
Identification of Aging and Young Subtypes for Predicting Colorectal Cancer Prognosis and Immunotherapy Responses
Abstract
:1. Introduction
2. Results
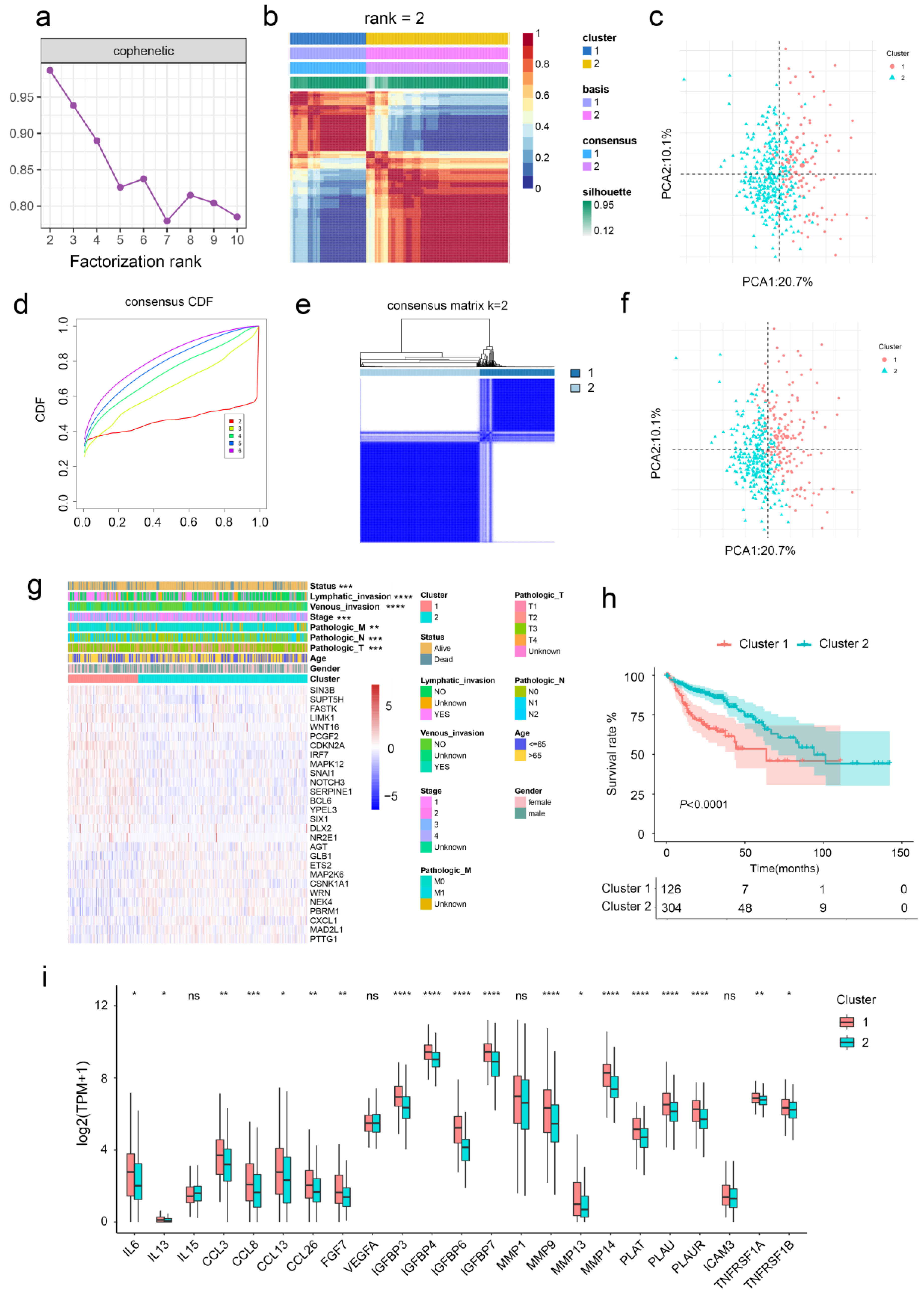
2.1. Aging-Related Genes-Based Clustering and Subtypes Identification
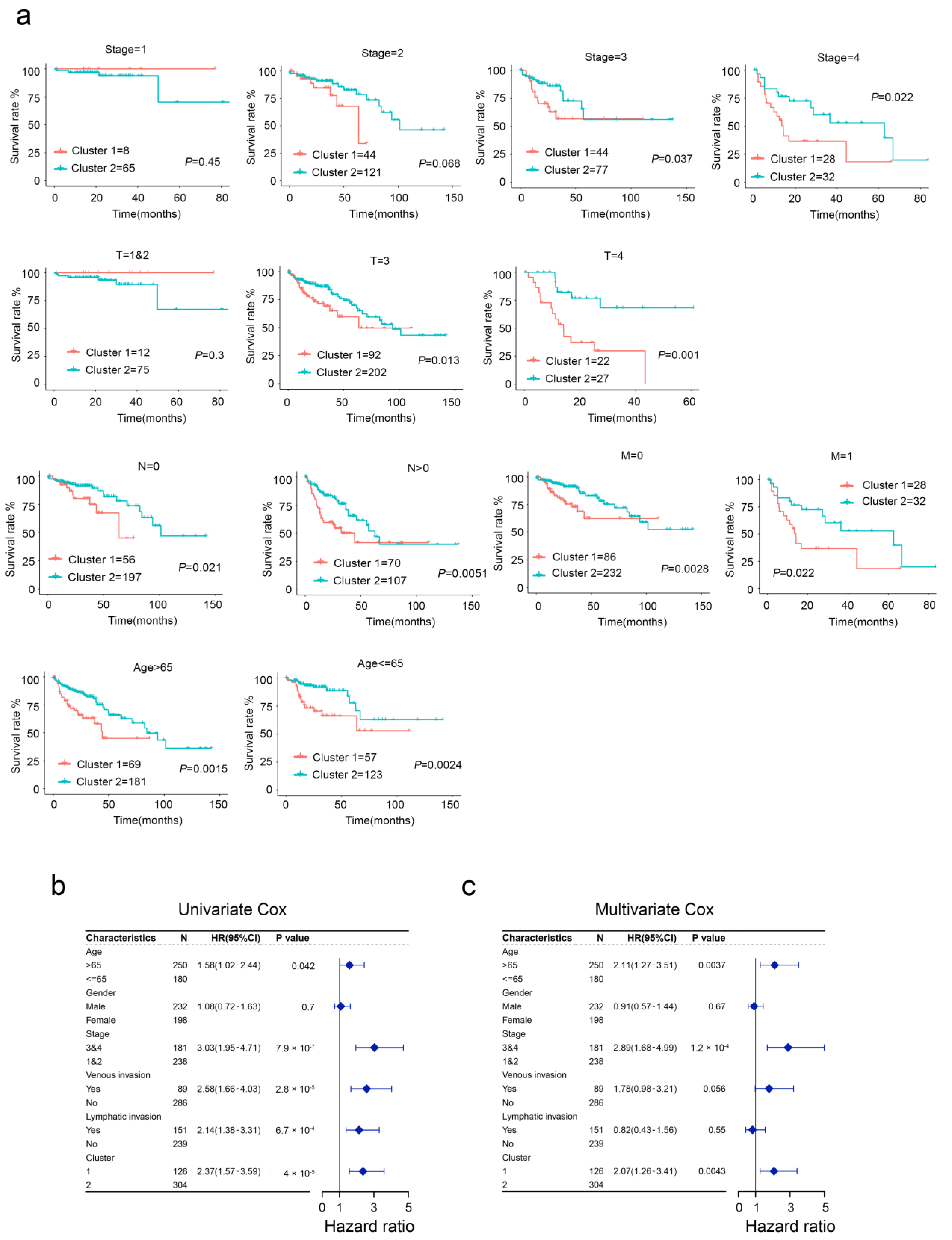
2.2. Clinical Characteristics of Aging-Related Genes-Based Subtypes
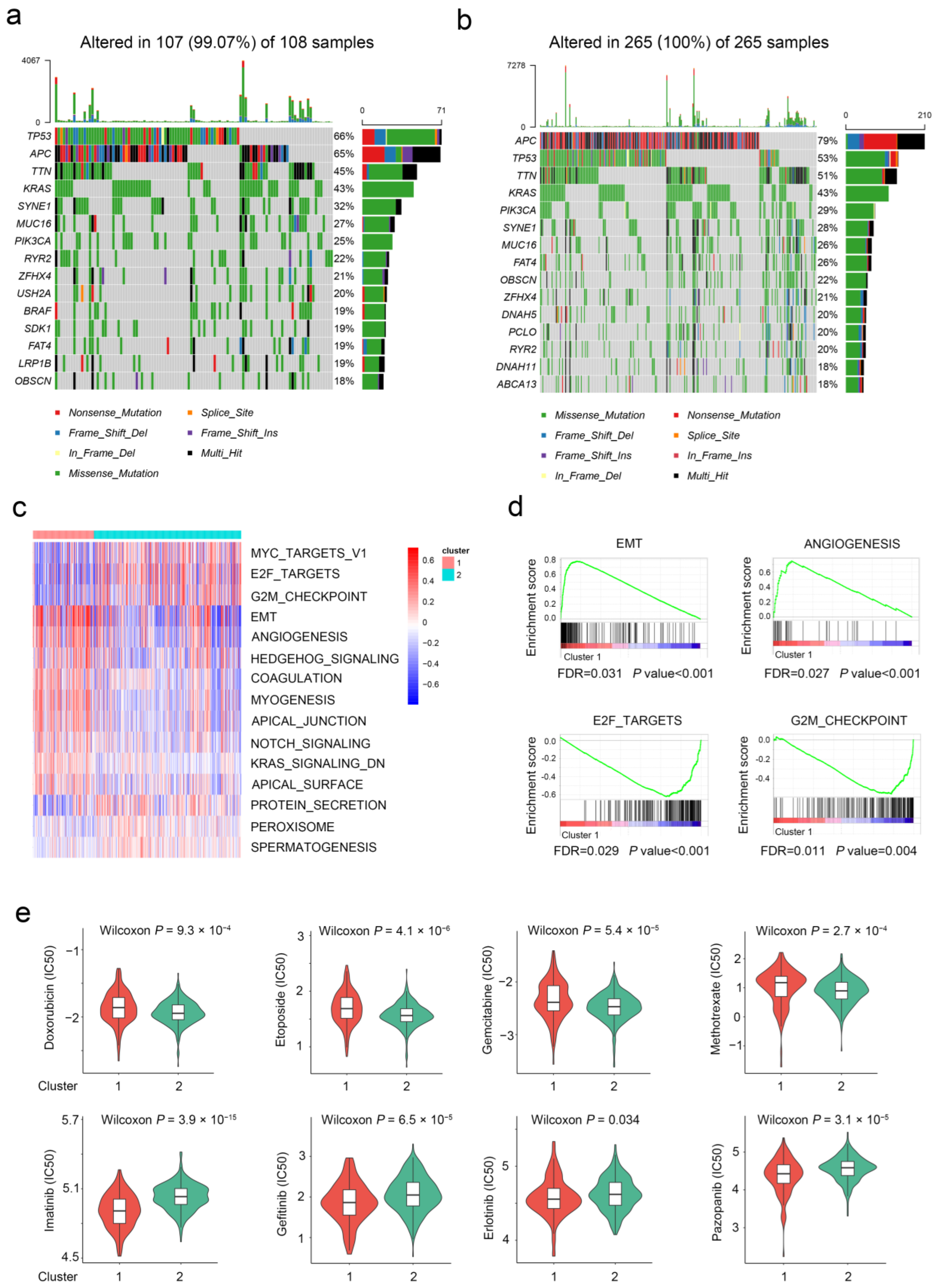
2.3. The Difference in Molecular Characteristics between Aging-Related Genes-Based Subtypes
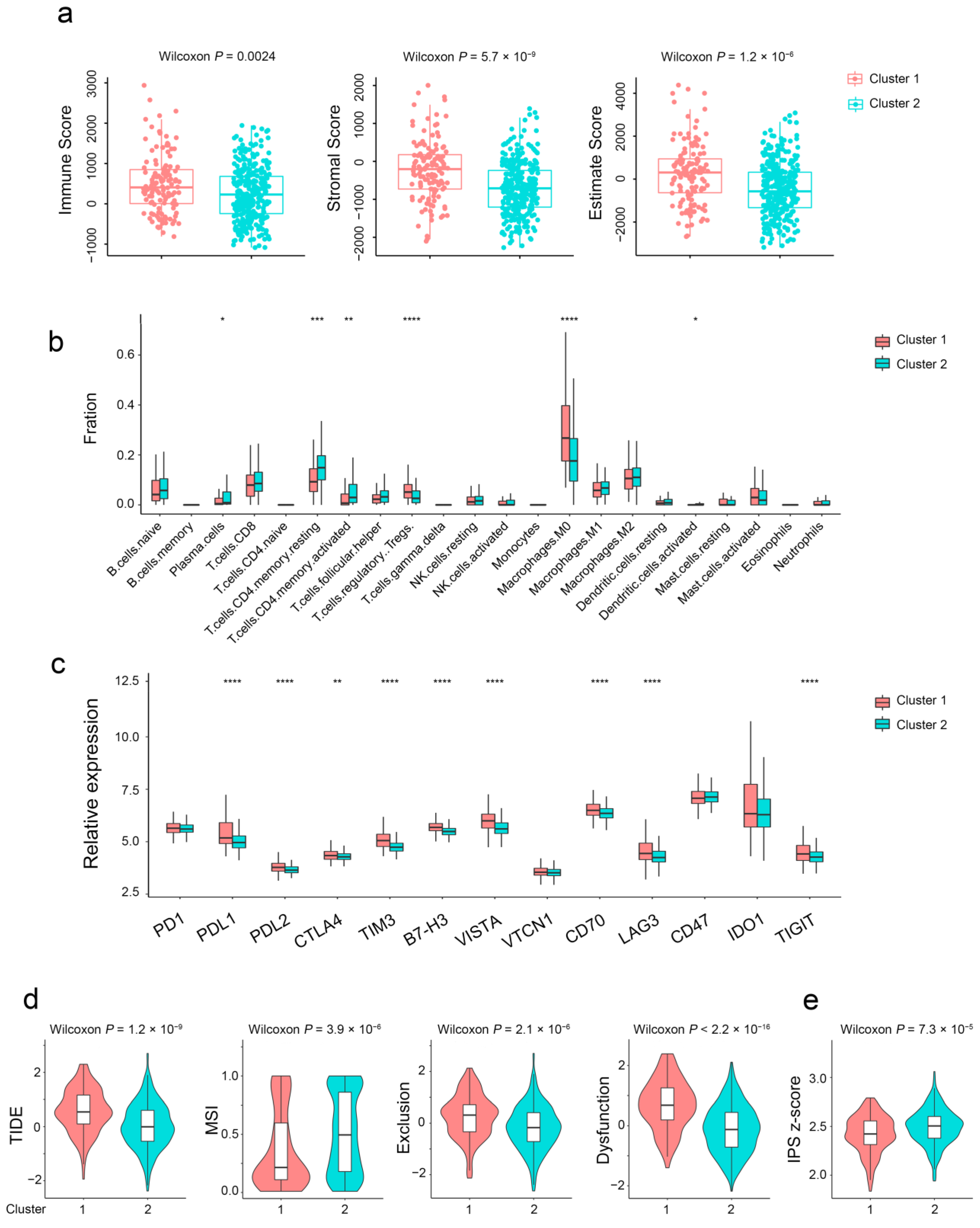
2.4. Aging-Related Genes-Based Subtypes Show Different Immune Features
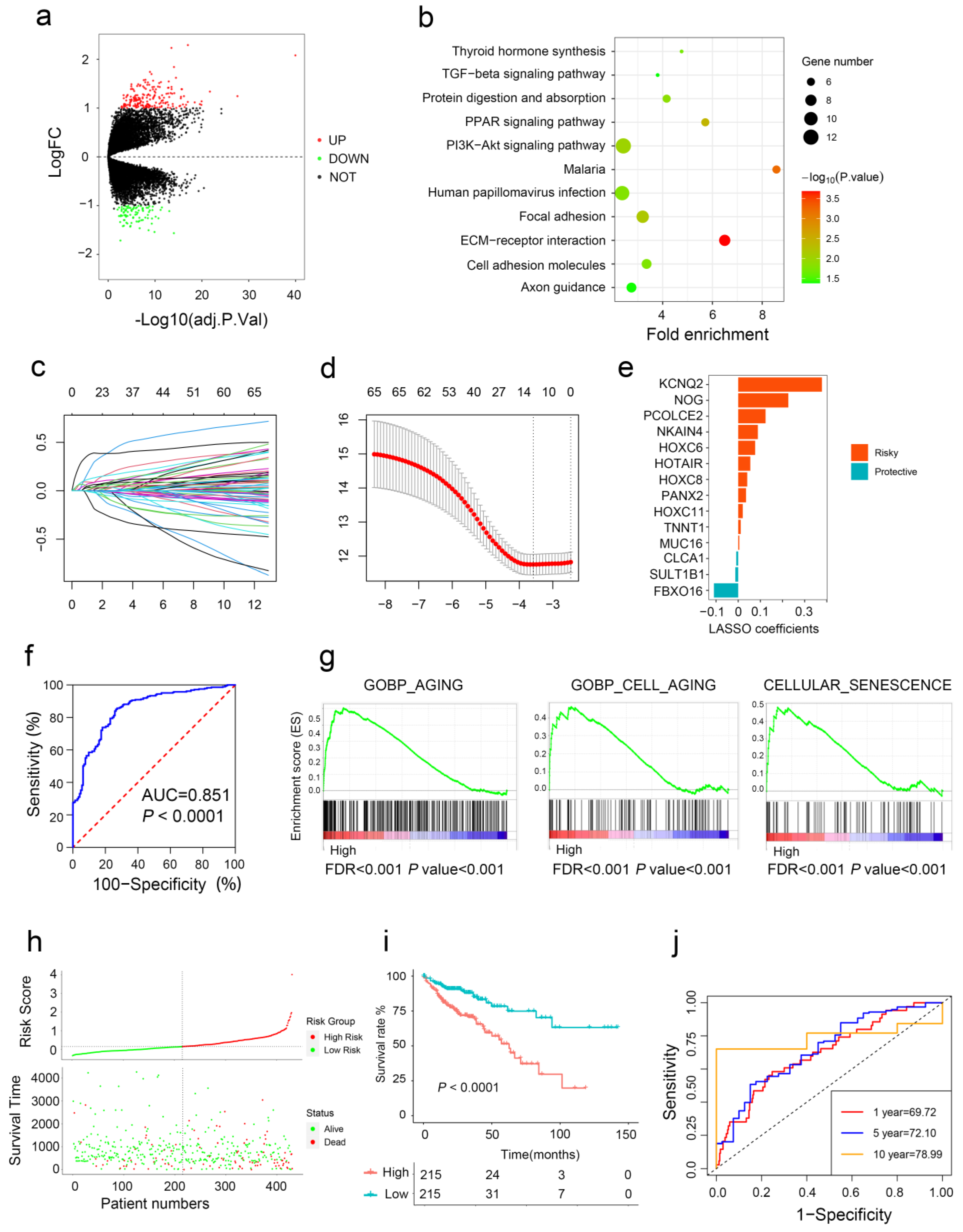
2.5. Aging-Related Gene Signature Is a Prediction Tool for CRC Prognosis
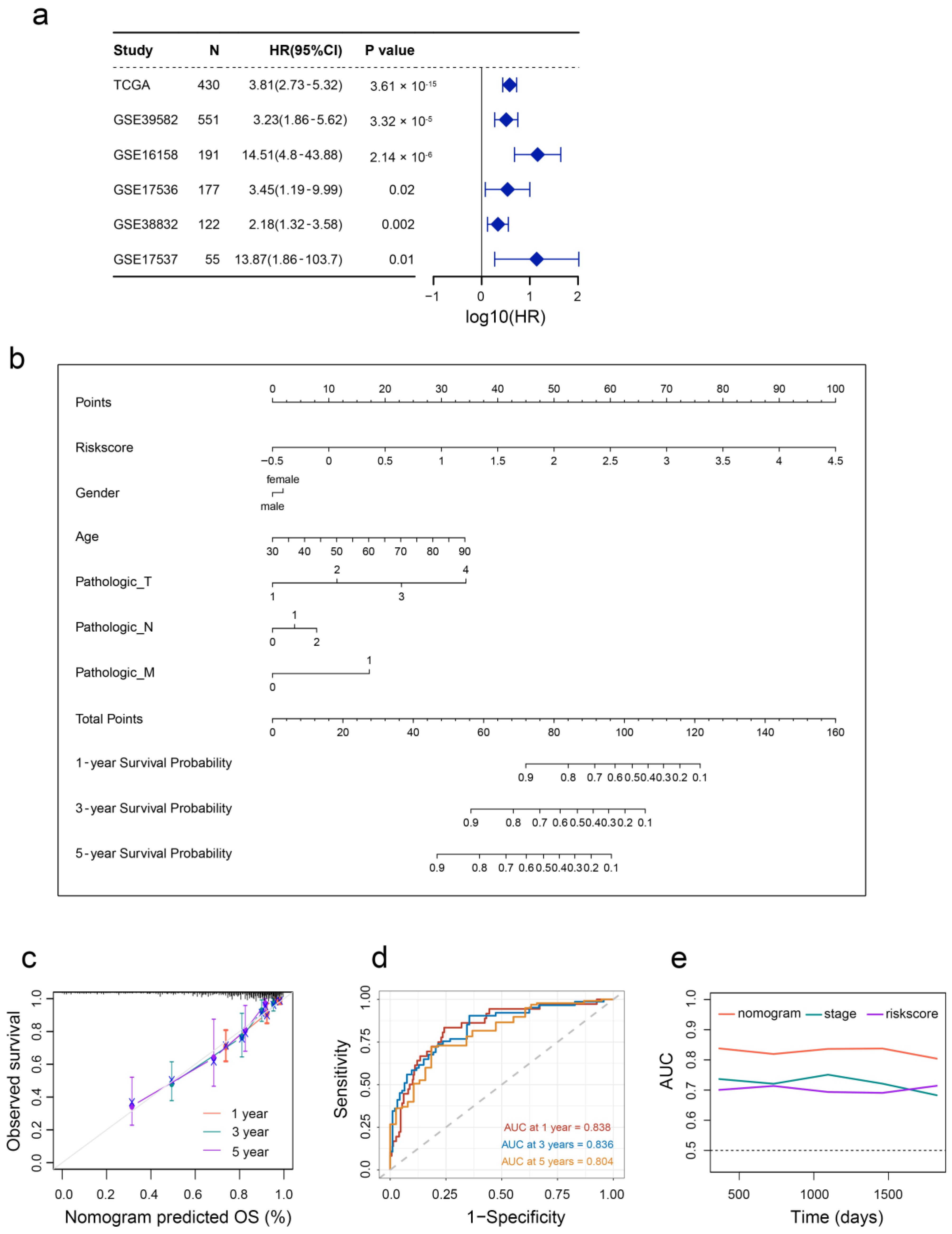
2.6. Aging-Based Nomogram Improves Survival Prediction for CRC
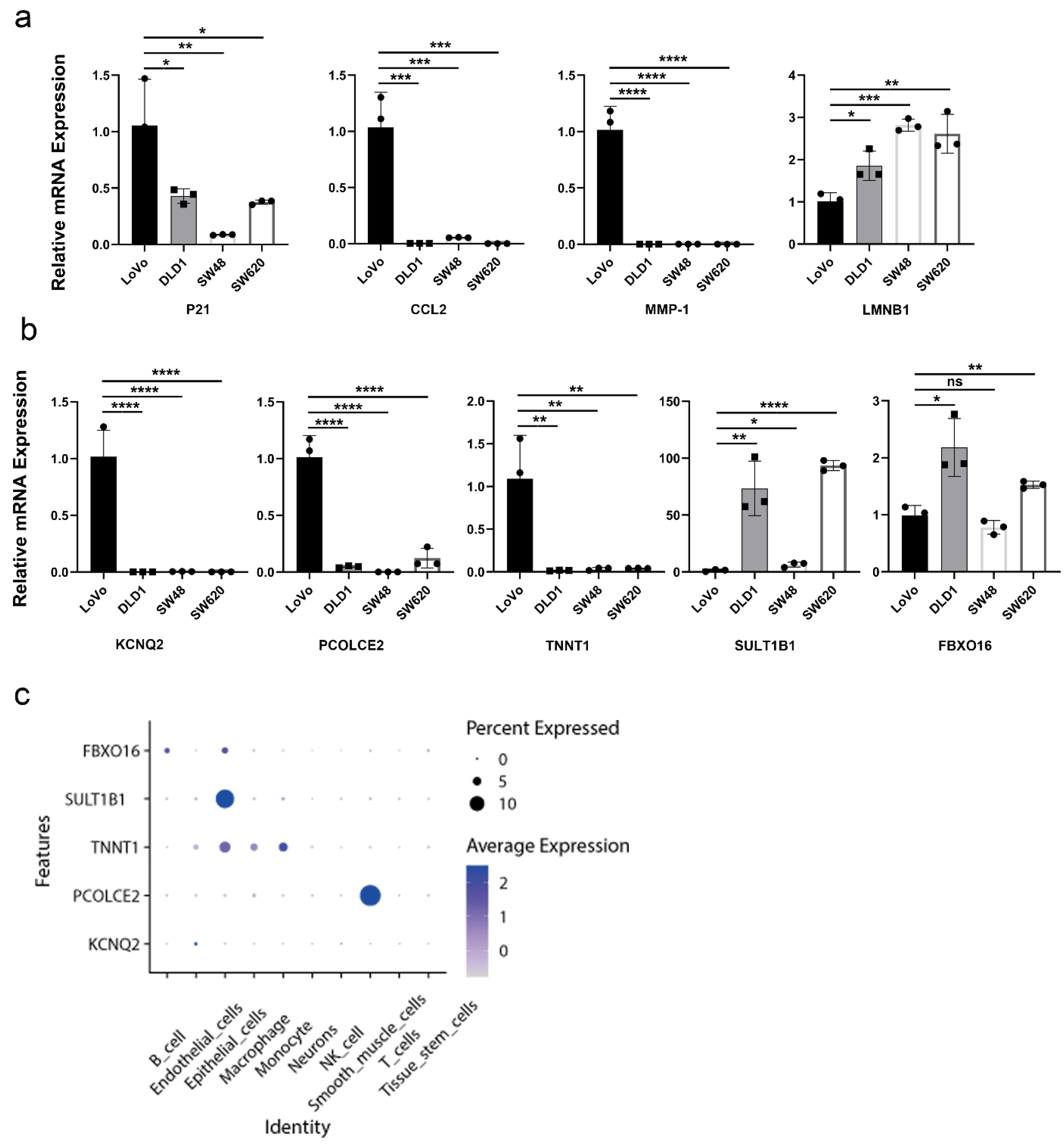
2.7. Validation of Aging-Related Genes
3. Discussion
4. Materials and Methods
4.1. Data Preprocessing
4.2. Unsupervised Clustering Analysis of Aging-Related Genes
4.3. Clinicopathological Features between the Aging and Young Subtypes
4.4. Immune Infiltration Analysis
4.5. Differentially Expressed Genes (DEGs) Identification
4.6. Construction of Aging-Related Gene Signature
4.7. Quantitative Real-Time Polymerase Chain Reaction PCR (RT-qPCR)
4.8. Single-Cell RNA-Seq Data Analysis
4.9. Additional Bioinformatic and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Ostos, L.C.; Lannon, W.A.; Grotzinger, C.; Del Rio, M.; et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bramsen, J.B.; Rasmussen, M.H.; Ongen, H.; Mattesen, T.B.; Orntoft, M.W.; Arnadottir, S.S.; Sandoval, J.; Laguna, T.; Vang, S.; Oster, B.; et al. Molecular-Subtype-Specific Biomarkers Improve Prediction of Prognosis in Colorectal Cancer. Cell Rep. 2017, 19, 1268–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Chen, J.; Yang, Y.; Hu, W. Hypoxia Correlates with Poor Survival and M2 Macrophage Infiltration in Colorectal Cancer. Front. Oncol. 2020, 10, 566430. [Google Scholar] [CrossRef]
- Lin, D.; Fan, W.; Zhang, R.; Zhao, E.; Li, P.; Zhou, W.; Peng, J.; Li, L. Molecular subtype identification and prognosis stratification by a metabolism-related gene expression signature in colorectal cancer. J. Transl. Med. 2021, 19, 279. [Google Scholar] [CrossRef]
- Lai, M.; Liu, Q.; Chen, W.; Qi, X.; Yang, J.; Jiang, L.; Yuan, M.; Liu, Z.; He, Q.; Cao, J.; et al. Identification and Validation of Two Heterogeneous Molecular Subtypes and a Prognosis Predictive Model for Hepatocellular Carcinoma Based on Pyroptosis. Oxid. Med. Cell. Longev. 2022, 2022, 8346816. [Google Scholar] [CrossRef]
- Soldevilla, B.; Carretero-Puche, C.; Gomez-Lopez, G.; Al-Shahrour, F.; Riesco, M.C.; Gil-Calderon, B.; Alvarez-Vallina, L.; Espinosa-Olarte, P.; Gomez-Esteves, G.; Rubio-Cuesta, B.; et al. The correlation between immune subtypes and consensus molecular subtypes in colorectal cancer identifies novel tumour microenvironment profiles, with prognostic and therapeutic implications. Eur. J. Cancer. 2019, 123, 118–129. [Google Scholar] [CrossRef] [Green Version]
- Smetana, K., Jr.; Lacina, L.; Szabo, P.; Dvorankova, B.; Broz, P.; Sedo, A. Ageing as an Important Risk Factor for Cancer. Anticancer Res. 2016, 36, 5009–5017. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ma, L.; Pei, X.; Wang, H.; Tang, X.; Pei, J.F.; Ding, Y.N.; Qu, S.; Wei, Z.Y.; Wang, H.Y.; et al. Comprehensive assessment of cellular senescence in the tumor microenvironment. Brief. Bioinform. 2022, 23, bbac118. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Wang, X.; Xu, Z.; Wang, Z.; Liu, T.; Cao, Z.; Feng, X.; Gao, Y.; He, J. Identification and validation of cellular senescence patterns to predict clinical outcomes and immunotherapeutic responses in lung adenocarcinoma. Cancer Cell Int. 2021, 21, 652. [Google Scholar] [CrossRef] [PubMed]
- Kellers, F.; Fernandez, A.; Konukiewitz, B.; Schindeldecker, M.; Tagscherer, K.E.; Heintz, A.; Jesinghaus, M.; Roth, W.; Foersch, S. Senescence-Associated Molecules and Tumor-Immune-Interactions as Prognostic Biomarkers in Colorectal Cancer. Front. Med. (Lausanne) 2022, 9, 865230. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.W.; Kim, Y.H.; Oh, S.Y.; Suh, K.W.; Kim, Y.S.; Lee, G.Y.; Yoon, J.E.; Park, S.S.; Lee, Y.K.; Park, Y.J.; et al. Senescent Tumor Cells Build a Cytokine Shield in Colorectal Cancer. Adv. Sci. (Weinh.) 2021, 8, 2002497. [Google Scholar] [CrossRef]
- Yue, T.; Chen, S.; Zhu, J.; Guo, S.; Huang, Z.; Wang, P.; Zuo, S.; Liu, Y. The aging-related risk signature in colorectal cancer. Aging-Us 2021, 13, 7330–7349. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Crescenzi, E.; De Palma, R.; Leonardi, A. Senescence and NF kappa B A Trojan horse in tumors? Oncoimmunology 2012, 1, 1594–1597. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Noma, K.; Ohara, T.; Kashima, H.; Katsura, Y.; Sato, H.; Komoto, S.; Katsube, R.; Ninomiya, T.; Tazawa, H.; et al. Cancer-Associated Fibroblasts Affect Intratumoral CD8(+) and FoxP3(+) T Cells Via IL6 in the Tumor Microenvironment. Clin. Cancer Res. 2018, 24, 4820–4833. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Wang, X.; Wang, Z.; Shao, F.; Yang, Y.; Cao, Z.; Feng, X.; Gao, Y.; He, J. Comprehensive Analysis Uncovers Prognostic and Immunogenic Characteristics of Cellular Senescence for Lung Adenocarcinoma. Front. Cell Dev. Biol. 2021, 9, 780461. [Google Scholar] [CrossRef]
- Sato, H.; Takino, T.; Miyamori, H. Roles of membrane-type matrix metalloproteinase-1 in tumor invasion and etastasis. Cancer Sci. 2005, 96, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, N.; Gheldof, A.; Tatari, M.; Christofori, G. EMT as the ultimate survival mechanism of cancer cells. Semin. Cancer Biol. 2012, 22, 194–207. [Google Scholar] [CrossRef]
- Seidl, C.; Panzitt, K.; Bertsch, A.; Brcic, L.; Schein, S.; Mack, M.; Leithner, K.; Prinz, F.; Olschewski, H.; Kornmueller, K.; et al. MicroRNA-182-5p regulates hedgehog signaling pathway and chemosensitivity of cisplatin-resistant lung adenocarcinoma cells via targeting GLI2. Cancer Lett. 2020, 469, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Lou, S.; Jiang, Z. PHLDA2 regulates EMT and autophagy in colorectal cancer via the PI3K/AKT signaling pathway. Aging-Us 2020, 12, 7985–8000. [Google Scholar] [CrossRef] [PubMed]
- Tamm, I.; Kikuchi, T.; Cardinale, I.; Krueger, J.G. Cell-adhesion-disrupting action of interleukin 6 in human ductal breast carcinoma cells. Proc. Natl. Acad. Sci. USA 1994, 91, 3329–3333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imran, S.A.M.; Yazid, M.D.; Idrus, R.B.H.; Maarof, M.; Nordin, A.; Razali, R.A.; Lokanathan, Y. Is There an Interconnection between Epithelial-Mesenchymal Transition (EMT) and Telomere Shortening in Aging? Int. J. Mol. Sci. 2021, 22, 3888. [Google Scholar] [CrossRef]
- Santos, F.; Moreira, C.; Nobrega-Pereira, S.; de Jesus, B.B. New Insights into the Role of Epithelial-Mesenchymal Transition during Aging. Int. J. Mol. Sci. 2019, 20, 891. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Li, Y.; Pan, Y.; Lan, X.; Song, F.; Sun, J.; Zhou, K.; Liu, X.; Ren, X.; Wang, F.; et al. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018, 9, 5395. [Google Scholar] [CrossRef] [Green Version]
- di Magliano, M.P.; Hebrok, M. Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 2003, 3, 903–911. [Google Scholar] [CrossRef]
- Kennedy, R.; Celis, E. Multiple roles for CD4(+) T cells in anti-tumor immune responses. Immunol. Rev. 2008, 222, 129–144. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Bi, X.G.; Xu, S.L.; Xiang, J. Tumor-infiltrating dendritic cell subsets of progressive or regressive tumors induce suppressive or protective immune responses. Cancer Res. 2005, 65, 4955–4962. [Google Scholar] [CrossRef] [Green Version]
- Xing, X.; Zou, Z.; He, C.; Hu, Z.; Liang, K.; Liang, W.; Wang, Y.; Du, X. Enhanced antitumor effect of cytotoxic T lymphocytes induced by dendritic cells pulsed with colorectal cancer cell lysate expressing alpha-Gal epitopes. Oncol. Lett. 2019, 18, 864–871. [Google Scholar]
- Liu, X.; Wu, S.; Yang, Y.; Zhao, M.; Zhu, G.; Hou, Z. The prognostic landscape of tumor-infiltrating immune cell and immunomodulators in lung cancer. Biomed. Pharmacother. 2017, 95, 55–61. [Google Scholar] [CrossRef]
- Zhang, Y.; Zou, J.; Chen, R. An M0 macrophage-related prognostic model for hepatocellular carcinoma. BMC Cancer 2022, 22, 791. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Wang, W.; Li, L.; Zhang, G.; Gao, Z.; Tang, Z.; Dang, X.; Wu, Y. Profiles of immune cell infiltration and immune-related genes in the tumor microenvironment of colorectal cancer. Biomed. Pharmacother. 2019, 118, 109228. [Google Scholar] [CrossRef]
- Olguin, J.E.; Medina-Andrade, I.; Rodriguez, T.; Rodriguez-Sosa, M.; Terrazas, L.I. Relevance of Regulatory T Cells during Colorectal Cancer Development. Cancers 2020, 12, 1888. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Tan, L.; Wang, L.; Zou, D.; Liu, J.; Lu, X.; Fu, D.; Wang, G.; Wang, L.; Wang, Z. The Expression Pattern of Hypoxia-Related Genes Predicts the Prognosis and Mediates Drug Resistance in Colorectal Cancer. Front. Cell Dev. Biol. 2022, 10, 814621. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Features | N | Cluster 1 | Cluster 2 | p Value |
|---|---|---|---|---|
| Age (years) | ||||
| ≤65 | 180 | 57 | 123 | 0.391 |
| >65 | 250 | 69 | 181 | |
| Gender | ||||
| Female | 198 | 59 | 139 | 0.915 |
| Male | 232 | 67 | 165 | |
| Status | ||||
| Alive | 336 | 85 | 251 | 0.0008 |
| Dead | 94 | 41 | 53 | |
| Lymphatic invasion | ||||
| Yes | 151 | 68 | 83 | <0.0001 |
| No | 239 | 47 | 192 | |
| Venous invasion | ||||
| Yes | 89 | 40 | 49 | 0.0001 |
| No | 286 | 66 | 220 | |
| Pathologic stage | ||||
| Stage1 + 2 | 238 | 52 | 186 | <0.0001 |
| Stage3 + 4 | 181 | 72 | 109 | |
| Metastasis | ||||
| M0 | 318 | 86 | 232 | 0.0034 |
| M1 | 60 | 28 | 32 | |
| Lymph node status | ||||
| N0 | 253 | 56 | 197 | 0.0002 |
| N1 + 2 + 3 | 177 | 70 | 107 | |
| Tumor stage | ||||
| T1 + T2 | 87 | 12 | 75 | 0.0007 |
| T3 + T4 | 320 | 103 | 217 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, L.; Xiaohalati, X.; Liu, F.; Liu, J.; Fu, H.; Zhang, Y.; Gao, J.; Tao, K.; Wang, G.; Wang, L.; et al. Identification of Aging and Young Subtypes for Predicting Colorectal Cancer Prognosis and Immunotherapy Responses. Int. J. Mol. Sci. 2023, 24, 1516. https://doi.org/10.3390/ijms24021516
Tan L, Xiaohalati X, Liu F, Liu J, Fu H, Zhang Y, Gao J, Tao K, Wang G, Wang L, et al. Identification of Aging and Young Subtypes for Predicting Colorectal Cancer Prognosis and Immunotherapy Responses. International Journal of Molecular Sciences. 2023; 24(2):1516. https://doi.org/10.3390/ijms24021516
Chicago/Turabian StyleTan, Lulu, Xiakeerzhati Xiaohalati, Feng Liu, Jia Liu, Haoyu Fu, Yang Zhang, Jinbo Gao, Kaixiong Tao, Guobin Wang, Lin Wang, and et al. 2023. "Identification of Aging and Young Subtypes for Predicting Colorectal Cancer Prognosis and Immunotherapy Responses" International Journal of Molecular Sciences 24, no. 2: 1516. https://doi.org/10.3390/ijms24021516