E.; Marotti, K.R.; Zurenko, G.E.; Munzner, J.B.; et al. Discovery and characterization of QPT-1, the progenitor of a new class of bacterial topoisomerase inhibitors. Antimicrob. Agents Chemother. 2008, 52, 2806–2812. [Google Scholar] [CrossRef] [Green Version]
- Bisacchi, G.S. Origins of the quinolone class of antibacterials: An expanded “discovery story” miniperspective. J. Med. Chem. 2015, 58, 4874–4882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoeffler, A.J.; Berger, J.M. DNA topoisomerases: Harnessing and constraining energy to govern chromosome topology. Q. Rev. Biophys. 2008, 41, 41–101. [Google Scholar] [CrossRef]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef]
- Bates, A.D.; Maxwell, A. DNA Topology; Oxford University Press: Cary, NC, USA, 2005. [Google Scholar]
- Bax, B.D.; Murshudov, G.; Maxwell, A.; Germe, T. DNA Topoisomerase Inhibitors: Trapping a DNA-Cleaving Machine in Motion. J. Mol. Biol. 2019, 431, 3427–3449. [Google Scholar] [CrossRef] [PubMed]
- Bush, N.G.; Diez-Santos, I.; Abbott, L.R.; Maxwell, A. Quinolones: Mechanism, lethality and their contributions to antibiotic resistance. Molecules 2020, 25, 5662. [Google Scholar] [CrossRef]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of quinolone action and resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef]
- Bax, B.D.; Chan, P.F.; Eggleston, D.S.; Fosberry, A.; Gentry, D.R.; Gorrec, F.; Giordano, I.; Hann, M.M.; Hennessy, A.; Hibbs, M.; et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Vanden Broeck, A.; Lotz, C.; Ortiz, J.; Lamour, V. Cryo-EM structure of the complete E. coli DNA gyrase nucleoprotein complex. Nat. Commun. 2019, 10, 4935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.S. Origin of diderm (Gram-negative) bacteria: Antibiotic selection pressure rather than endosymbiosis likely led to the evolution of bacterial cells with two membranes. Antonie Leeuwenhoek 2011, 100, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, M.S.; Paterson, D.L. Antibiotics in the clinical pipeline in October 2019. J. Antibiot. 2020, 73, 329–364. [Google Scholar] [CrossRef]
- Butler, M.S.; Blaskovich, M.A.; Cooper, M.A. Antibiotics in the clinical pipeline in 2013. J. Antibiot. 2013, 66, 571–591. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.S.; Cooper, M.A. Antibiotics in the clinical pipeline in 2011. J. Antibiot. 2011, 64, 413–425. [Google Scholar] [CrossRef]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef]
- Tommasi, R.; Brown, D.G.; Walkup, G.K.; Manchester, J.I.; Miller, A.A. ESKAPEing the labyrinth of antibacterial discovery. Nat. Rev. Drug Discov. 2015, 14, 529–542. [Google Scholar] [CrossRef]
- Scangarella-Oman, N.E.; Hossain, M.; Hoover, J.L.; Perry, C.R.; Tiffany, C.; Barth, A.; Dumont, E.F. Dose Selection for Phase III Clinical Evaluation of Gepotidacin (GSK2140944) in the Treatment of Uncomplicated Urinary Tract Infections. Antimicrob. Agents Chemother. 2022, 66, e01492-21. [Google Scholar] [CrossRef]
- Miles, T.J.; Hennessy, A.J.; Bax, B.; Brooks, G.; Brown, B.S.; Brown, P.; Cailleau, N.; Chen, D.; Dabbs, S.; Davies, D.T. Novel tricyclics (eg, GSK945237) as potent inhibitors of bacterial type IIA topoisomerases. Bioorg. Med. Chem. Lett. 2016, 26, 2464–2469. [Google Scholar] [CrossRef]
- Miles, T.J.; Hennessy, A.J.; Bax, B.; Brooks, G.; Brown, B.S.; Brown, P.; Cailleau, N.; Chen, D.; Dabbs, S.; Davies, D.T. Novel hydroxyl tricyclics (eg, GSK966587) as potent inhibitors of bacterial type IIA topoisomerases. Bioorg. Med. Chem. Lett. 2013, 23, 5437–5441. [Google Scholar] [CrossRef] [PubMed]
- Gibson, E.G.; Bax, B.; Chan, P.F.; Osheroff, N. Mechanistic and Structural Basis for the Actions of the Antibacterial Gepotidacin against Staphylococcus aureus Gyrase. ACS Infect. Dis. 2019, 5, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Kolaric, A.; Germe, T.; Hrast, M.; Stevenson, C.E.M.; Lawson, D.M.; Burton, N.P.; Voros, J.; Maxwell, A.; Minovski, N.; Anderluh, M. Potent DNA gyrase inhibitors bind asymmetrically to their target using symmetrical bifurcated halogen bonds. Nat. Commun. 2021, 12, 150. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Kaelin, D.E.; Wu, J.; Miesel, L.; Tan, C.M.; Black, T.; Nargund, R.; Meinke, P.T.; Olsen, D.B.; Lagrutta, A.; et al. Tricyclic 1,5-naphthyridinone oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad-spectrum antibacterial agents-SAR of left-hand-side moiety (Part-2). Bioorg. Med. Chem. Lett. 2015, 25, 1831–1835. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Kaelin, D.E.; Wu, J.; Miesel, L.; Tan, C.M.; Meinke, P.T.; Olsen, D.; Lagrutta, A.; Bradley, P.; Lu, J.; et al. Oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad spectrum antibacterial agents. ACS Med. Chem. Lett. 2014, 5, 609–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tashiro, S.; Mihara, T.; Sasaki, M.; Shimamura, C.; Shimamura, R.; Suzuki, S.; Yoshikawa, M.; Hasegawa, T.; Enoki, Y.; Taguchi, K. Oral fidaxomicin versus vancomycin for the treatment of Clostridioides difficile infection: A systematic review and meta-analysis of randomized controlled trials. J. Infect. Chemother. 2022, 28, 1536–1545. [Google Scholar] [CrossRef]
- Butler, M.S.; Blaskovich, M.A.; Cooper, M.A. Antibiotics in the clinical pipeline at the end of 2015. J. Antibiot. 2017, 70, 3–24. [Google Scholar] [CrossRef]
- Lockshon, D.; Morris, D.R. Sites of reaction of Escherichia coli DNA gyrase on pBR322 in vivo as revealed by oxolinic acid-induced plasmid linearization. J. Mol. Biol. 1985, 181, 63–74. [Google Scholar] [CrossRef]
- Leo, E.; Gould, K.A.; Pan, X.-S.; Capranico, G.; Sanderson, M.R.; Palumbo, M.; Fisher, L.M. Novel symmetric and asymmetric DNA scission determinants for Streptococcus pneumoniae topoisomerase IV and gyrase are clustered at the DNA breakage site. J. Biol. Chem. 2005, 280, 14252–14263. [Google Scholar] [CrossRef] [Green Version]
- Wohlkonig, A.; Chan, P.F.; Fosberry, A.P.; Homes, P.; Huang, J.; Kranz, M.; Leydon, V.R.; Miles, T.J.; Pearson, N.D.; Perera, R.L.; et al. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol. 2010, 17, 1152–1153. [Google Scholar] [CrossRef]
- Chan, P.F.; Srikannathasan, V.; Huang, J.; Cui, H.; Fosberry, A.P.; Gu, M.; Hann, M.M.; Hibbs, M.; Homes, P.; Ingraham, K.; et al. Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nat. Commun. 2015, 6, 10048. [Google Scholar] [CrossRef] [Green Version]
- Blower, T.R.; Williamson, B.H.; Kerns, R.J.; Berger, J.M. Crystal structure and stability of gyrase-fluoroquinolone cleaved complexes from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2016, 113, 1706–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laponogov, I.; Sohi, M.K.; Veselkov, D.A.; Pan, X.S.; Sawhney, R.; Thompson, A.W.; McAuley, K.E.; Fisher, L.M.; Sanderson, M.R. Structural insight into the quinolone-DNA cleavage complex of type IIA topoisomerases. Nat. Struct. Mol. Biol. 2009, 16, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Laponogov, I.; Pan, X.S.; Veselkov, D.A.; McAuley, K.E.; Fisher, L.M.; Sanderson, M.R. Structural basis of gate-DNA breakage and resealing by type II topoisomerases. PLoS ONE 2010, 5, e11338. [Google Scholar] [CrossRef]
- Srikannathasan, V.; Wohlkonig, A.; Shillings, A.; Singh, O.; Chan, P.F.; Huang, J.; Gwynn, M.N.; Fosberry, A.P.; Homes, P.; Hibbs, M.; et al. Crystallization and preliminary X-ray crystallographic analysis of covalent DNA cleavage complexes of Staphyloccocus Aureus DNA Gyrase with QPT-1, Moxilfloxacin and Etoposide. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2015, 71, 1242–1246. [Google Scholar] [CrossRef] [Green Version]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Basarab, G.S.; Ghorpade, S.; Gibhard, L.; Mueller, R.; Njoroge, M.; Peton, N.; Govender, P.; Massoudi, L.M.; Robertson, G.T.; Lenaerts, A.J. Spiropyrimidinetriones: A class of DNA gyrase inhibitors with activity against Mycobacterium tuberculosis and without cross-resistance to fluoroquinolones. Antimicrob. Agents Chemother. 2022, 66, e02192-21. [Google Scholar] [CrossRef]
- Govender, P.; Müller, R.; Singh, K.; Reddy, V.; Eyermann, C.J.; Fienberg, S.; Ghorpade, S.R.; Koekemoer, L.; Myrick, A.; Schnappinger, D. Spiropyrimidinetrione DNA Gyrase Inhibitors with Potent and Selective Antituberculosis Activity. J. Med. Chem. 2022, 65, 6903–6925. [Google Scholar] [CrossRef] [PubMed]
- Al-Kadmy, I.M.; Ibrahim, S.A.; Al-Saryi, N.; Aziz, S.N.; Besinis, A.; Hetta, H.F. Prevalence of genes involved in colistin resistance in Acinetobacter baumannii: First report from Iraq. Microb. Drug Resist. 2020, 26, 616–622. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst. D Biol. Cryst. 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Aldred, K.J.; McPherson, S.A.; Turnbough, C.L., Jr.; Kerns, R.J.; Osheroff, N. Topoisomerase IV-quinolone interactions are mediated through a water-metal ion bridge: Mechanistic basis of quinolone resistance. Nucleic Acids Res. 2013, 41, 4628–4639. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; McPherson, S.A.; Wang, P.; Kerns, R.J.; Graves, D.E.; Turnbough, C.L., Jr.; Osheroff, N. Drug interactions with Bacillus anthracis topoisomerase IV: Biochemical basis for quinolone action and resistance. Biochemistry 2012, 51, 370–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alm, R.A.; Lahiri, S.D.; Kutschke, A.; Otterson, L.G.; McLaughlin, R.E.; Whiteaker, J.D.; Lewis, L.A.; Su, X.; Huband, M.D.; Gardner, H.; et al. Characterization of the Novel DNA Gyrase Inhibitor AZD0914: Low Resistance Potential and Lack of Cross-Resistance in Neisseria gonorrhoeae. Antimicrob. Agents Chemother. 2015, 59, 1478–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bax, B.; Chung, C.W.; Edge, C. Getting the chemistry right: Protonation, tautomers and the importance of H atoms in biological chemistry. Acta Cryst. D Struct. Biol. 2017, 73, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Fogg, J.M.; Randall, G.L.; Pettitt, B.M.; Sumners, W.L.D.; Harris, S.A.; Zechiedrich, L. Bullied no more: When and how DNA shoves proteins around. Q. Rev. Biophys. 2012, 45, 257–299. [Google Scholar] [CrossRef] [Green Version]
- Gibson, E.G.; Oviatt, A.A.; Cacho, M.; Neuman, K.C.; Chan, P.F.; Osheroff, N. Bimodal actions of a naphthyridone/aminopiperidine-based antibacterial that targets gyrase and topoisomerase IV. Biochemistry 2019, 58, 4447–4455. [Google Scholar] [CrossRef]
- Sutormin, D.; Rubanova, N.; Logacheva, M.; Ghilarov, D.; Severinov, K. Single-nucleotide-resolution mapping of DNA gyrase cleavage sites across the Escherichia coli genome. Nucleic Acids Res. 2019, 47, 1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamson, P.C.; Lin, E.Y.; Ha, S.M.; Klausner, J.D. Using a public database of Neisseria gonorrhoeae genomes to detect mutations associated with zoliflodacin resistance. J. Antimicrob. Chemother. 2021, 76, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Foerster, S.; Drusano, G.; Golparian, D.; Neely, M.; Piddock, L.J.V.; Alirol, E.; Unemo, M. In vitro antimicrobial combination testing of and evolution of resistance to the first-in-class spiropyrimidinetrione zoliflodacin combined with six therapeutically relevant antimicrobials for Neisseria gonorrhoeae. J. Antimicrob. Chemother. 2019, 74, 3521–3529. [Google Scholar] [CrossRef] [PubMed]
- Foerster, S.; Golparian, D.; Jacobsson, S.; Hathaway, L.J.; Low, N.; Shafer, W.M.; Althaus, C.L.; Unemo, M. Genetic resistance determinants, in vitro time-kill curve analysis and pharmacodynamic functions for the novel topoisomerase II inhibitor ETX0914 (AZD0914) in Neisseria gonorrhoeae. Front. Microbiol. 2015, 6, 1377. [Google Scholar] [CrossRef] [Green Version]
- Scangarella-Oman, N.E.; Ingraham, K.A.; Tiffany, C.A.; Tomsho, L.; Van Horn, S.F.; Mayhew, D.N.; Perry, C.R.; Ashton, T.C.; Dumont, E.F.; Huang, J.; et al. In Vitro Activity and Microbiological Efficacy of Gepotidacin from a Phase 2, Randomized, Multicenter, Dose-Ranging Study in Patients with Acute Bacterial Skin and Skin Structure Infections. Antimicrob. Agents Chemother. 2020, 64, e01302-19. [Google Scholar] [CrossRef] [Green Version]
- Hooper, D.C.; Rubinstein, E. (Eds.) Quinolone Antimicrobial Agents; ASM Press: Washington, DC, USA, 2003. [Google Scholar]
- Coelho, J.M.; Turton, J.F.; Kaufmann, M.E.; Glover, J.; Woodford, N.; Warner, M.; Palepou, M.-F.; Pike, R.; Pitt, T.L.; Patel, B.C. Occurrence of carbapenem-resistant Acinetobacter baumannii clones at multiple hospitals in London and Southeast England. J. Clin. Microbiol. 2006, 44, 3623–3627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damier-Piolle, L.; Magnet, S.; Brémont, S.; Lambert, T.; Courvalin, P. AdeIJK, a resistance-nodulation-cell division pump effluxing multiple antibiotics in Acinetobacter baumannii. Antimicrob. Agents Chemother. 2008, 52, 557–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colclough, A.L.; Alav, I.; Whittle, E.E.; Pugh, H.L.; Darby, E.M.; Legood, S.W.; McNeil, H.E.; Blair, J.M. RND efflux pumps in Gram-negative bacteria; regulation, structure and role in antibiotic resistance. Future Microbiol. 2020, 15, 143–157. [Google Scholar] [CrossRef]
- Lowrence, R.C.; Subramaniapillai, S.G.; Ulaganathan, V.; Nagarajan, S. Tackling drug resistance with efflux pump inhibitors: From bacteria to cancerous cells. Crit. Rev. Microbiol. 2019, 45, 334–353. [Google Scholar] [CrossRef]
- Imai, Y.; Hauk, G.; Quigley, J.; Liang, L.; Son, S.; Ghiglieri, M.; Gates, M.F.; Morrissette, M.; Shahsavari, N.; Niles, S. Evybactin is a DNA gyrase inhibitor that selectively kills Mycobacterium tuberculosis. Nat. Chem. Biol. 2022, 18, 1236–1244. [Google Scholar] [CrossRef]
- Chan, P.F.; Germe, T.; Bax, B.D.; Huang, J.; Thalji, R.K.; Bacqué, E.; Checchia, A.; Chen, D.; Cui, H.; Ding, X. Thiophene antibacterials that allosterically stabilize DNA-cleavage complexes with DNA gyrase. Proc. Natl. Acad. Sci. USA 2017, 114, E4492–E4500. [Google Scholar] [CrossRef] [Green Version]
- Thalji, R.K.; Raha, K.; Andreotti, D.; Checchia, A.; Cui, H.; Meneghelli, G.; Profeta, R.; Tonelli, F.; Tommasi, S.; Bakshi, T.; et al. Structure-guided design of antibacterials that allosterically inhibit DNA gyrase. Bioorg. Med. Chem. Lett. 2019, 29, 1407–1412. [Google Scholar] [CrossRef] [Green Version]
- Gubaev, A.; Weidlich, D.; Klostermeier, D. DNA gyrase with a single catalytic tyrosine can catalyze DNA supercoiling by a nicking-closing mechanism. Nucleic Acids Res. 2016, 44, 10354–10366. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.; Beilsten-Edmands, J.; Devenish, N.; Gerstel, M.; Gildea, R.J.; McDonagh, D.; Pascal, E.; Waterman, D.G.; Williams, B.H.; Evans, G. DIALS as a toolkit. Protein Sci. 2022, 31, 232–250. [Google Scholar] [CrossRef] [PubMed]
- Beilsten-Edmands, J.; Winter, G.; Gildea, R.; Parkhurst, J.; Waterman, D.; Evans, G. Scaling diffraction data in the DIALS software package: Algorithms and new approaches for multi-crystal scaling. Acta Crystallogr. Sect. D Struct. Biol. 2020, 76, 385–399. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.; Waterman, D.G.; Parkhurst, J.M.; Brewster, A.S.; Gildea, R.J.; Gerstel, M.; Fuentes-Montero, L.; Vollmar, M.; Michels-Clark, T.; Young, I.D.; et al. DIALS: Implementation and evaluation of a new integration package. Acta Cryst. D Struct. Biol. 2018, 74, 85–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Cryst. 2013, 69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Poon, B.K.; Read, R.J.; Sobolev, O.V.; Terwilliger, T.C.; Urzhumtsev, A.; Adams, P.D. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Cryst. D Struct. Biol. 2018, 74, 531–544. [Google Scholar] [CrossRef] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Cryst. D Biol. Cryst. 2012, 68, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Kovalevskiy, O.; Nicholls, R.A.; Long, F.; Carlon, A.; Murshudov, G.N. Overview of refinement procedures within REFMAC5: Utilizing data from different sources. Acta Cryst. D Struct. Biol. 2018, 74, 215–227. [Google Scholar] [CrossRef]
- Long, F.; Nicholls, R.A.; Emsley, P.; Graaeulis, S.; Merkys, A.; Vaitkus, A.; Murshudov, G.N. AceDRG: A stereochemical description generator for ligands. Acta Cryst. D Struct. Biol. 2017, 73, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, K.S.D.; Gurusaran, M.; Satheesh, S.N.; Radha, P.; Pavithra, S.; Thulaa Tharshan, K.P.S.; Helliwell, J.R.; Sekar, K. Online_DPI: A web server to calculate the diffraction precision index for a protein structure. J. Appl. Crystallogr. 2015, 48, 939–942. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Cryst. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Cryst. D Biol. Cryst. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- International Organization for Standardization. Susceptibility Testing of Infectious Agents and Evaluation of Performance of Antimicrobial Susceptibility Test Devices—Part 1: Broth Micro-Dilution Reference Method for Testing the In Vitro Activity of Antimicrobial Agents against Rapidly Growing Aerobic Bacteria Involved in Infectious Diseases; International Organization for Standardization: Geneva, Switzerland, 2019. [Google Scholar]
- EUCAST. MIC Determination of Non-fastidious and Fastidious Organisms. 2022. Available online: https://www.eucast.org/ast_of_bacteria/mic_determination (accessed on 9 November 2022).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Species | MIC (µg/mL) |
|---|---|---|
| Zoliflodacin | A. baumannii BCC 807 (UK OXA-23 clone) | 4 |
| Zoliflodacin | A. baumannii BCC 810 (South East OXA-23 clone) | 4 |
| QPT-1 QPT-1 | A. baumannii BM4454A. baumannii BM4454 (ΔadeABC ΔadeIJK) * | 16 0.125 |
| ZOL-2.8 | |
|---|---|
| PDB code | 8BP2 |
| Diffraction source | I24, DLS |
| Wavelength (Å) | 0.9999 |
| Resolution range (Å) | 24.85–2.78 (2.83–2.78) * |
| Space group | P61 |
| Unit cell | 94.54, 94.54, 417.13, 90, 90, 120 |
| Total reflections | 974,457 (46,716) * |
| Unique reflections | 52,704 (2554) * |
| Multiplicity | 18.5 (18.3) * |
| Completeness (%) | 100.0 (100.0) * |
| Mean I/sigma(I) | 5.9 (0.4) * |
| Wilson Bfactor | 62.4 |
| Rmerge | 0.286 (3.872) * |
| Rmeas | 0.294 (3.980) * |
| Rpim | 0.067 (1.297) * |
| CC1/2 | 0.997 (0.318) * |
| ZOL-2.8 | |
|---|---|
| PDB code | 8BP2 |
| Resolution range (Å) | 24.85–2.80 (2.87–2.80) |
| Completeness (%) | 99.41 (94.60) |
| No. of reflections, working set | 48,785 (3421) |
| No. of reflections, test set | 2538 (185) |
| Final Rcryst | 0.1957 (0.372) |
| Final Rfree | 0.2375 (0.374) |
| Cruickshank DPI (Å) * | 0.325 |
| No. of non-H atoms (total) | 11,713 |
| Protein | 10,574 |
| DNA | 801 |
| Zoliflodacin | 70 |
| Other ligands (Mn, glycerol etc.) | 42 |
| Water molecules | 226 |
| RMS deviations | |
| Bonds (Å) | 0.009 |
| Angles (°) | 1.569 |
| Average B factors (Å2) | |
| Protein | 96.024 |
| DNA | 85.103 |
| Zoliflodacin | 84.491 |
| Other ligands (Mn, glycerol etc.) | 88.911 |
| Waters ** | 75.396 |
| Ramachandran plot | |
| Favored regions | 96% |
| Additionally allowed | 4% |
| Outliers | 0% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, H.; Lipka-Lloyd, M.; Warren, A.J.; Hughes, N.; Holmes, J.; Burton, N.P.; Mahenthiralingam, E.; Bax, B.D. A 2.8 Å Structure of Zoliflodacin in a DNA Cleavage Complex with Staphylococcus aureus DNA Gyrase. Int. J. Mol. Sci. 2023, 24, 1634. https://doi.org/10.3390/ijms24021634
Morgan H, Lipka-Lloyd M, Warren AJ, Hughes N, Holmes J, Burton NP, Mahenthiralingam E, Bax BD. A 2.8 Å Structure of Zoliflodacin in a DNA Cleavage Complex with Staphylococcus aureus DNA Gyrase. International Journal of Molecular Sciences. 2023; 24(2):1634. https://doi.org/10.3390/ijms24021634
Chicago/Turabian StyleMorgan, Harry, Magdalena Lipka-Lloyd, Anna J. Warren, Naomi Hughes, John Holmes, Nicolas P. Burton, Eshwar Mahenthiralingam, and Ben D. Bax. 2023. "A 2.8 Å Structure of Zoliflodacin in a DNA Cleavage Complex with Staphylococcus aureus DNA Gyrase" International Journal of Molecular Sciences 24, no. 2: 1634. https://doi.org/10.3390/ijms24021634