

Population Genomic Analyses Suggest a Hybrid Origin, Cryptic Sexuality, and Decay of Genes Regulating Seed Development for the Putatively Strictly Asexual Kingdonia uniflora (Circaeasteraceae, Ranunculales)
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
2.1. Sequence Data Processing
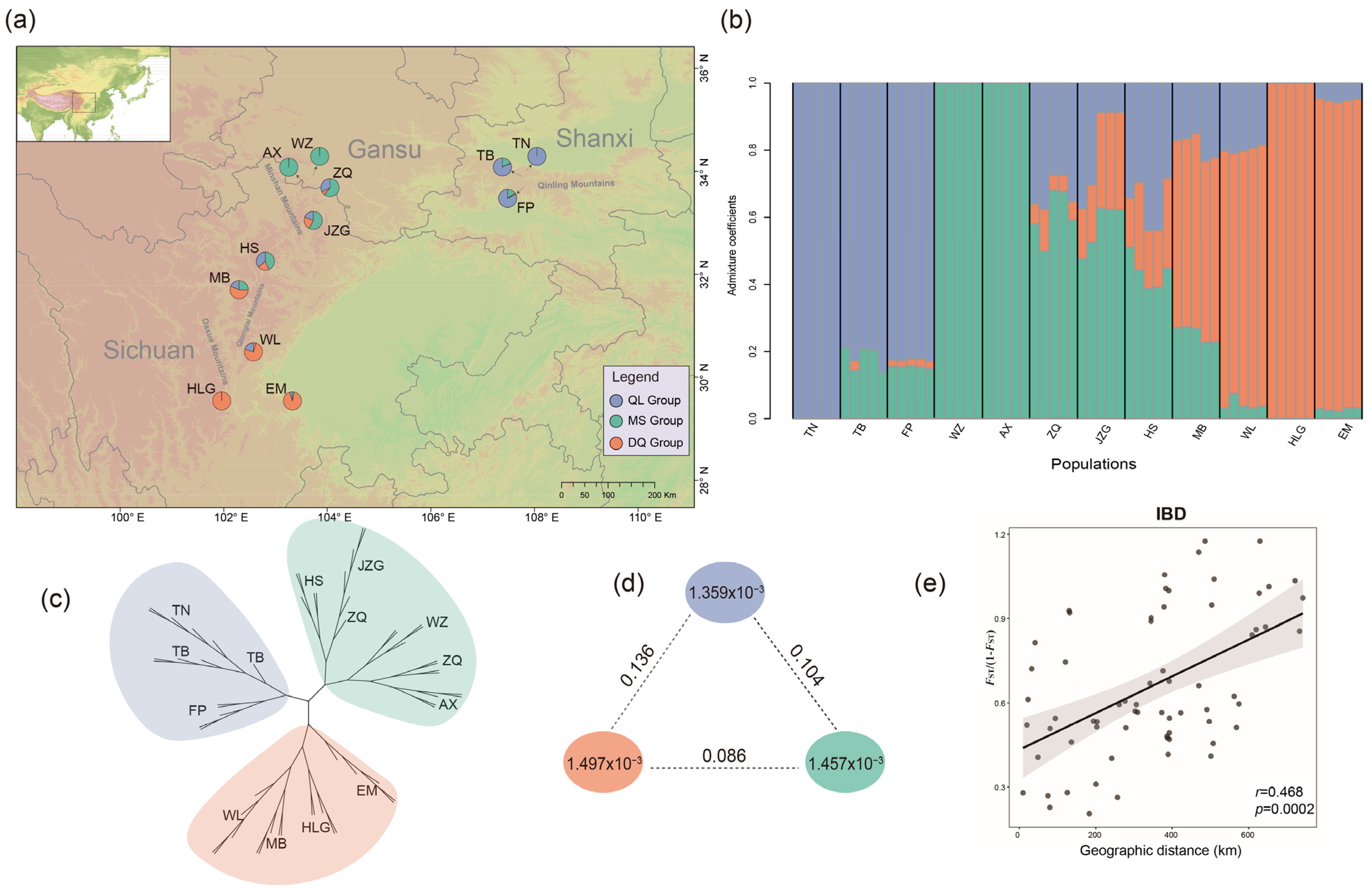
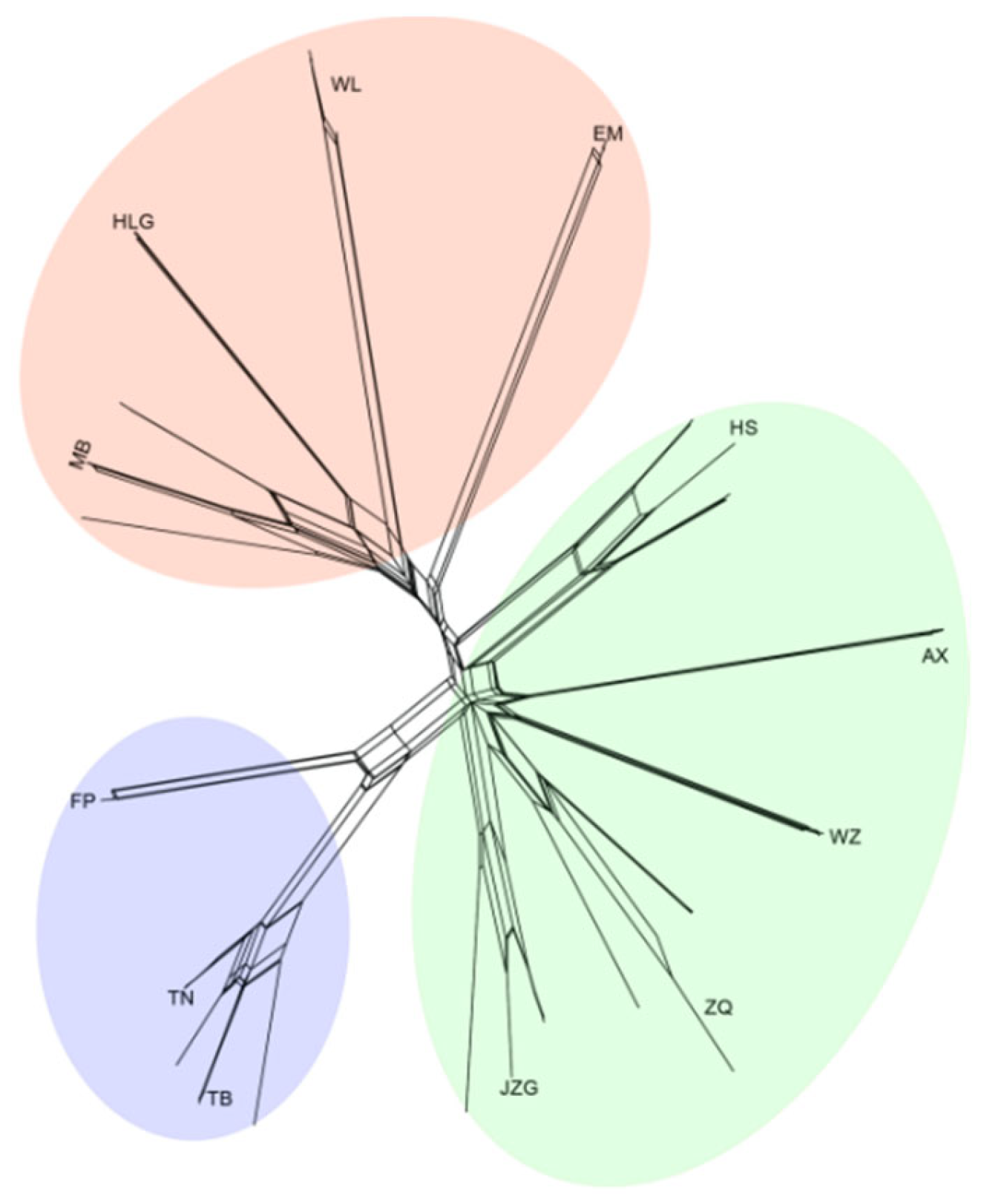
2.2. Genetic Diversity and Population Structure
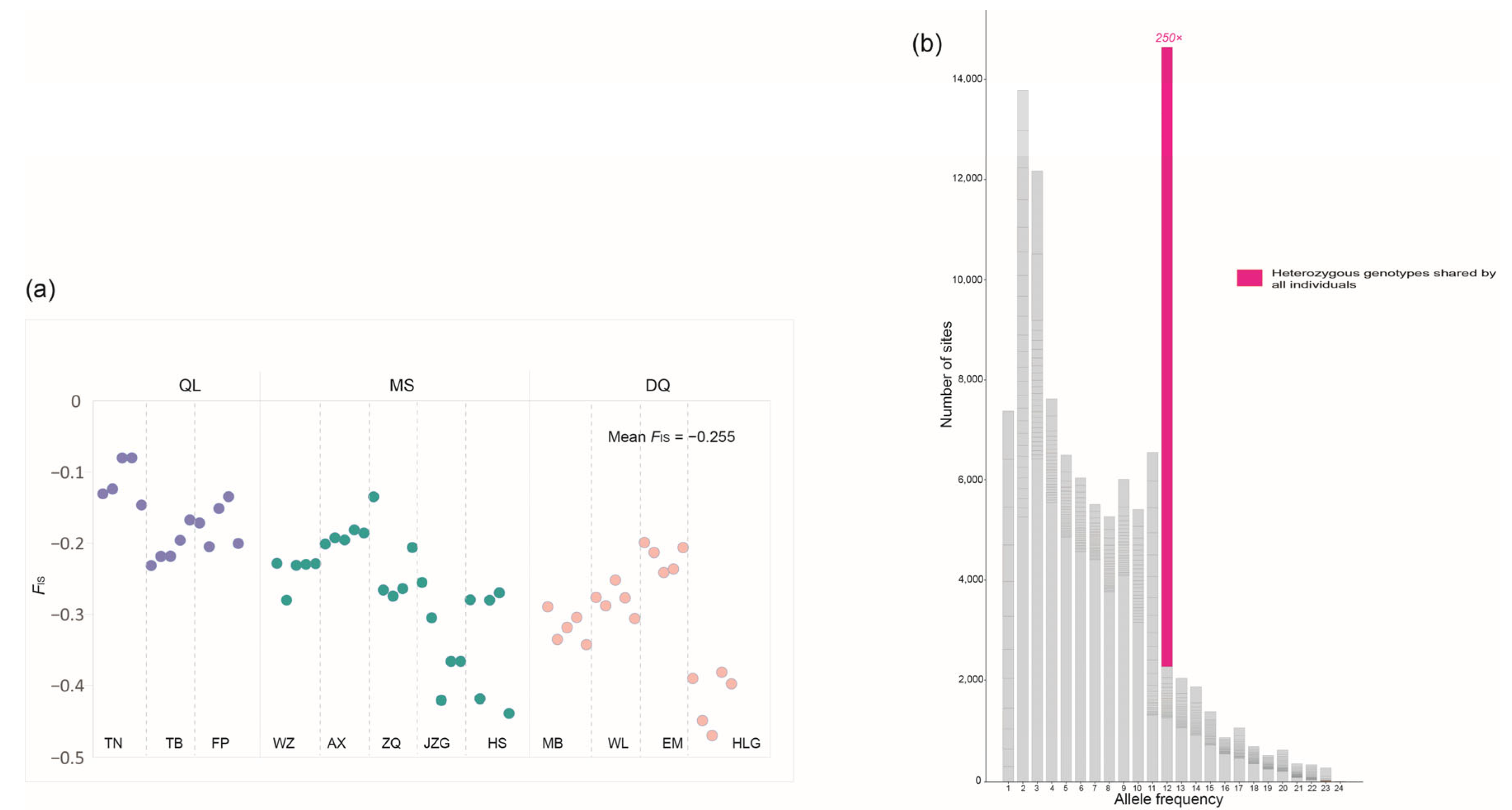
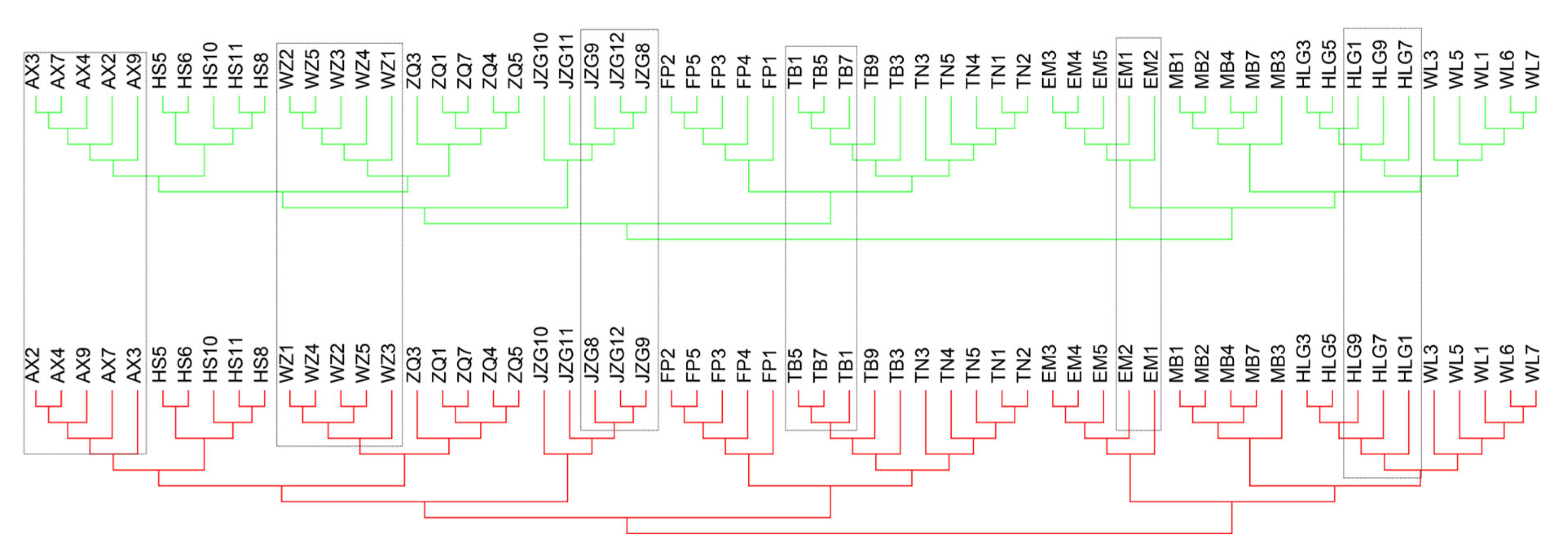
2.3. Detecting Predicted Genomic Signatures of Asexuality and Signs of Sexual Reproduction
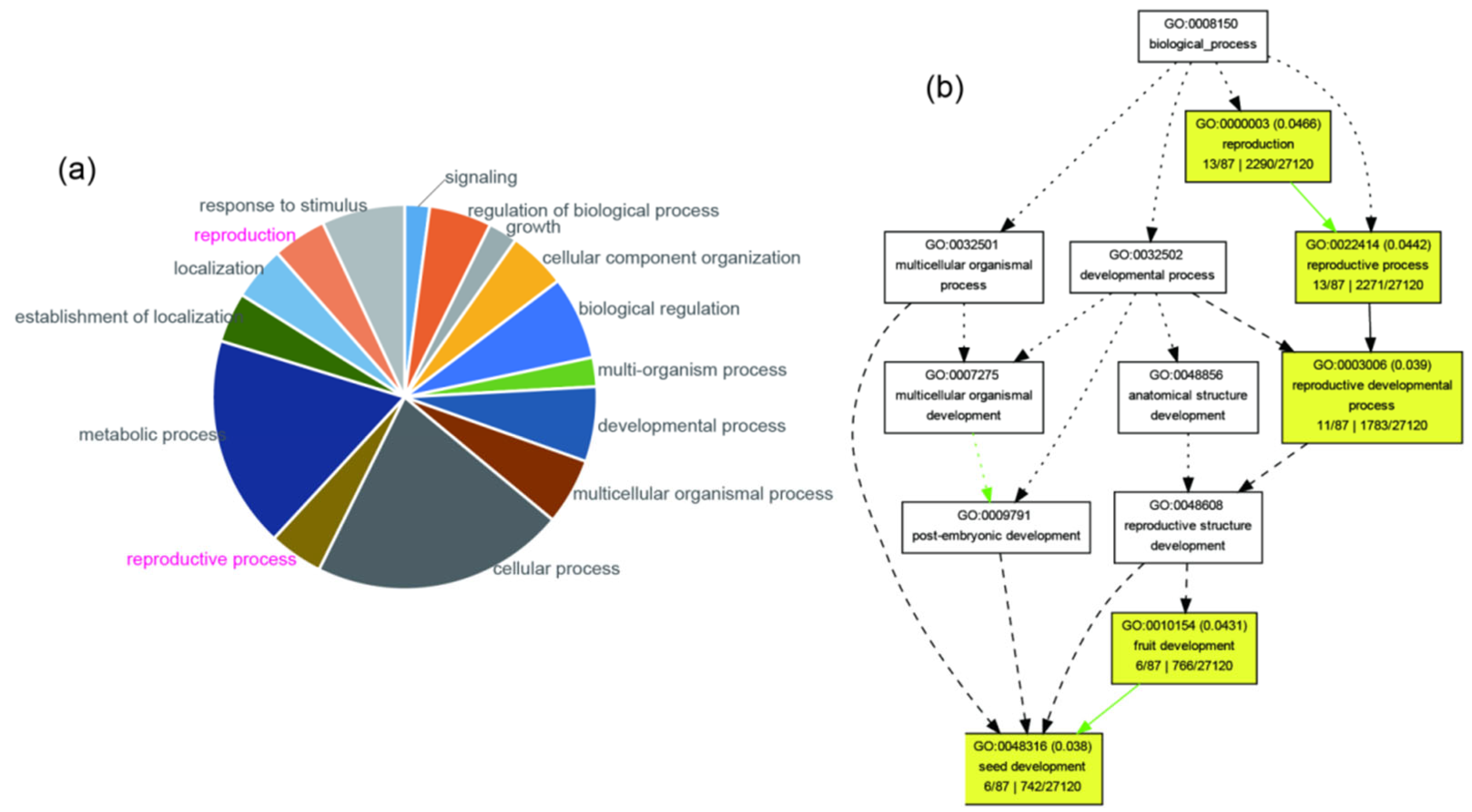
2.4. Detecting and Annotating Genes Showing Elevated Genetic Differentiation and Containing Premature Stop Codons
3. Discussion
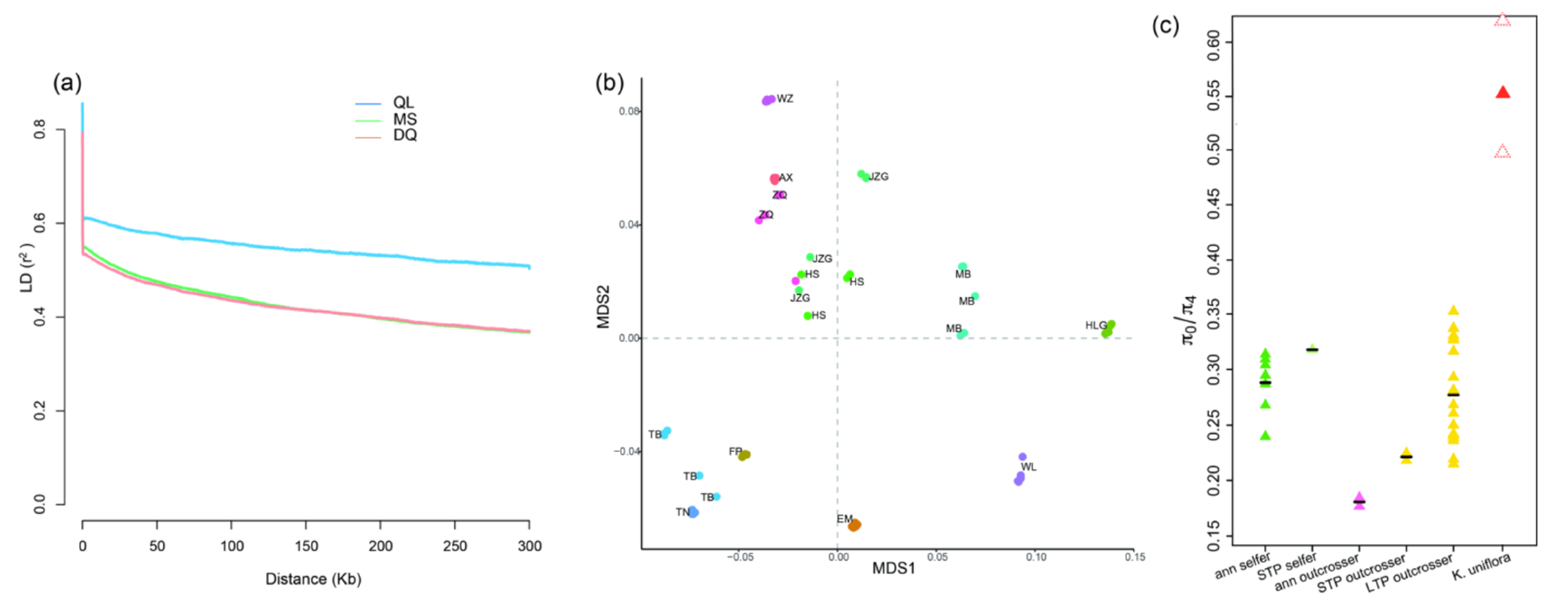
3.1. Kingdonia uniflora Is Characterized by High Allelic Heterozygosity, Slow Linkage Disequilibrium Decay and Elevated πN/πS Ratio, and Shows Signs of DNA Recombination
3.2. Genes Regulating Seed Development Show Signs of Decay
4. Methods and Materials
4.1. Sample Collection and Resequencing
4.2. SNP Calling and Quality Control
4.3. Genetic Diversity and Population Structure
4.4. Detecting Predicted Genomic Signatures of Asexuality and Signs of Sexual Reproduction
4.5. Detecting and Annotating Genes Showing Elevated Genetic Differentiation and Containing Premature Stop Codons
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mogie, M. The Evolution of Asexual Reproduction in Plants; Chapman & Hall: London, UK, 1992. [Google Scholar]
- Corley, L.S.; Blankenship, J.R.; Moore, A.J. Genetic variation and asexual reproduction in the facultatively parthenogenetic cockroach Nauphoeta cinerea: Implications for the evolution of sex. J. Evol. Biol. 2001, 14, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Normark, B.B.; Moran, N.A. Testing for the accumulation of deleterious mutations in asexual eukaryote genomes using molecular sequences. J. Nat. Hist. 2000, 34, 1719–1729. [Google Scholar] [CrossRef]
- Ament-Velásquez, S.L.; Figuet, E.; Ballenghien, M.; Zattara, E.E.; Norenburg, J.L.; Fernández-Álvarez, F.A.; Bierne, J.; Bierne, N.; Galtier, N. Population genomics of sexual and asexual lineages in fissiparous ribbon worms (Lineus, Nemertea): Hybridization, polyploidy and the Meselson effect. Mol. Ecol. 2016, 25, 3356–3369. [Google Scholar] [CrossRef] [PubMed]
- Muller, H. The relation of recombination to mutational advance. Mutat. Res. Mol. Mech. Mutagen. 1964, 1, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. The evolutionary advantage of recombination. Genetics 1974, 78, 737–756. [Google Scholar] [CrossRef] [PubMed]
- Maynard Smith, J. The Evolution of Sex; Cambridge University Press: Cambridge, UK, 1978. [Google Scholar]
- Zimmer, C. Origins. On the origin of sexual reproduction. Science 2009, 324, 1254–1256. [Google Scholar] [CrossRef]
- Heethoff, M.; Domes, K.; Laumann, M.; Maraun, M.; Norton, R.A.; Scheu, S. High genetic divergences indicate ancient separation of parthenogenetic lineages of the oribatid mite Platynothrus peltifer (Acari, Oribatida). J. Evol. Biol. 2007, 20, 392–402. [Google Scholar] [CrossRef]
- Schön, I.; Rossetti, G.; Martens, K. Darwinulid Ostracods: Ancient Asexual Scandals or Scandalous Gossip? In Lost Sex; Springer: Cham, Switzerland, 2009; pp. 217–240. [Google Scholar] [CrossRef]
- Brandt, A.; Van, P.T.; Bluhm, C.; Anselmetti, Y.; Dumas, Z.; Figuet, E.; François, C.M.; Galtier, N.; Heimburger, B.; Jaron, K.S.; et al. Haplotype divergence supports long-term asexuality in the oribatid mite Oppiella nova. Proc. Natl. Acad. Sci. USA 2021, 118, e2101485118. [Google Scholar] [CrossRef]
- Schurko, A.M.; Neiman, M.; Logsdon, J.M., Jr. Signs of sex: What we know and how we know it. Trends Ecol. Evol. 2009, 24, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Birky, C.W.; Wolf, C.; Maughan, H.; Herbertson, L.; Henry, E. Speciation and selection without sex. Hydrobiologia 2005, 546, 29–45. [Google Scholar] [CrossRef] [Green Version]
- Gladyshev, E.; Meselson, M. Extreme resistance of bdelloid rotifers to ionizing radiation. Proc. Natl. Acad. Sci. USA 2008, 105, 5139–5144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunt, D.H. Genetic tests of ancient asexuality in root knot nematodes reveal recent hybrid origins. BMC Evol. Biol. 2008, 8, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Signorovitch, A.; Hur, J.; Gladyshev, E.; Meselson, M. Allele sharing and evidence for sexuality in a mitochondrial clade of bdelloid rotifers. Genetics 2015, 200, 581–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwander, T. Evolution: The end of an ancient asexual scandal. Curr. Biol. 2016, 26, R233–R235. [Google Scholar] [CrossRef] [Green Version]
- Laine, V.N.; Sackton, T.; Meselson, M. Sexual reproduction in bdelloid rotifers. bioRxiv 2020. [Google Scholar] [CrossRef]
- Simion, P.; Narayan, J.; Houtain, A.; Derzelle, A.; Baudry, L.; Nicolas, E.; Arora, R.; Cariou, M.; Cruaud, C.; Gaudray, F.R.; et al. Chromosome-level genome assembly reveals homologous chromosomes and recombination in asexual rotifer Adineta vaga. Sci. Adv. 2021, 7, eabg4216. [Google Scholar] [CrossRef] [PubMed]
- Neiman, M.; Meirmans, S.; Meirmans, P.G. What can asexual lineage age tell us about the maintenance of sex? Ann. N. Y. Acad. Sci. 2009, 1168, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Birky, C.W. Heterozygosity, Heteromorphy, and Phylogenetic trees in asexual eukaryotes. Genetics 1996, 144, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Mark Welch, D.; Meselson, M. evidence for the evolution of bdelloid rotifers without sexual reproduction or genetic exchange. Science 2000, 288, 1211–1215. [Google Scholar] [CrossRef]
- Beck, J.B.; Alexander, P.J.; Allphin, L.; Al-Shehbaz, I.A.; Rushworth, C.; Bailey, C.D.; Windham, M.D. Does hybridization drive the transition to asexuality in diploid Boechera? Evolution 2012, 66, 985–995. [Google Scholar] [CrossRef]
- Jaron, K.S.; Bast, J.; Nowell, R.W.; Ranallo-Benavidez, T.R.; Robinson-Rechavi, M.; Schwander, T. Genomic features of parthenogenetic animals. J. Hered. 2021, 112, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Balloux, F.; Lehmann, L.; de Meeûs, T. The Population genetics of clonal and partially clonal diploids. Genetics 2003, 164, 1635–1644. [Google Scholar] [CrossRef]
- De Meeûs, T.; Balloux, F. Clonal reproduction and linkage disequilibrium in diploids: A simulation study. Infect. Genet. Evol. 2004, 4, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Henry, L.; Schwander, T.; Crespi, B.J. Deleterious mutation accumulation in asexual Timema stick insects. Mol. Biol. Evol. 2012, 29, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Lovell, J.T.; Grogan, K.; Sharbel, T.F.; McKay, J.K. Mating system and environmental variation drive patterns of adaptation in Boechera spatifolia (Brassicaceae). Mol. Ecol. 2014, 23, 4486–4497. [Google Scholar] [CrossRef] [PubMed]
- Normark, B.B.; Judson, O.P.; Moran, N.A. Genomic signatures of ancient asexual lineages. Biol. J. Linn. Soc. 2003, 79, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Schurko, A.M.; Logsdon, J.M., Jr. Using a meiosis detection toolkit to investigate ancient asexual “scandals” and the evolution of sex. Bioessays 2008, 30, 579–589. [Google Scholar] [CrossRef] [PubMed]
- The Angiosperm Phylogeny Group. An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Sanchez, E.; Rodríguez-Gómez, F.; Sosa, V. Refugia and geographic barriers of populations of the desert poppy, Hunnemannia fumariifolia (Papaveraceae). Org. Divers. Evol. 2012, 12, 133–143. [Google Scholar] [CrossRef]
- Sun, Y.; Deng, T.; Zhang, A.; Moore, M.J.; Landis, J.B.; Lin, N.; Zhang, H.; Zhang, X.; Huang, J.; Zhang, X.; et al. Genome sequencing of the endangered kingdonia uniflora (Circaeasteraceae, Ranunculales) reveals potential mechanisms of evolutionary specialization. iScience 2020, 23, 101124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, Y.; Landis, J.B.; Zhang, J.; Yang, L.; Lin, N.; Zhang, H.; Guo, R.; Li, L.; Zhang, Y.; et al. Genomic insights into adaptation to heterogeneous environments for the ancient relictual Circaeaster agrestis (Circaeasteraceae, Ranunculales). New Phytol. 2020, 228, 285–301. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Ren, L.; Li, Z.; Ren, Y. Studies on vegetative reproduction pattern of Kingdonia uniflora. Acta Bot. Boreali-Occident. Sin. 2000, 20, 432–435. [Google Scholar]
- Li, J.; Zhang, W.; Li, H. Research on distribution pattern of rare and endangered plant Kingdonia uniflora population. J. Northwest For. Univ. 2003, 18, 38–40. [Google Scholar]
- Xu, J.; Cao, B.; Bai, C. Prediction of potential suitable distribution of endangered plant Kingdonia uniflora in China with MaxEnt. Chin. J. Ecol. 2015, 34, 3354–3359. [Google Scholar]
- Li, J.; Zhao, J.; Li, L. Property of sexual reproduction of Kingdonia uniflora population in Mountain Taibai. J. Northwest Sci-Tech Univ. Agric. For. 2004, 32, 89–92. [Google Scholar]
- He, H.X. Pollination Biology and Breeding System of the Endangered Chinese Endemic Kingdonia uniflora. Master’s Thesis, Shaanxi Normal University, Xi’an, China, 2006. [Google Scholar]
- Chifman, J.; Kubatko, L. Quartet inference from snp data under the coalescent model. Bioinformatics 2014, 30, 3317–3324. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Schwander, T.; Henry, L.; Crespi, B.J. Molecular evidence for ancient asexuality in timema stick insects. Curr. Biol. 2011, 21, 1129–1134. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Glémin, S.; Lascoux, M. Genetic diversity and the efficacy of purifying selection across plant and animal species. Mol. Biol. Evol. 2017, 34, 1417–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartfield, M. On the origin of asexual species by means of hybridization and drift. Mol. Ecol. 2016, 25, 3264–3265. [Google Scholar] [CrossRef] [Green Version]
- Pellino, M.; Hojsgaard, D.; Schmutzer, T.; Scholz, U.; Hörandl, E.; Vogel, H.; Sharbel, T.F. Asexual genome evolution in the apomictic Ranunculus auricomus complex: Examining the effects of hybridization and mutation accumulation. Mol. Ecol. 2013, 22, 5908–5921. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Li, Z.-J.; Lei, Y. Acene and seed abortion contribute to the rarity of Kingdonia uniflora. Isr. J. Plant Sci. 2003, 51, 39–44. [Google Scholar] [CrossRef]
- Ceplitis, A. Coalescence times and the Meselson effect in asexual eukaryotes. Genet. Res. 2003, 82, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Lunt, D.H.; Kumar, S.; Koutsovoulos, G.; Blaxter, M.L. The complex hybrid origins of the root knot nematodes revealed through comparative genomics. Peer J. 2014, 2, e356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flint-Garcia, S.A.; Thornsberry, J.M.; Buckler, E.S. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 2003, 54, 357–374. [Google Scholar] [CrossRef] [Green Version]
- Simko, I.; Haynes, K.G.; Jones, R.W. Assessment of linkage disequilibrium in potato genome with single nucleotide polymorphism markers. Genetics 2006, 173, 2237–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordborg, M.; Hu, T.T.; Ishino, Y.; Jhaveri, J.; Toomajian, C.; Zheng, H.; Bakker, E.; Calabrese, P.; Gladstone, J.; Goyal, R.; et al. The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol. 2005, 3, e196. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Plagnol, V.; Hu, T.T.; Toomajian, C.; Clark, R.M.; Ossowski, S.; Ecker, J.; Weigel, D.; Nordborg, M. Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat. Genet. 2007, 39, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Branca, A.; Paape, T.D.; Zhou, P.; Briskine, R.; Farmer, A.D.; Mudge, J.; Bharti, A.K.; Woodward, J.E.; May, G.D.; Gentzbittel, L.; et al. Whole-genome nucleotide diversity, recombination, and linkage disequilibrium in the model legume Medicago truncatula. Proc. Natl. Acad. Sci. USA 2011, 108, E864–E870. [Google Scholar] [CrossRef] [Green Version]
- Ho, E.K.H.; Bartkowska, M.; Wright, S.I.; Agrawal, A.F. Population genomics of the facultatively asexual duckweed Spirodela polyrhiza. New Phytol. 2019, 224, 1361–1371. [Google Scholar] [CrossRef]
- Foxe, J.P.; Slotte, T.; Stahl, E.A.; Neuffer, B.; Hurka, H.; Wright, S.I. Recent speciation associated with the evolution of selfing in Capsella. Proc. Natl. Acad. Sci. USA 2009, 106, 5241–5245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, T.F.C.; Richards, S.; Stone, E.A.; Barbadilla, A.; Ayroles, J.F.; Zhu, D.; Casillas, S.; Han, Y.; Magwire, M.M.; Cridland, J.M.; et al. The Drosophila melanogaster genetic reference panel. Nature 2012, 482, 173–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordo, I.; Charlesworth, B. Genetic linkage and molecular evolution. Curr. Biol. 2001, 11, R684–R686. [Google Scholar] [CrossRef] [PubMed]
- Otto, S.P. Selective interference and the evolution of sex. J. Hered. 2021, 112, 9–18. [Google Scholar] [CrossRef]
- Kawecki, T.J.; Ebert, D. Conceptual issues in local adaptation. Ecol. Lett. 2004, 7, 1225–1241. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:https://arxiv.org/abs/1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Frichot, E.; François, O. LEA: An R package for landscape and ecological association studies. Methods Ecol. Evol. 2015, 6, 925–929. [Google Scholar] [CrossRef]
- Pritchard, J.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Reaz, R.; Bayzid, M.S.; Rahman, M.S. Accurate phylogenetic tree reconstruction from quartets: A heuristic approach. PLoS ONE 2014, 9, e104008. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Goudet, J. Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef]
- R Core Team. R Version 3.6.1: A Language and Environment for Statistical Computing; R Foundation for Statistical Computting: Vienna, Austria, 2014. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Vakhrusheva, O.A.; Mnatsakanova, E.A.; Galimov, Y.R.; Neretina, T.V.; Gerasimov, E.S.; Naumenko, S.A.; Ozerova, S.G.; Zalevsky, A.O.; Yushenova, I.A.; Rodriguez, F.; et al. Genomic signatures of recombination in a natural population of the bdelloid rotifer Adineta vaga. Nat. Commun. 2020, 11, 6421. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Foll, M.; Gaggiotti, O.E. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Chen, H.; He, Y.; Xia, R. TBtools, a Toolkit for biologists integrating various biological data handling tools with a user-friendly interface. bioRxiv 2018. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source of Variation | Sum of Squares | Variance Components | Percentage Variation | Fixation Indices | p-Values |
|---|---|---|---|---|---|
| Among groups | 166,030.43 | 993.35 | 5.06 | 0.05 | 0.00 |
| Among populations within groups | 396,979.34 | 3879.21 | 19.76 | 0.21 | 0.00 |
| Among individuals within populations | 255,202.00 | −9443.63 | −48.10 | −0.64 | 1.00 |
| Within individuals | 1,452,238.00 | 24,203.97 | 123.28 | −0.23 | 1.00 |
| GO Term | Class | Description | Significant | Annotated | p-Value |
|---|---|---|---|---|---|
| GO:0048316 | P | seed development | 6 | 742 | 0.038 |
| GO:0003006 | P | reproductive developmental process | 11 | 1783 | 0.039 |
| GO:0010154 | P | fruit development | 6 | 766 | 0.043 |
| GO:0022414 | P | reproductive process | 13 | 2271 | 0.044 |
| GO:0000003 | P | reproduction | 13 | 2290 | 0.047 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Zhang, X.; Zhang, A.; Landis, J.B.; Zhang, H.; Sun, H.; Xiang, Q.-Y.; Wang, H. Population Genomic Analyses Suggest a Hybrid Origin, Cryptic Sexuality, and Decay of Genes Regulating Seed Development for the Putatively Strictly Asexual Kingdonia uniflora (Circaeasteraceae, Ranunculales). Int. J. Mol. Sci. 2023, 24, 1451. https://doi.org/10.3390/ijms24021451
Sun Y, Zhang X, Zhang A, Landis JB, Zhang H, Sun H, Xiang Q-Y, Wang H. Population Genomic Analyses Suggest a Hybrid Origin, Cryptic Sexuality, and Decay of Genes Regulating Seed Development for the Putatively Strictly Asexual Kingdonia uniflora (Circaeasteraceae, Ranunculales). International Journal of Molecular Sciences. 2023; 24(2):1451. https://doi.org/10.3390/ijms24021451
Chicago/Turabian StyleSun, Yanxia, Xu Zhang, Aidi Zhang, Jacob B. Landis, Huajie Zhang, Hang Sun, Qiu-Yun (Jenny) Xiang, and Hengchang Wang. 2023. "Population Genomic Analyses Suggest a Hybrid Origin, Cryptic Sexuality, and Decay of Genes Regulating Seed Development for the Putatively Strictly Asexual Kingdonia uniflora (Circaeasteraceae, Ranunculales)" International Journal of Molecular Sciences 24, no. 2: 1451. https://doi.org/10.3390/ijms24021451