

Assessing the Role of Aquaporin 4 in Skeletal Muscle Function


{kind=link}
Abstract
:1. Introduction
2. Discovery of Orthogonal Array Particles (OAPs)
3. Characterizing Aquaporin 4 in the Skeletal Muscle
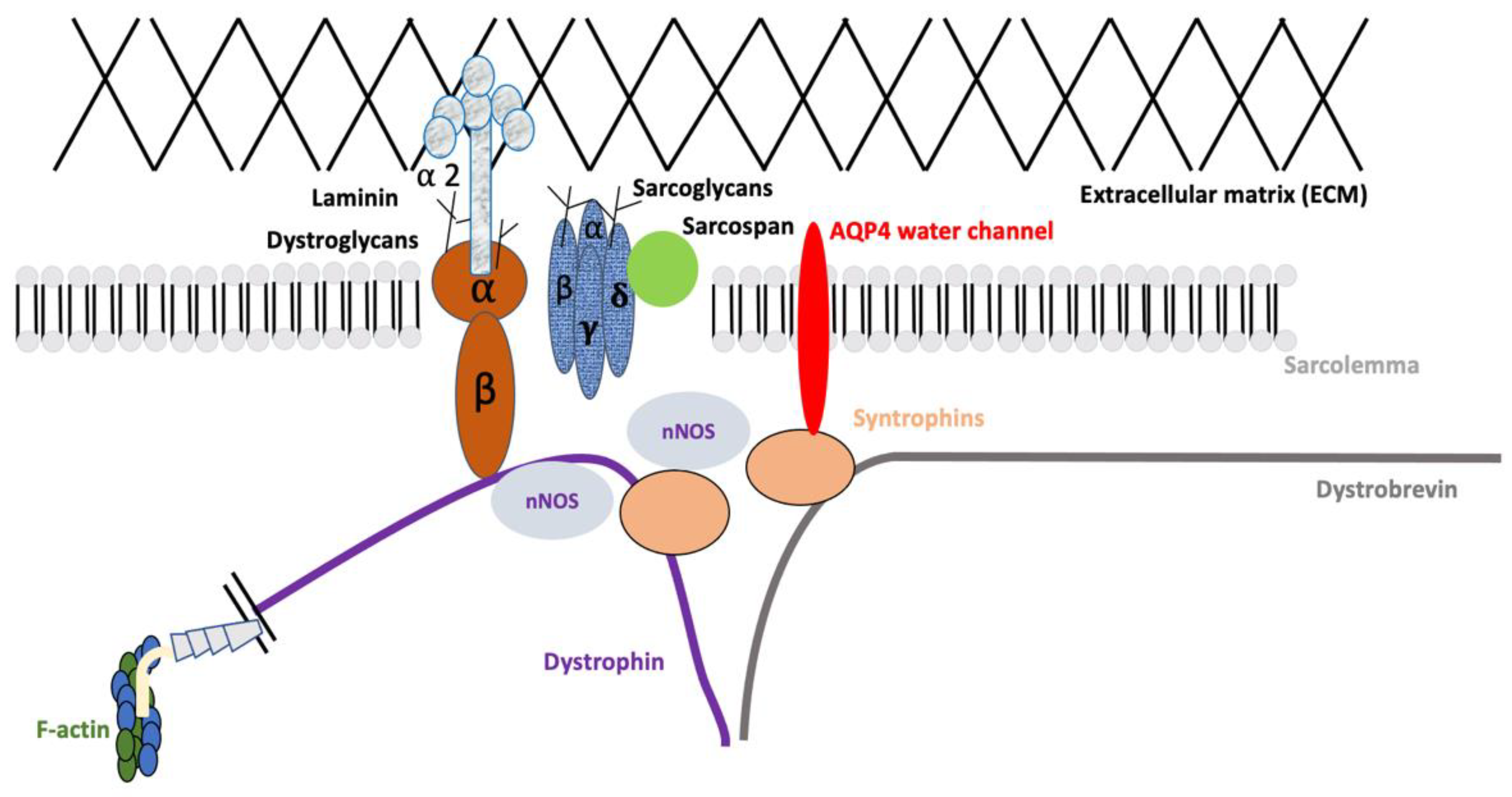
Association of AQP4 with the Dystrophin-Associated Glycoprotein Complex (DAPC)
4. Relevance of Aquaporins in Duchenne Muscular Dystrophy (DMD) and Other Neuromuscular Disorders (NMDs)
4.1. Duchenne Muscular Dystrophy (DMD)
4.2. Findings in DMD Models and Patients
4.3. Findings in Patients of Other NMDs
5. Effects of Exercise on AQP4 Expression in the Skeletal Muscle
6. AQP1 Acts as a Potential Compensator for AQP4 Loss
7. Effects of Skeletal Muscle Atrophy and Denervation on the Expressions of AQP4 and AQP1
8. Expression of Other AQPs in the Skeletal Muscle
9. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Frigeri, A.; Nicchia, G.P.; Balena, R.; Nico, B.; Svelto, M. Aquaporins in skeletal muscle: Reassessment of the functional role of aquaporin-4. FASEB J. 2004, 18, 905–907. [Google Scholar] [CrossRef] [PubMed]
- Neering, I.R.; Quesenberry, L.A.; Morris, V.A.; Taylor, S.R. Nonuniform volume changes during muscle contraction. Biophys. J. 1991, 59, 926–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markou, A.; Unger, L.; Abir-Awan, M.; Saadallah, A.; Halsey, A.; Balklava, Z.; Conner, M.; Tornroth-Horsefield, S.; Greenhill, S.D.; Conner, A.; et al. Molecular mechanisms governing aquaporin relocalisation. Biochim. Biophys. Acta Biomembr. 2022, 1864, 183853. [Google Scholar] [CrossRef]
- Wakayama, Y.; Inoue, M.; Kojima, H.; Jimi, T.; Shibuya, S.; Hara, H.; Oniki, H. Expression and localization of aquaporin 7 in normal skeletal myofiber. Cell Tissue Res. 2004, 316, 123–129. [Google Scholar] [CrossRef]
- Denker, B.M.; Smith, B.L.; Kuhajda, F.P.; Agre, P. Identification, purification, and partial characterization of a novel Mr 28,000 integral membrane protein from erythrocytes and renal tubules. J. Biol. Chem. 1988, 263, 15634–15642. [Google Scholar] [CrossRef]
- Preston, G.M.; Agre, P. Isolation of the cDNA for erythrocyte integral membrane protein of 28 kilodaltons: Member of an ancient channel family. Proc. Natl. Acad. Sci. USA 1991, 88, 11110–11114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preston, G.M.; Carroll, T.P.; Guggino, W.B.; Agre, P. Appearance of water channels in Xenopus oocytes expressing red cell CHIP28 protein. Science 1992, 256, 385–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakayama, Y. Aquaporin expression in normal and pathological skeletal muscles: A brief review with focus on AQP4. J. Biomed. Biotechnol. 2010, 2010, 731569. [Google Scholar] [CrossRef] [Green Version]
- Madeira, A.; Moura, T.F.; Soveral, G. Detecting Aquaporin Function and Regulation. Front. Chem. 2016, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.B.; Skach, W.R.; Verkman, A.S. Functional independence of monomeric CHIP28 water channels revealed by expression of wild-type mutant heterodimers. J. Biol. Chem. 1994, 269, 10417–10422. [Google Scholar] [CrossRef]
- Gomes, D.; Agasse, A.; Thiebaud, P.; Delrot, S.; Geros, H.; Chaumont, F. Aquaporins are multifunctional water and solute transporters highly divergent in living organisms. Biochim. Biophys. Acta 2009, 1788, 1213–1228. [Google Scholar] [CrossRef] [Green Version]
- Aaij, R.; Adeva, B.; Adinolfi, M.; Ajaltouni, Z.; Akar, S.; Albrecht, J.; Alessio, F.; Alexander, M.; Alfonso Albero, A.; Ali, S.; et al. Search for Dark Photons Produced in 13 TeV pp Collisions. Phys. Rev. Lett. 2018, 120, 061801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondy, C.; Chin, E.; Smith, B.L.; Preston, G.M.; Agre, P. Developmental gene expression and tissue distribution of the CHIP28 water-channel protein. Proc. Natl. Acad. Sci. USA 1993, 90, 4500–4504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, T.; Yang, B.; Gillespie, A.; Carlson, E.J.; Epstein, C.J.; Verkman, A.S. Severely impaired urinary concentrating ability in transgenic mice lacking aquaporin-1 water channels. J. Biol. Chem. 1998, 273, 4296–4299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, L.S.; Choi, M.; Fernandez, P.C.; Cartron, J.P.; Agre, P. Defective urinary concentrating ability due to a complete deficiency of aquaporin-1. N. Engl. J. Med. 2001, 345, 175–179. [Google Scholar] [CrossRef]
- Verkman, A.S. Physiological importance of aquaporin water channels. Ann. Med. 2002, 34, 192–200. [Google Scholar] [CrossRef]
- Nielsen, S.; Smith, B.L.; Christensen, E.I.; Agre, P. Distribution of the aquaporin CHIP in secretory and resorptive epithelia and capillary endothelia. Proc. Natl. Acad. Sci. USA 1993, 90, 7275–7279. [Google Scholar] [CrossRef] [Green Version]
- Verkman, A.S. Aquaporin water channels and endothelial cell function. J. Anat. 2002, 200, 617–627. [Google Scholar] [CrossRef]
- Folkesson, H.G.; Matthay, M.A.; Hasegawa, H.; Kheradmand, F.; Verkman, A.S. Transcellular water transport in lung alveolar epithelium through mercury-sensitive water channels. Proc. Natl. Acad. Sci. USA 1994, 91, 4970–4974. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, H.; Lian, S.C.; Finkbeiner, W.E.; Verkman, A.S. Extrarenal tissue distribution of CHIP28 water channels by in situ hybridization and antibody staining. Am. J. Physiol. 1994, 266, C893–C903. [Google Scholar] [CrossRef]
- Effros, R.M.; Darin, C.; Jacobs, E.R.; Rogers, R.A.; Krenz, G.; Schneeberger, E.E. Water transport and the distribution of aquaporin-1 in pulmonary air spaces. J. Appl. Physiol. 1997, 83, 1002–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gresz, V.; Kwon, T.H.; Hurley, P.T.; Varga, G.; Zelles, T.; Nielsen, S.; Case, R.M.; Steward, M.C. Identification and localization of aquaporin water channels in human salivary glands. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G247–G254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, P.T.; Ferguson, C.J.; Kwon, T.H.; Andersen, M.L.; Norman, A.G.; Steward, M.C.; Nielsen, S.; Case, R.M. Expression and immunolocalization of aquaporin water channels in rat exocrine pancreas. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G701–G709. [Google Scholar] [CrossRef] [Green Version]
- Benga, G. The first discovered water channel protein, later called aquaporin 1: Molecular characteristics, functions and medical implications. Mol. Asp. Med. 2012, 33, 518–534. [Google Scholar] [CrossRef] [PubMed]
- Holmes, R.P. The role of renal water channels in health and disease. Mol. Asp. Med. 2012, 33, 547–552. [Google Scholar] [CrossRef]
- Nielsen, S.; Nagelhus, E.A.; Amiry-Moghaddam, M.; Bourque, C.; Agre, P.; Ottersen, O.P. Specialized membrane domains for water transport in glial cells: High-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J. Neurosci. 1997, 17, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Longatti, P.L.; Basaldella, L.; Orvieto, E.; Fiorindi, A.; Carteri, A. Choroid plexus and aquaporin-1: A novel explanation of cerebrospinal fluid production. Pediatr. Neurosurg. 2004, 40, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Nagelhus, E.A.; Amiry-Moghaddam, M.; Agre, P.; Ottersen, O.P.; Nielsen, S. Ontogeny of water transport in rat brain: Postnatal expression of the aquaporin-4 water channel. Eur. J. Neurosci. 1999, 11, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Frigeri, A.; Nicchia, G.P.; Verbavatz, J.M.; Valenti, G.; Svelto, M. Expression of aquaporin-4 in fast-twitch fibers of mammalian skeletal muscle. J. Clin. Investig. 1998, 102, 695–703. [Google Scholar] [CrossRef] [Green Version]
- Branton, D. Fracture faces of frozen membranes. Proc. Natl. Acad. Sci. USA 1966, 55, 1048–1056. [Google Scholar] [CrossRef]
- Verbavatz, J.M.; Ma, T.; Gobin, R.; Verkman, A.S. Absence of orthogonal arrays in kidney, brain and muscle from transgenic knockout mice lacking water channel aquaporin-4. J. Cell Sci. 1997, 110 Pt 22, 2855–2860. [Google Scholar] [CrossRef]
- Frigeri, A.; Gropper, M.A.; Umenishi, F.; Kawashima, M.; Brown, D.; Verkman, A.S. Localization of MIWC and GLIP water channel homologs in neuromuscular, epithelial and glandular tissues. J. Cell Sci. 1995, 108 Pt 9, 2993–3002. [Google Scholar] [CrossRef]
- Yang, B.; Brown, D.; Verkman, A.S. The mercurial insensitive water channel (AQP-4) forms orthogonal arrays in stably transfected Chinese hamster ovary cells. J. Biol. Chem. 1996, 271, 4577–4580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirken, S.M.; Fischbeck, K.H. Freeze-fracture studies of denervated and tenotomized rat muscle. J. Neuropathol. Exp. Neurol. 1985, 44, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Rash, J.E.; Yasumura, T.; Hudson, C.S.; Agre, P.; Nielsen, S. Direct immunogold labeling of aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc. Natl. Acad. Sci. USA 1998, 95, 11981–11986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, S.W.Y.; Inoue, M. Identification of aquaporin 4 molecule in the replica of normal rat sarcolemma: Fracture-label immnoelectron microscopic study. Ann. Neurol. 1999, 46, 460. [Google Scholar]
- Shibuya, S.; Wakayama, Y. Changes in muscle plasma membranes in young mice with X chromosome-linked muscular dystrophy: A freeze-fracture study. Neuropathol. Appl. Neurobiol. 1991, 17, 335–344. [Google Scholar] [CrossRef]
- Wakayama, Y.; Okayasu, H.; Shibuya, S.; Kumagai, T. Duchenne dystrophy: Reduced density of orthogonal array subunit particles in muscle plasma membrane. Neurology 1984, 34, 1313–1317. [Google Scholar] [CrossRef]
- Hasegawa, H.; Ma, T.; Skach, W.; Matthay, M.A.; Verkman, A.S. Molecular cloning of a mercurial-insensitive water channel expressed in selected water-transporting tissues. J. Biol. Chem. 1994, 269, 5497–5500. [Google Scholar] [CrossRef]
- Lu, M.; Lee, M.D.; Smith, B.L.; Jung, J.S.; Agre, P.; Verdijk, M.A.; Merkx, G.; Rijss, J.P.; Deen, P.M. The human AQP4 gene: Definition of the locus encoding two water channel polypeptides in brain. Proc. Natl. Acad. Sci. USA 1996, 93, 10908–10912. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.S.; Bhat, R.V.; Preston, G.M.; Guggino, W.B.; Baraban, J.M.; Agre, P. Molecular characterization of an aquaporin cDNA from brain: Candidate osmoreceptor and regulator of water balance. Proc. Natl. Acad. Sci. USA 1994, 91, 13052–13056. [Google Scholar] [CrossRef] [PubMed]
- Basco, D.; Nicchia, G.P.; D′Alessandro, A.; Zolla, L.; Svelto, M.; Frigeri, A. Absence of aquaporin-4 in skeletal muscle alters proteins involved in bioenergetic pathways and calcium handling. PLoS ONE 2011, 6, e19225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishido, M.; Nakamura, T. The expression of aquaporin-4 is regulated based on innervation in skeletal muscles. J. Muscle Res. Cell Motil. 2018, 39, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Vizzaccaro, E.; Terracciano, C.; Rastelli, E.; Massa, R. Aquaporin 4 expression in human skeletal muscle fiber types. Muscle Nerve 2018, 57, 856–858. [Google Scholar] [CrossRef]
- Rossi, A.; Pisani, F.; Nicchia, G.P.; Svelto, M.; Frigeri, A. Evidences for a leaky scanning mechanism for the synthesis of the shorter M23 protein isoform of aquaporin-4: Implication in orthogonal array formation and neuromyelitis optica antibody interaction. J. Biol. Chem. 2010, 285, 4562–4569. [Google Scholar] [CrossRef] [Green Version]
- Moe, S.E.; Sorbo, J.G.; Sogaard, R.; Zeuthen, T.; Petter Ottersen, O.; Holen, T. New isoforms of rat Aquaporin-4. Genomics 2008, 91, 367–377. [Google Scholar] [CrossRef] [Green Version]
- De Bellis, M.; Pisani, F.; Mola, M.G.; Basco, D.; Catalano, F.; Nicchia, G.P.; Svelto, M.; Frigeri, A. A novel human aquaporin-4 splice variant exhibits a dominant-negative activity: A new mechanism to regulate water permeability. Mol. Biol. Cell 2014, 25, 470–480. [Google Scholar] [CrossRef]
- De Bellis, M.; Pisani, F.; Mola, M.G.; Rosito, S.; Simone, L.; Buccoliero, C.; Trojano, M.; Nicchia, G.P.; Svelto, M.; Frigeri, A. Translational readthrough generates new astrocyte AQP4 isoforms that modulate supramolecular clustering, glial endfeet localization, and water transport. Glia 2017, 65, 790–803. [Google Scholar] [CrossRef]
- Grady, R.M.; Zhou, H.; Cunningham, J.M.; Henry, M.D.; Campbell, K.P.; Sanes, J.R. Maturation and maintenance of the neuromuscular synapse: Genetic evidence for roles of the dystrophin--glycoprotein complex. Neuron 2000, 25, 279–293. [Google Scholar] [CrossRef] [Green Version]
- Neely, J.D.; Amiry-Moghaddam, M.; Ottersen, O.P.; Froehner, S.C.; Agre, P.; Adams, M.E. Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc. Natl. Acad. Sci. USA 2001, 98, 14108–14113. [Google Scholar] [CrossRef] [Green Version]
- Au, C.G.; Butler, T.L.; Egan, J.R.; Cooper, S.T.; Lo, H.P.; Compton, A.G.; North, K.N.; Winlaw, D.S. Changes in skeletal muscle expression of AQP1 and AQP4 in dystrophinopathy and dysferlinopathy patients. Acta Neuropathol. 2008, 116, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Gaver, M.G.; Campbell, K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 1990, 345, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Aslesh, T.; Erkut, E.; Yokota, T. Restoration of dystrophin expression and correction of Duchenne muscular dystrophy by genome editing. Expert Opin. Biol. Ther. 2021, 21, 1049–1061. [Google Scholar] [CrossRef]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Juan-Mateu, J.; Gonzalez-Quereda, L.; Rodriguez, M.J.; Baena, M.; Verdura, E.; Nascimento, A.; Ortez, C.; Baiget, M.; Gallano, P. DMD Mutations in 576 Dystrophinopathy Families: A Step Forward in Genotype-Phenotype Correlations. PLoS ONE 2015, 10, e0135189. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Marden, F.A.; Connolly, A.M.; Siegel, M.J.; Rubin, D.A. Compositional analysis of muscle in boys with Duchenne muscular dystrophy using MR imaging. Skelet. Radiol. 2005, 34, 140–148. [Google Scholar] [CrossRef]
- Zhou, L.; Lu, H. Targeting fibrosis in Duchenne muscular dystrophy. J. Neuropathol. Exp. Neurol. 2010, 69, 771–776. [Google Scholar] [CrossRef]
- Veltrop, M.; van Vliet, L.; Hulsker, M.; Claassens, J.; Brouwers, C.; Breukel, C.; van der Kaa, J.; Linssen, M.M.; den Dunnen, J.T.; Verbeek, S.; et al. A dystrophic Duchenne mouse model for testing human antisense oligonucleotides. PLoS ONE 2018, 13, e0193289. [Google Scholar] [CrossRef]
- Manzur, A.Y.; Kinali, M.; Muntoni, F. Update on the management of Duchenne muscular dystrophy. Arch. Dis. Child. 2008, 93, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Zubrzycka-Gaarn, E.E.; Bulman, D.E.; Karpati, G.; Burghes, A.H.; Belfall, B.; Klamut, H.J.; Talbot, J.; Hodges, R.S.; Ray, P.N.; Worton, R.G. The Duchenne muscular dystrophy gene product is localized in sarcolemma of human skeletal muscle. Nature 1988, 333, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, C.G. The dystrophinopathies: An alternative to the structural hypothesis. Neurobiol. Dis. 1998, 5, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakayama, Y.; Jimi, T.; Misugi, N.; Kumagai, T.; Miyake, S.; Shibuya, S.; Miike, T. Dystrophin immunostaining and freeze-fracture studies of muscles of patients with early stage amyotrophic lateral sclerosis and Duchenne muscular dystrophy. J. Neurol. Sci. 1989, 91, 191–205. [Google Scholar] [CrossRef]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef] [Green Version]
- Tracey, I.; Dunn, J.F.; Radda, G.K. Brain metabolism is abnormal in the mdx model of Duchenne muscular dystrophy. Brain 1996, 119 Pt 3, 1039–1044. [Google Scholar] [CrossRef] [Green Version]
- Frigeri, A.; Nicchia, G.P.; Nico, B.; Quondamatteo, F.; Herken, R.; Roncali, L.; Svelto, M. Aquaporin-4 deficiency in skeletal muscle and brain of dystrophic mdx mice. FASEB J. 2001, 15, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Frigeri, A.; Nicchia, G.P.; Repetto, S.; Bado, M.; Minetti, C.; Svelto, M. Altered aquaporin-4 expression in human muscular dystrophies: A common feature? FASEB J. 2002, 16, 1120–1122. [Google Scholar] [CrossRef]
- Adams, M.E.; Mueller, H.A.; Froehner, S.C. In vivo requirement of the alpha-syntrophin PDZ domain for the sarcolemmal localization of nNOS and aquaporin-4. J. Cell Biol. 2001, 155, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Yokota, T.; Miyagoe, Y.; Hosaka, Y.; Tsukita, K.; Kameya, S.; Shibuya, S.; Matsuda, R.; Wakayama, Y.; Takeda, S.I. Aquaporin-4 is absent at the sarcolemma and at perivascular astrocyte endfeet in α1-syntrophin knockout mice. Proc. Japan. Acad. 2000, 76, 22–27. [Google Scholar] [CrossRef]
- Compton, A.G.; Cooper, S.T.; Hill, P.M.; Yang, N.; Froehner, S.C.; North, K.N. The syntrophin-dystrobrevin subcomplex in human neuromuscular disorders. J. Neuropathol. Exp. Neurol. 2005, 64, 350–361. [Google Scholar] [CrossRef] [Green Version]
- Wakayama, Y.; Jimi, T.; Inoue, M.; Kojima, H.; Murahashi, M.; Kumagai, T.; Yamashita, S.; Hara, H.; Shibuya, S. Reduced aquaporin 4 expression in the muscle plasma membrane of patients with Duchenne muscular dystrophy. Arch. Neurol. 2002, 59, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakayama, Y.; Jimi, T.; Inoue, M.; Kojima, H.; Yamashita, S.; Kumagai, T.; Murahashi, M.; Hara, H.; Shibuya, S. Altered aquaporin 4 expression in muscles of Fukuyama-type congenital muscular dystrophy. Virchows Arch. 2003, 443, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Nakahori, Y.; Miyake, M.; Matsumura, K.; Kondo-Iida, E.; Nomura, Y.; Segawa, M.; Yoshioka, M.; Saito, K.; Osawa, M.; et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 1998, 394, 388–392. [Google Scholar] [CrossRef]
- Fukuyama, Y.; Osawa, M.; Suzuki, H. Congenital progressive muscular dystrophy of the Fukuyama type-clinical, genetic and pathological considerations. Brain Dev. 1981, 3, 1–29. [Google Scholar] [CrossRef]
- Nakano, I.; Funahashi, M.; Takada, K.; Toda, T. Are breaches in the glia limitans the primary cause of the micropolygyria in Fukuyama-type congenital muscular dystrophy (FCMD)? Pathological study of the cerebral cortex of an FCMD fetus. Acta Neuropathol. 1996, 91, 313–321. [Google Scholar] [CrossRef]
- Yamamoto, T.; Shibata, N.; Kanazawa, M.; Kobayashi, M.; Komori, T.; Kondo, E.; Saito, K.; Osawa, M. Early ultrastructural changes in the central nervous system in Fukuyama congenital muscular dystrophy. Ultrastruct. Pathol. 1997, 21, 355–360. [Google Scholar] [CrossRef]
- Liu, J.; Aoki, M.; Illa, I.; Wu, C.; Fardeau, M.; Angelini, C.; Serrano, C.; Urtizberea, J.A.; Hentati, F.; Hamida, M.B.; et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 1998, 20, 31–36. [Google Scholar] [CrossRef]
- Wakayama, Y.; Inoue, M.; Kojima, H.; Yamashita, S.; Shibuya, S.; Jimi, T.; Hara, H.; Matsuzaki, Y.; Oniki, H.; Kanagawa, M.; et al. Reduced expression of sarcospan in muscles of Fukuyama congenital muscular dystrophy. Histol. Histopathol. 2008, 23, 1425–1438. [Google Scholar] [CrossRef]
- Wakayama, Y.; Inoue, M.; Kojima, H.; Jimi, T.; Yamashita, S.; Kumagai, T.; Shibuya, S.; Hara, H.; Oniki, H. Altered alpha1-syntrophin expression in myofibers with Duchenne and Fukuyama muscular dystrophies. Histol. Histopathol. 2006, 21, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Laval, S.H.; Bushby, K.M. Limb-girdle muscular dystrophies--from genetics to molecular pathology. Neuropathol. Appl. Neurobiol. 2004, 30, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Crosbie, R.H.; Dovico, S.A.; Flanagan, J.D.; Chamberlain, J.S.; Ownby, C.L.; Campbell, K.P. Characterization of aquaporin-4 in muscle and muscular dystrophy. FASEB J. 2002, 16, 943–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assereto, S.; Mastrototaro, M.; Stringara, S.; Gazzerro, E.; Broda, P.; Nicchia, G.P.; Svelto, M.; Bruno, C.; Nigro, V.; Lisanti, M.P.; et al. Aquaporin-4 expression is severely reduced in human sarcoglycanopathies and dysferlinopathies. Cell Cycle 2008, 7, 2199–2207. [Google Scholar] [CrossRef] [Green Version]
- Basco, D.; Blaauw, B.; Pisani, F.; Sparaneo, A.; Nicchia, G.P.; Mola, M.G.; Reggiani, C.; Svelto, M.; Frigeri, A. AQP4-dependent water transport plays a functional role in exercise-induced skeletal muscle adaptations. PLoS ONE 2013, 8, e58712. [Google Scholar] [CrossRef]
- Crenshaw, A.G.; Thornell, L.E.; Friden, J. Intramuscular pressure, torque and swelling for the exercise-induced sore vastus lateralis muscle. Acta Physiol. Scand. 1994, 152, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Greenleaf, J.E.; Van Beaumont, W.; Brock, P.J.; Morse, J.T.; Mangseth, G.R. Plasma volume and electrolyte shifts with heavy exercise in sitting and supine positions. Am. J. Physiol. 1979, 236, R206–R214. [Google Scholar] [CrossRef]
- Lindinger, M.I.; Spriet, L.L.; Hultman, E.; Putman, T.; McKelvie, R.S.; Lands, L.C.; Jones, N.L.; Heigenhauser, G.J. Plasma volume and ion regulation during exercise after low- and high-carbohydrate diets. Am. J. Physiol. 1994, 266, R1896–R1906. [Google Scholar] [CrossRef]
- Leinonen, H.; Juntunen, J.; Somer, H.; Rapola, J. Capillary circulation and morphology in Duchenne muscular dystrophy. Eur. Neurol. 1979, 18, 249–255. [Google Scholar] [CrossRef]
- Jimi, T.; Wakayama, Y.; Inoue, M.; Kojima, H.; Oniki, H.; Matsuzaki, Y.; Shibuya, S.; Hara, H.; Takahashi, J. Aquaporin 1: Examination of its expression and localization in normal human skeletal muscle tissue. Cells Tissues Organs 2006, 184, 181–187. [Google Scholar] [CrossRef]
- Au, C.G.; Cooper, S.T.; Lo, H.P.; Compton, A.G.; Yang, N.; Wintour, E.M.; North, K.N.; Winlaw, D.S. Expression of aquaporin 1 in human cardiac and skeletal muscle. J. Mol. Cell. Cardiol. 2004, 36, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Jain, R.K.; Witwer, B.; Brown, D. Water channel (aquaporin 1) expression and distribution in mammary carcinomas and glioblastomas. Microvasc. Res. 1999, 58, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, S.; Papadopoulos, M.C.; Davies, D.C.; Bell, B.A.; Krishna, S. Increased aquaporin 1 water channel expression in human brain tumours. Br. J. Cancer 2002, 87, 621–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saadoun, S.; Papadopoulos, M.C.; Hara-Chikuma, M.; Verkman, A.S. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature 2005, 434, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Frigeri, A.; Ribatti, D.; Nicchia, G.P.; Nico, B.; Ria, R.; Svelto, M.; Dammacco, F. Microvessel overexpression of aquaporin 1 parallels bone marrow angiogenesis in patients with active multiple myeloma. Br. J. Haematol. 2001, 113, 415–421. [Google Scholar] [CrossRef]
- Straino, S.; Germani, A.; Di Carlo, A.; Porcelli, D.; De Mori, R.; Mangoni, A.; Napolitano, M.; Martelli, F.; Biglioli, P.; Capogrossi, M.C. Enhanced arteriogenesis and wound repair in dystrophin-deficient mdx mice. Circulation 2004, 110, 3341–3348. [Google Scholar] [CrossRef] [Green Version]
- Ishido, M.; Nakamura, T. Aquaporin-4 Protein Is Stably Maintained in the Hypertrophied Muscles by Functional Overload. Acta Histochem. Cytochem. 2016, 49, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Benfenati, V.; Caprini, M.; Dovizio, M.; Mylonakou, M.N.; Ferroni, S.; Ottersen, O.P.; Amiry-Moghaddam, M. An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 2563–2568. [Google Scholar] [CrossRef] [Green Version]
- Mola, M.G.; Sparaneo, A.; Gargano, C.D.; Spray, D.C.; Svelto, M.; Frigeri, A.; Scemes, E.; Nicchia, G.P. The speed of swelling kinetics modulates cell volume regulation and calcium signaling in astrocytes: A different point of view on the role of aquaporins. Glia 2016, 64, 139–154. [Google Scholar] [CrossRef] [Green Version]
- Kunert-Keil, C.; Bisping, F.; Kruger, J.; Brinkmeier, H. Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genom. 2006, 7, 159. [Google Scholar] [CrossRef] [Green Version]
- Pritschow, B.W.; Lange, T.; Kasch, J.; Kunert-Keil, C.; Liedtke, W.; Brinkmeier, H. Functional TRPV4 channels are expressed in mouse skeletal muscle and can modulate resting Ca2+ influx and muscle fatigue. Pflug. Arch. 2011, 461, 115–122. [Google Scholar] [CrossRef]
- Ishido, M.; Nakamura, T. Marked decrease of aquaporin-4 protein is independent of the changes in alpha1-syntrophin and TRPV4 levels in response to denervation-induced muscle atrophy in vivo. J. Muscle Res. Cell Motil. 2017, 38, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Fanzani, A.; Conraads, V.M.; Penna, F.; Martinet, W. Molecular and cellular mechanisms of skeletal muscle atrophy: An update. J. Cachexia Sarcopenia Muscle 2012, 3, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Kalyani, R.R.; Corriere, M.; Ferrucci, L. Age-related and disease-related muscle loss: The effect of diabetes, obesity, and other diseases. Lancet Diabetes Endocrinol. 2014, 2, 819–829. [Google Scholar] [CrossRef] [Green Version]
- Wing, S.S.; Goldberg, A.L. Glucocorticoids activate the ATP-ubiquitin-dependent proteolytic system in skeletal muscle during fasting. Am. J. Physiol. 1993, 264, E668–E676. [Google Scholar] [CrossRef]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.W.; Kim, J.Y.; Yoon, J.P.; Suh, D.W.; Yeo, W.J.; Lee, Y.S. Atrogin1-induced loss of aquaporin 4 in myocytes leads to skeletal muscle atrophy. Sci. Rep. 2020, 10, 14189. [Google Scholar] [CrossRef]
- Dibas, A.; Yang, M.H.; He, S.; Bobich, J.; Yorio, T. Changes in ocular aquaporin-4 (AQP4) expression following retinal injury. Mol. Vis. 2008, 14, 1770–1783. [Google Scholar] [PubMed]
- Basco, D.; Nicchia, G.P.; Desaphy, J.F.; Camerino, D.C.; Frigeri, A.; Svelto, M. Analysis by two-dimensional Blue Native/SDS-PAGE of membrane protein alterations in rat soleus muscle after hindlimb unloading. Eur. J. Appl. Physiol. 2010, 110, 1215–1224. [Google Scholar] [CrossRef]
- Jimi, T.; Wakayama, Y.; Matsuzaki, Y.; Hara, H.; Inoue, M.; Shibuya, S. Reduced expression of aquaporin 4 in human muscles with amyotrophic lateral sclerosis and other neurogenic atrophies. Pathol. Res. Pr. 2004, 200, 203–209. [Google Scholar] [CrossRef]
- Munsat, T.L.; Davies, K.E. International SMA consortium meeting. (26–28 June 1992, Bonn, Germany). Neuromuscul. Disord. 1992, 2, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Melki, J.; Lefebvre, S.; Burglen, L.; Burlet, P.; Clermont, O.; Millasseau, P.; Reboullet, S.; Benichou, B.; Zeviani, M.; Le Paslier, D.; et al. De novo and inherited deletions of the 5q13 region in spinal muscular atrophies. Science 1994, 264, 1474–1477. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Egginton, S.; Hudlicka, O.; Brown, M.D.; Walter, H.; Weiss, J.B.; Bate, A. Capillary growth in relation to blood flow and performance in overloaded rat skeletal muscle. J. Appl. Physiol. 1998, 85, 2025–2032. [Google Scholar] [CrossRef] [Green Version]
- Wagatsuma, A.; Osawa, T. Time course of changes in angiogenesis-related factors in denervated muscle. Acta Physiol. 2006, 187, 503–509. [Google Scholar] [CrossRef]
- Ishido, M.; Nakamura, T. Time course changes in AQP4 expression patterns in progressive skeletal muscle atrophy during the early stage of denervation. J. Musculoskelet. Neuronal. Interact. 2020, 20, 114–120. [Google Scholar] [PubMed]
- Wang, W.; Hart, P.S.; Piesco, N.P.; Lu, X.; Gorry, M.C.; Hart, T.C. Aquaporin expression in developing human teeth and selected orofacial tissues. Calcif. Tissue Int. 2003, 72, 222–227. [Google Scholar] [CrossRef]
- Wakayama, Y.; Jimi, T.; Inoue, M.; Kojima, H.; Shibuya, S.; Murahashi, M.; Hara, H.; Oniki, H. Expression of aquaporin 3 and its localization in normal skeletal myofibres. Histochem. J. 2002, 34, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Wakayama, Y.; Kojima, H.; Shibuya, S.; Jimi, T.; Hara, H.; Iijima, S.; Masaki, H.; Oniki, H.; Matsuzaki, Y. Aquaporin 9 expression and its localization in normal skeletal myofiber. J. Mol. Histol. 2009, 40, 165–170. [Google Scholar] [CrossRef]
- Hwang, S.M.; Lee, R.H.; Song, J.M.; Yoon, S.; Kim, Y.S.; Lee, S.J.; Kang, S.K.; Jung, J.S. Expression of aquaporin-5 and its regulation in skeletal muscle cells. Exp. Mol. Med. 2002, 34, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Echevarria, M.; Windhager, E.E.; Tate, S.S.; Frindt, G. Cloning and expression of AQP3, a water channel from the medullary collecting duct of rat kidney. Proc. Natl. Acad. Sci. USA 1994, 91, 10997–11001. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K.; Sasaki, S.; Fushimi, K.; Uchida, S.; Kuwahara, M.; Saito, H.; Furukawa, T.; Nakajima, K.; Yamaguchi, Y.; Gojobori, T.; et al. Molecular cloning and expression of a member of the aquaporin family with permeability to glycerol and urea in addition to water expressed at the basolateral membrane of kidney collecting duct cells. Proc. Natl. Acad. Sci. USA 1994, 91, 6269–6273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, T.; Frigeri, A.; Hasegawa, H.; Verkman, A.S. Cloning of a water channel homolog expressed in brain meningeal cells and kidney collecting duct that functions as a stilbene-sensitive glycerol transporter. J. Biol. Chem. 1994, 269, 21845–21849. [Google Scholar] [CrossRef] [PubMed]
- Inase, N.; Fushimi, K.; Ishibashi, K.; Uchida, S.; Ichioka, M.; Sasaki, S.; Marumo, F. Isolation of human aquaporin 3 gene. J. Biol. Chem. 1995, 270, 17913–17916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbrey, J.M.; Gorelick-Feldman, D.A.; Kozono, D.; Praetorius, J.; Nielsen, S.; Agre, P. Aquaglyceroporin AQP9: Solute permeation and metabolic control of expression in liver. Proc. Natl. Acad. Sci. USA 2003, 100, 2945–2950. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, R.M.; Granner, D.K. Regulation of gene expression by insulin. Physiol. Rev. 1996, 76, 1109–1161. [Google Scholar] [CrossRef]
- O’Brien, R.M.; Streeper, R.S.; Ayala, J.E.; Stadelmaier, B.T.; Hornbuckle, L.A. Insulin-regulated gene expression. Biochem. Soc. Trans. 2001, 29, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Skowronski, M.T.; Lebeck, J.; Rojek, A.; Praetorius, J.; Fuchtbauer, E.M.; Frokiaer, J.; Nielsen, S. AQP7 is localized in capillaries of adipose tissue, cardiac and striated muscle: Implications in glycerol metabolism. Am. J. Physiol. Ren. Physiol. 2007, 292, F956–F965. [Google Scholar] [CrossRef] [Green Version]
- Qiu, S.; Mintz, J.D.; Salet, C.D.; Han, W.; Giannis, A.; Chen, F.; Yu, Y.; Su, Y.; Fulton, D.J.; Stepp, D.W. Increasing muscle mass improves vascular function in obese (db/db) mice. J. Am. Heart Assoc. 2014, 3, e000854. [Google Scholar] [CrossRef] [Green Version]
- Acevedo, L.M.; Raya, A.I.; Rios, R.; Aguilera-Tejero, E.; Rivero, J.L. Obesity-induced discrepancy between contractile and metabolic phenotypes in slow- and fast-twitch skeletal muscles of female obese Zucker rats. J. Appl. Physiol. 2017, 123, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Holmang, A.; Brzezinska, Z.; Bjorntorp, P. Effects of hyperinsulinemia on muscle fiber composition and capitalization in rats. Diabetes 1993, 42, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Klueber, K.M.; Feczko, J.D.; Schmidt, G.; Watkins, J.B., 3rd. Skeletal muscle in the diabetic mouse: Histochemical and morphometric analysis. Anat. Rec. 1989, 225, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Wakayama, Y.; Hirako, S.; Ohtaki, H.; Arata, S.; Jimi, T.; Honda, K. Histopathological and aquaporin7 mRNA expression analyzes in the skeletal and cardiac muscles of obese db/db mice. J. Vet. Med. Sci. 2021, 83, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Raina, S.; Preston, G.M.; Guggino, W.B.; Agre, P. Molecular cloning and characterization of an aquaporin cDNA from salivary, lacrimal, and respiratory tissues. J. Biol. Chem. 1995, 270, 1908–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Dong, M.; Dong, H.; Wang, W.; Sun, W.; Hao, Y.; Jiao, Y.; Cui, L.; Jiao, J. Reduced sarcolemmal aquaporin 4 expression can support the differential diagnosis of neuromyelitis optica spectrum disorders. J. Neuroimmunol. 2020, 339, 577121. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Takahashi, T.; Aoki, M.; Misu, T.; Konohana, S.; Okumura, T.; Takahashi, H.; Kameya, S.; Yamaki, K.; Kumagai, T.; et al. Neuromyelitis optica preceded by hyperCKemia episode. Neurology 2010, 74, 1543–1545. [Google Scholar] [CrossRef]
- He, D.; Li, Y.; Dai, Q.; Zhang, Y.; Xu, Z.; Li, Y.; Cai, G.; Chu, L. Myopathy associated with neuromyelitis optica spectrum disorders. Int. J. Neurosci. 2016, 126, 863–866. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aslesh, T.; Al-aghbari, A.; Yokota, T. Assessing the Role of Aquaporin 4 in Skeletal Muscle Function. Int. J. Mol. Sci. 2023, 24, 1489. https://doi.org/10.3390/ijms24021489
Aslesh T, Al-aghbari A, Yokota T. Assessing the Role of Aquaporin 4 in Skeletal Muscle Function. International Journal of Molecular Sciences. 2023; 24(2):1489. https://doi.org/10.3390/ijms24021489
Chicago/Turabian StyleAslesh, Tejal, Ammar Al-aghbari, and Toshifumi Yokota. 2023. "Assessing the Role of Aquaporin 4 in Skeletal Muscle Function" International Journal of Molecular Sciences 24, no. 2: 1489. https://doi.org/10.3390/ijms24021489