

Identification of Potential Druggable Targets and Structure-Based Virtual Screening for Drug-like Molecules against the Shrimp Pathogen Enterocytozoon hepatopenaei
Abstract
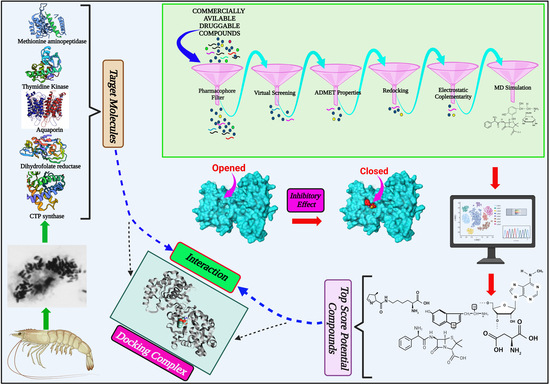
:
1. Introduction
2. Results
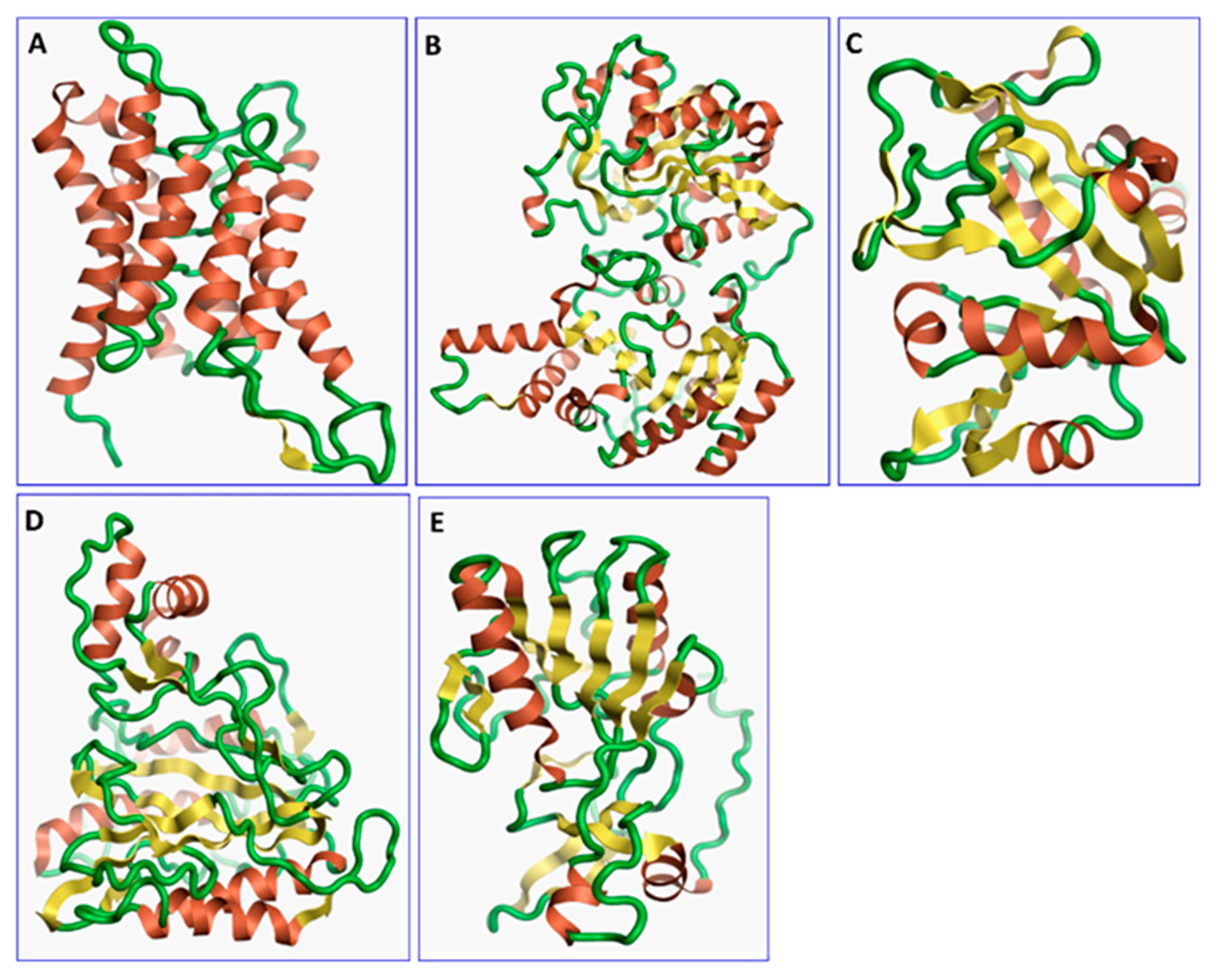
2.1. Identification of Potential Druggable Target Proteins and Generation of Three-Dimensional Models
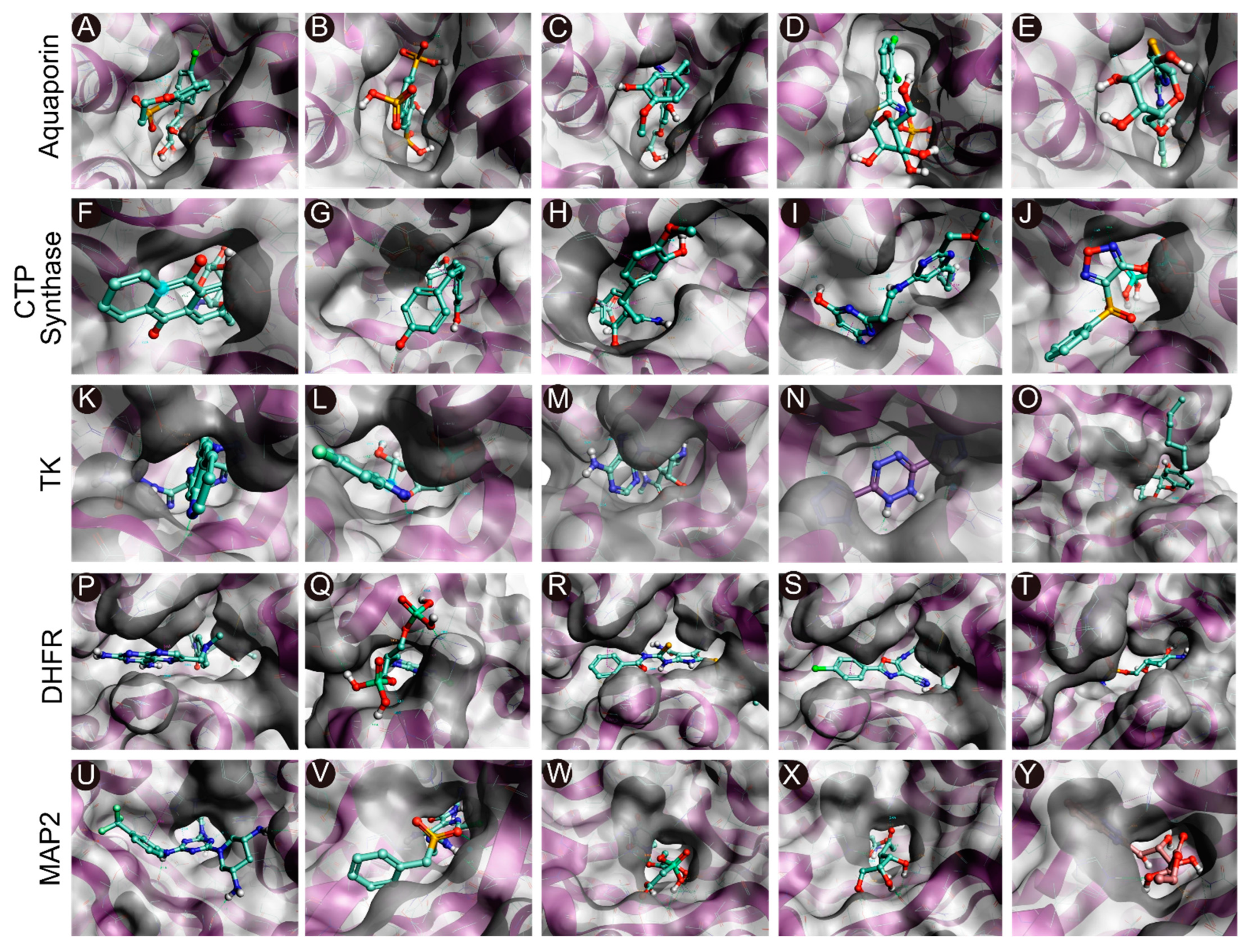
2.2. Identification of Pockets Based on Conserved Amino Acid Residues in Active Sites
2.3. Docking-Based Virtual Screening
2.4. Prediction of ADMET Properties
2.5. EC-Based Screening
2.6. Analysis of Protein-Ligand Interactions
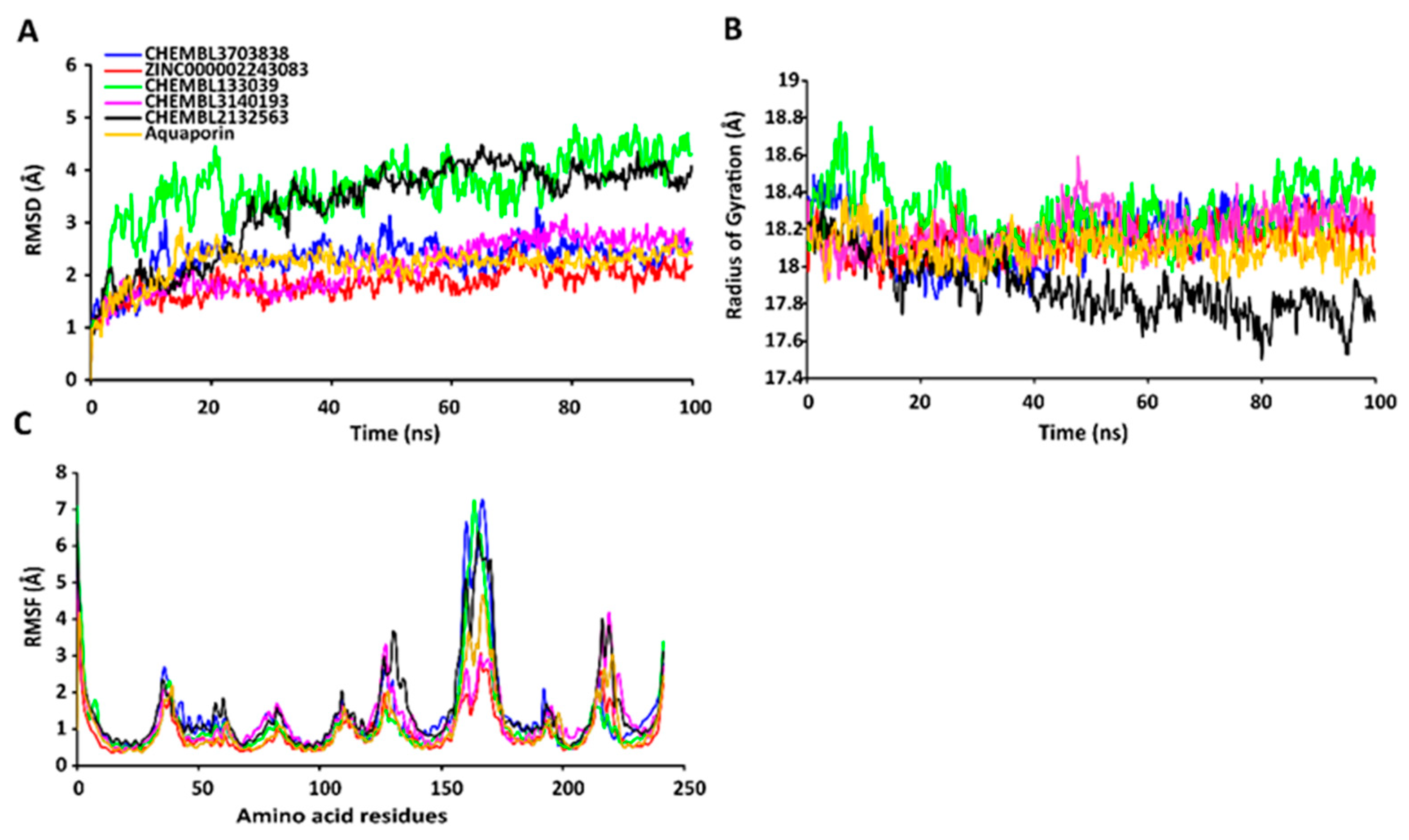
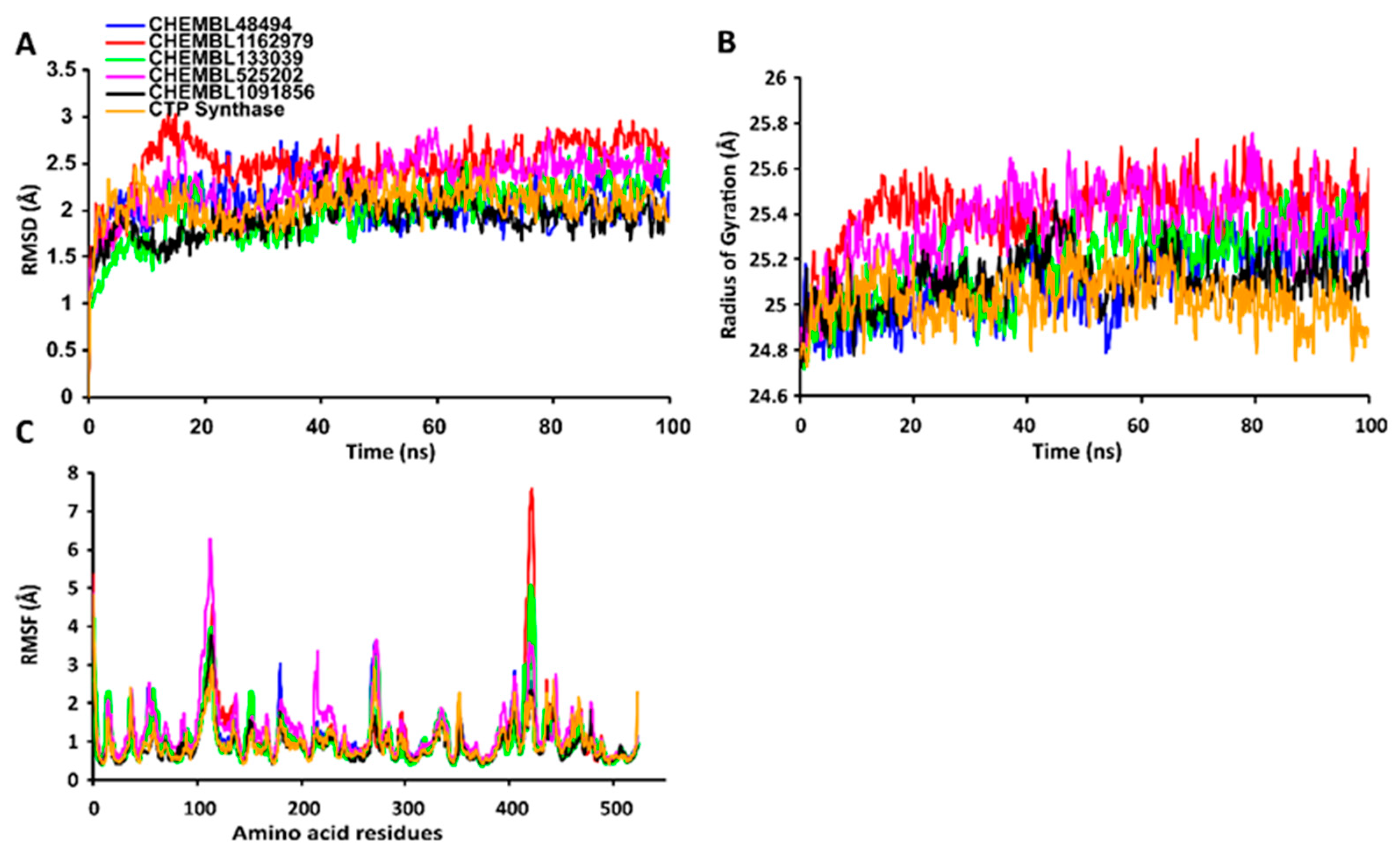
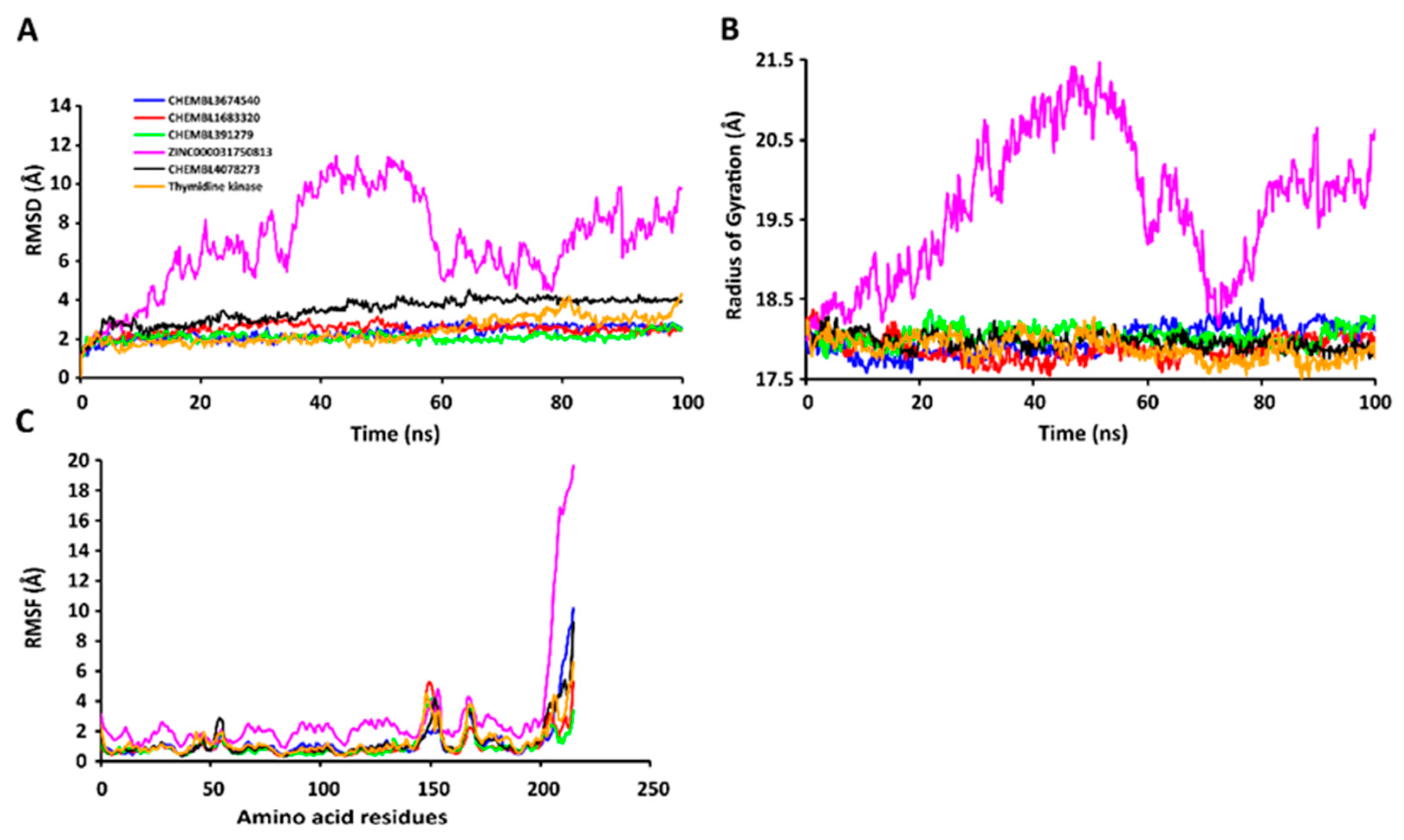
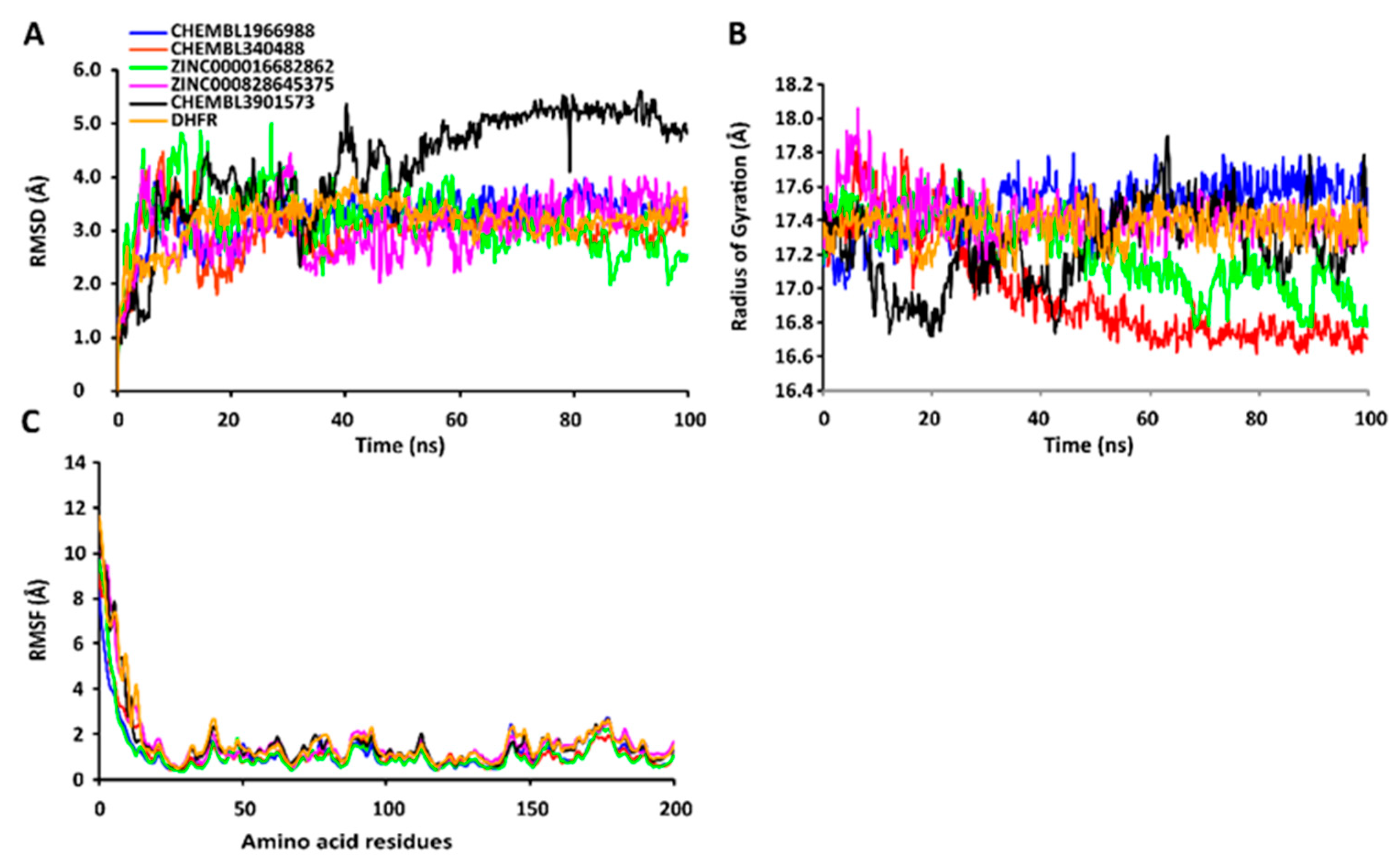
2.7. MD Simulations of Protein-Ligand Complexes
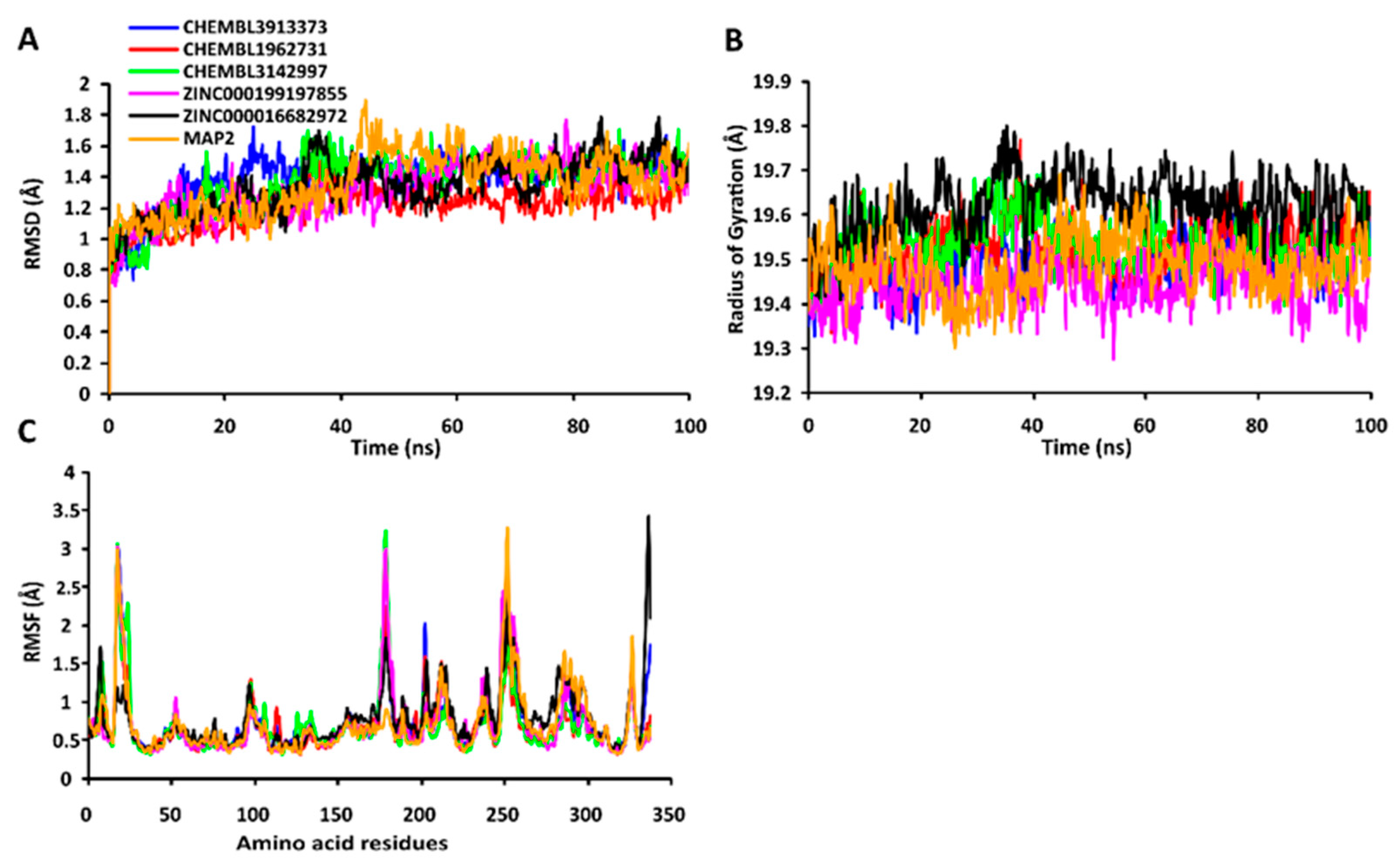
2.7.1. RMSD Values of the Cα Backbone of Target Proteins
2.7.2. RgValues of the Cα Backbone of Target Proteins
2.7.3. RMSF Values of the Cα Backbone of Target Proteins
2.7.4. Dynamic Cross-Correlation and PCA
2.8. Determination of Binding Free Energies of Protein-Ligand Complexes
3. Discussion
4. Materials and Methods
4.1. Identification of Potential Druggable Target Proteins
4.2. Identification of Druggable Pockets
4.3. Structure-Based VS
4.4. Prediction of ADMET Properties
4.5. Screening Based on EC
4.6. MD Simulations of Protein-Ligand Complexes
4.7. Determination of Free Energies of Protein-Ligand Complexes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salachan, P.V.; Jaroenlak, P.; Thitamadee, S.; Itsathitphaisarn, O.; Sritunyalucksana, K. Laboratory cohabitation challenge model for shrimp hepatopancreatic microsporidiosis (HPM) caused by Enterocytozoonhepatopenaei (EHP). BMC Vet. Res. 2016, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Tourtip, S.; Wongtripop, S.; Stentiford, G.D.; Bateman, K.S.; Sriurairatana, S.; Chavadej, J.; Sritunyalucksana, K.; Withyachumnarnkul, B. Enterocytozoonhepatopenaei sp. nov. (Microsporida: Enterocytozoonidae), a parasite of the black tiger shrimp Penaeus monodon (Decapoda: Penaeidae): Fine structure and phylogenetic relationships. J. Invertebr. Pathol. 2009, 102, 21–29. [Google Scholar] [CrossRef]
- Tang, K.F.; Han, J.E.; Aranguren, L.F.; White-Noble, B.; Schmidt, M.M.; Piamsomboon, P.; Risdiana, E.; Hanggono, B. Dense populations of the microsporidian Enterocytozoonhepatopenaei (EHP) in feces of Penaeus vannamei exhibiting white feces syndrome and pathways of their transmission to healthy shrimp. J. Invertebr. Pathol. 2016, 140, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aranguren Caro, L.F.; Alghamdi, F.; De Belder, K.; Lin, J.; Mai, H.N.; Millabas, J.; Alrehaili, Y.; Alazwari, A.; Algetham, S.; Dhar, A.K. The effect of salinity on Enterocytozoonhepatopenaei infection in Penaeus vannamei under experimental conditions. BMC Vet. Res. 2021, 17, 65. [Google Scholar] [CrossRef]
- Tangprasittipap, A.; Srisala, J.; Chouwdee, S.; Somboon, M.; Chuchird, N.; Limsuwan, C.; Srisuvan, T.; Flegel, T.W.; Sritunyalucksana, K. The microsporidian Enterocytozoonhepatopenaei is not the cause of white feces syndrome in whiteleg shrimp Penaeus (Litopenaeus) vannamei. BMC Vet. Res. 2013, 9, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiredu Boakye, D.; Jaroenlak, P.; Prachumwat, A.; Williams, T.A.; Bateman, K.S.; Itsathitphaisarn, O.; Sritunyalucksana, K.; Paszkiewicz, K.H.; Moore, K.A.; Stentiford, G.D. Decay of the glycolytic pathway and adaptation to intranuclear parasitism within Enterocytozoonidae microsporidia. Environ. Microbiol. 2017, 19, 2077–2089. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, J.; Liao, G.; Hu, M.; Zhang, Q.; Meng, X.; Li, T.; Long, M.; Fan, X.; Yu, Q.; et al. Down-Regulation of Lipid Metabolism in the Hepatopancreas of Shrimp Litopenaeusvannamei upon Light and Heavy Infection of Enterocytozoonhepatopenaei: A Comparative Proteomic Study. Int. J. Mol. Sci. 2022, 23, 11574. [Google Scholar] [CrossRef]
- Trentmann, O.; Horn, M.; van Scheltinga, A.C.T.; Neuhaus, H.E.; Haferkamp, I. Enlightening energy parasitism by analysis of an ATP/ADP transporter from chlamydiae. PLoS Biol. 2007, 5, e231. [Google Scholar]
- Karthikeyan, K.; Sudhakaran, R. Experimental horizontal transmission of Enterocytozoonhepatopenaei in post-larvae of whiteleg shrimp, Litopenaeusvannamei. J. Fish Dis. 2019, 42, 397–404. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput.-Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Dasgupta, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Maneeruttanarungroj, C.; Nichols, S.E.; Lyons, T.M.; Tirado-Rives, J.; Jorgensen, W.L.; Yuthavong, Y.; Anderson, K.S. Exploiting structural analysis, in silico screening, and serendipity to identify novel inhibitors of drug-resistant falciparum malaria. ACS Chem. Biol. 2009, 4, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Salin, N.H.; Noordin, R.; Al-Najjar, B.O.; Kamarulzaman, E.E.; Yunus, M.H.; Karim, I.Z.A.; Nasim, N.N.M.; Zakaria, I.I.; Wahab, H.A. Identification of potential dual-targets anti-Toxoplasma gondii compounds through structure-based virtual screening and in-vitro studies. PLoS ONE 2020, 15, e0225232. [Google Scholar] [CrossRef]
- Ferreira, L.L.; Ferreira, R.S.; Palomino, D.L.; Andricopulo, A.D. Structure-based virtual screening and biochemical evaluation for the identification of novel trypanosoma brucei aldolase inhibitors. Curr. Top. Med. Chem. 2018, 18, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Maia, M.; Raimundo e Silva, J.P.; de Lima Nunes, T.A.; Saraiva de Sousa, J.M.; Soares Rodrigues, G.C.; Messias Monteiro, A.F.; Fechine Tavares, J.; da Franca Rodrigues, K.A.; Mendonça-Junior, F.J.B.; Scotti, L.; et al. Virtual screening and the in vitro assessment of the antileishmanial activity of lignans. Molecules 2020, 25, 2281. [Google Scholar] [CrossRef] [PubMed]
- Shinn, A.P.; Pratoomyot, J.; Griffiths, D.; Trong, T.Q.; Vu, N.T.; Jiravanichpaisal, P.; Briggs, M. Asian shrimp production and the economic costs of disease. Asian Fish. Sci. 2018, 31, 29–58. [Google Scholar] [CrossRef]
- Kim, B.S.; Jang, G.I.; Kim, S.M.; Kim, Y.S.; Jeon, Y.G.; Oh, Y.K.; Hwang, J.Y.; Kwon, M.G. First Report of Enterocytozoonhepatopenaei Infection in Pacific Whiteleg Shrimp (Litopenaeusvannamei) Cultured in Korea. Animals 2021, 11, 3150. [Google Scholar] [CrossRef]
- Narvaez-Ortiz, H.Y.; Lopez, A.J.; Gupta, N.; Zimmermann, B.H. A CTP synthase undergoing stage-specific spatial expression is essential for the survival of the intracellular parasite Toxoplasma gondii. Front. Cell. Infect. Microbiol. 2018, 8, 83. [Google Scholar] [CrossRef] [Green Version]
- Kassel, K.M.; Higgins, M.J.; Hines, M.; Graves, L.M. Regulation of human cytidine triphosphate synthetase 2 by phosphorylation. J. Biol. Chem. 2010. 285, 33727–33736. [CrossRef] [Green Version]
- Denecke, J.; Kranz, C. Hypoglycosylation due to dolichol metabolism defects. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 888–895. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.F.; Carman, G.M. CTP synthetase and its role in phospholipid synthesis in the yeast Saccharomyces cerevisiae. Prog. Lipid Res. 2008, 47, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Sellmeier, M.; Weinhold, B.; Münster-Kühnel, A. CMP-Sialic acid synthetase: The point of constriction in the sialylation pathway. In Sialo Glyco Chemistry and Biology, I. Topics in Current Chemistry; Gerardy-Schahn, R., Delannoy, P., Itzstein, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 139–167. [Google Scholar]
- Hofer, A.; Steverding, D.; Chabes, A.; Brun, R.; Thelander, L. Trypanosoma brucei CTP synthetase: A target for the treatment of African sleeping sickness. Proc. Natl. Acad. Sci. USA 2001, 98, 6412–6416. [Google Scholar] [CrossRef]
- Hendriks, E.F.; O’Sullivan, W.J.; Stewart, T.S. Molecular cloning and characterization of the Plasmodium falciparum cytidine triphosphate synthetase gene. Biochim. Biophys. Acta. 1998, 1399, 213–218. [Google Scholar]
- Jimenez, B.M.; O’Sullivan, W.J. CTP synthetase and enzymes of pyrimidine ribonucleotide metabolism in Giardia intestinalis. Int. J. Parasitol. 1994, 24, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Guo, C.J.; Chang, C.C.; Zhong, J.; Hu, H.H.; Lu, G.M.; Liu, J.L. Structural basis for ligand binding modes of CTP synthase. Proc. Natl. Acad. Sci. USA 2021, 118, e2026621118. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.O.O.; Crispim, M.; Silber, A.M.; Damasceno, F.S. Glutamine analogues impair cell proliferation, the intracellular cycle and metacyclogenesis in Trypanosoma cruzi. Molecules 2020, 25, 1628. [Google Scholar]
- Dize, D.; Tata, R.B.; Keumoe, R.; KouipouToghueo, R.M.; Tchatat, M.B.; Njanpa, C.N.; Tchuenguia, V.C.; Yamthe, L.T.; Fokou, P.V.T.; Laleu, B.; et al. Preliminary Structure–Activity Relationship Study of the MMV Pathogen Box Compound MMV675968 (2, 4-Diaminoquinazoline) Unveils Novel Inhibitors of Trypanosoma brucei brucei. Molecules 2022, 27, 6574. [Google Scholar] [CrossRef]
- Chen, M.J.; Shimada, T.; Moulton, A.D.; Cline, A.; Humphries, R.K.; Maizel, J.; Nienhuis, A.W. The functional human dihydrofolate reductase gene. J. Biol. Chem. 1984, 259, 3933–3943. [Google Scholar]
- Klon, A.E.; Héroux, A.; Ross, L.J.; Pathak, V.; Johnson, C.A.; Piper, J.R.; Borhani, D.W. Atomic structures of human dihydrofolate reductase complexed with NADPH and two lipophilic antifolates at 1.09 Å and 1.05 Å resolution. J. Mol. Biol. 2002, 320, 677–693. [Google Scholar] [CrossRef]
- Pelphrey, P.M.; Popov, V.M.; Joska, T.M.; Beierlein, J.M.; Bolstad, E.S.; Fillingham, Y.A.; Wright, D.L.; Anderson, A.C. Highly efficient ligands for dihydrofolate reductase from Cryptosporidium hominis and Toxoplasma gondii inspired by structural analysis. J. Med. Chem. 2007, 50, 940–950. [Google Scholar] [CrossRef]
- Ferrari, S.; Morandi, F.; Motiejunas, D.; Nerini, E.; Henrich, S.; Luciani, R.; Venturelli, A.; Lazzari, S.; Calo, S.; Gupta, S.; et al. Virtual screening identification of nonfolate compounds, including a CNS drug, as antiparasitic agents inhibiting pteridine reductase. J. Med. Chem. 2011, 54, 211–221. [Google Scholar] [CrossRef]
- Lam, T.; Hilgers, M.T.; Cunningham, M.L.; Kwan, B.P.; Nelson, K.J.; Brown-Driver, V.; Ong, V.; Trzoss, M.; Hough, G.; Shaw, K.J.; et al. Structure-based design of new dihydrofolate reductase antibacterial agents: 7-(benzimidazol-1-yl)-2, 4-diaminoquinazolines. J. Med. Chem. 2014, 57, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Santucci, M.; Luciani, R.; Gianquinto, E.; Pozzi, C.; Pisa, F.D.; delloIacono, L.; Landi, G.; Tagliazucchi, L.; Mangani, S.; Spyrakis, F.; et al. Repurposing the trypanosomatidicgskkinetobox for the inhibition of parasitic pteridine and dihydrofolate reductases. Pharmaceuticals 2021, 14, 1246. [Google Scholar] [CrossRef] [PubMed]
- Solbiati, J.; Chapman-Smith, A.; Miller, J.L.; Miller, C.G.; Cronan, J.E., Jr. Processing of the N termini of nascent polypeptide chains requires deformylation prior to methionine removal. J. Mol. Biol. 1999, 290, 607–614. [Google Scholar] [CrossRef]
- Tucker, L.A.; Zhang, Q.; Sheppard, G.S.; Lou, P.; Jiang, F.; McKeegan, E.; Lesniewski, R.; Davidsen, S.K.; Bell, R.L.; Wang, J. Ectopic expression of methionine aminopeptidase-2 causes cell transformation and stimulates proliferation. Oncogene 2008, 27, 3967–3976. [Google Scholar] [CrossRef]
- Katinka, M.D.; Duprat, S.; Cornillot, E.; Metenier, G.; Thomarat, F.; Prensier, G.; Barbe, V.; Peyretaillade, E.; Brottier, P.; Wincker, P.; et al. Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature 2001, 414, 450–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Huang, H.; Cali, A.; Takvorian, P.M.; Feng, X.; Zhou, G.; Weiss, L.M. Investigations into microsporidian methionine aminopeptidase type 2: A therapeutic target for microsporidiosis. Folia Parasitol. 2005, 52, 182. [Google Scholar] [CrossRef] [Green Version]
- Alvarado, J.J.; Nemkal, A.; Sauder, J.M.; Russell, M.; Akiyoshi, D.E.; Shi, W.; Almo, S.C.; Weiss, L.M. Structure of a microsporidian methionine aminopeptidase type 2 complexed with fumagillin and TNP-470. Mol. Biochem. Parasitol. 2009, 168, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Padia, J.; Kulakova, L.; Galkin, A.; Herzberg, O. Discovery and preclinical development of antigiardiasisfumagillol derivatives. Antimicrob. Agents Chemother. 2020, 64, e00582-20. [Google Scholar] [CrossRef]
- Marino, J.P.; Fisher, P.W.; Hofmann, G.A.; Kirkpatrick, R.B.; Janson, C.A.; Johnson, R.K.; Ma, C.; Mattern, M.; Meek, T.D.; Ryan, M.D.; et al. Highly potent inhibitors of methionine aminopeptidase-2 based on a 1, 2, 4-triazole pharmacophore. J. Med. Chem. 2007, 50, 3777–3785. [Google Scholar] [CrossRef]
- Morgen, M.; Jöst, C.; Malz, M.; Janowski, R.; Niessing, D.; Klein, C.D.; Gunkel, N.; Miller, A.K. Spiroepoxytriazoles are fumagillin-like irreversible inhibitors of MetAP2 with potent cellular activity. ACS Chem. Biol. 2016, 11, 1001–1011. [Google Scholar] [CrossRef]
- Wei, J.; Fei, Z.; Pan, G.; Weiss, L.M.; Zhou, Z. Current therapy and therapeutic targets for microsporidiosis. Front. Microbiol. 2022, 13, 835390. [Google Scholar] [CrossRef] [PubMed]
- Reichard, P. Interactions between deoxyribonucleotide and DNA synthesis. Annu. Rev. Biochem. 1988, 57, 349–374. [Google Scholar] [CrossRef] [PubMed]
- Timm, J.; Bosch-Navarrete, C.; Recio, E.; Nettleship, J.E.; Rada, H.; Gonzalez-Pacanowska, D.; Wilson, K.S. Structural and kinetic characterization of thymidine kinase from Leishmania major. PLoS Negl. Trop. Dis. 2015, 9, e0003781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, M.; Timm, J.; Castillo-Acosta, V.M.; Ruiz-Pérez, L.M.; Balzarini, T.; Nettleship, J.E.; Bird, L.E.; Rada, H.; Wilson, K.S.; González-Pacanowska, D. Cell cycle regulation and novel structural features of thymidine kinase, an essential enzyme in Trypanosoma brucei. Mol. Microbiol. 2016, 102, 365–385. [Google Scholar] [CrossRef] [PubMed]
- Skovgaard, T.; Uhlin, U.; Munch-Petersen, B. Comparative active-site mutation study of human and Caenorhabditis elegans thymidine kinase 1. FEBS J. 2012, 279, 1777–1787. [Google Scholar] [CrossRef]
- Birringer, M.S.; Claus, M.T.; Folkers, G.; Kloer, D.P.; Schulz, G.E.; Scapozza, L. Structure of a type II thymidine kinase with bound dTTP. FEBS Lett. 2005, 579, 1376–1382. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Carrero-Lérida, J.; Silva, A.P.; Whittingham, J.L.; Brannigan, J.A.; Ruiz-Pérez, L.M.; Read, K.D.; Wilson, K.S.; González-Pacanowska, D.; Gilbert, I.H. Synthesis and evaluation of α-thymidine analogues as novel antimalarials. J. Med. Chem. 2012, 55, 10948–10957. [Google Scholar] [CrossRef]
- Sykes, M.L.; Hilko, D.H.; Kung, L.I.; Poulsen, S.A.; Avery, V.M. Investigation of pyrimidine nucleoside analogues as chemical probes to assess compound effects on the proliferation of Trypanosoma cruzi intracellular parasites. PLoS Negl. Trop. Dis. 2020, 14, e0008068. [Google Scholar] [CrossRef] [Green Version]
- Enninful, K.S.; Kwofie, S.K.; Tetteh-Tsifoanya, M.; Lamptey, A.N.; Djameh, G.; Nyarko, S.; Ghansah, A.; Wilson, M.D. Targeting the Plasmodium falciparum’s Thymidylate Monophosphate Kinase for the Identification of novel antimalarial natural compounds. Front. Cell. Infect. Microbiol. 2022, 12, 868529. [Google Scholar] [CrossRef]
- Tani, K.; Fujiyoshi, Y. Water channel structures analysed by electron crystallography. Biochim. Biophys. Acta 2014, 1840, 1605–1613. [Google Scholar] [CrossRef]
- Abir-Awan, M.; Kitchen, P.; Salman, M.M.; Conner, M.T.; Conner, A.C.; Bill, R.M. Inhibitors of mammalian aquaporin water channels. Int. J. Mol. Sci. 2019, 20, 1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenc, D.; Song, J.; Almasalmeh, A.; Wu, B.; Beitz, E. The arginine-facing amino acid residue of the rat aquaporin 1 constriction determines solute selectivity according to its size and lipophilicity. Mol. Membr. Biol. 2014, 31, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Kozono, D.; Yasui, M.; King, L.S.; Agre, P. Aquaporin water channels: Atomic structure molecular dynamics meet clinical medicine. J. Clin. Investig. 2002, 109, 1395–1399. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Polonais, V.; Sugi, T.; Yakubu, R.; Takvorian, P.M.; Cali, A.; Maier, K.; Long, M.; Levy, M.; Tanowitz, H.B.; et al. The role of microsporidian polar tube protein 4 (PTP4) in host cell infection. PLoS Pathog. 2017, 13, e1006341. [Google Scholar] [CrossRef] [PubMed]
- Jaroenlak, P.; Cammer, M.; Davydov, A.; Sall, J.; Usmani, M.; Liang, F.X.; Ekiert, D.C.; Bhabha, G. 3-Dimensional organization and dynamics of the microsporidian polar tube invasion machinery. PLoS Pathog. 2020, 16, e1008738. [Google Scholar] [CrossRef]
- Frixione, E.; Ruiz, L.; Cerbon, J.; Undeen, A.H. Germination of Nosema algerae (Microspora) spores: Conditional inhibition by D2O, ethanol and Hg2+ suggests dependence of water influx upon membrane hydration and specific transmembrane pathways. J. Eukaryot. Microbiol. 1997, 44, 109–116. [Google Scholar] [CrossRef]
- Ho, J.D.; Yeh, R.; Sandstrom, A.; Chorny, I.; Harries, W.E.; Robbins, R.A.; Miercke, L.J.; Stroud, R.M. Crystal structure of human aquaporin 4 at 1.8 Å and its mechanism of conductance. Proc. Natl. Acad. Sci. USA 2009, 106, 7437–7442. [Google Scholar] [CrossRef] [Green Version]
- Yadav, D.K.; Kumar, S.; Choi, E.H.; Chaudhary, S.; Kim, M.H. Computational modeling on aquaporin-3 as skin cancer target: A virtual screening study. Front. Chem. 2020, 8, 250. [Google Scholar] [CrossRef] [Green Version]
- Kamegawa, A.; Hiroaki, Y.; Tani, K.; Fujiyoshi, Y. Two-dimensional crystal structure of aquaporin-4 bound to the inhibitor acetazolamide. J. Electron. Microsc. 2016, 65, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.Y.; Bachtiar, M.; Choo, C.C.; Lee, C.G. Comprehensive review of Hepatitis BV irus-associated hepatocellular carcinoma research through text mining and big data analytics. Biol. Rev. 2019, 94, 353–367. [Google Scholar] [CrossRef]
- Pu, J.; Yu, Y.; Liu, Y.; Tian, L.; Gui, S.; Zhong, X.; Fan, C.; Xu, S.; Song, X.; Liu, L.; et al. MENDA: A comprehensive curated resource of metabolic characterization in depression. Brief. Bioinform. 2020, 21, 1455–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Chistyakov, V.V.; Thornton, J.M. PDBSummore:new summaries and analysis of the known 3D structure of proteins and nucleic acids. Nucleic Acids Res. 2005, 33, 266–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006, 34, W116–W118. [Google Scholar] [CrossRef] [Green Version]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Pires, D.E.; Veloso, W.N.; Myung, Y.; Rodrigues, C.H.; Silk, M.; Rezende, P.M.; Silva, F.; Xavier, J.S.; Velloso, J.P.; da Silveira, C.H.; et al. EasyVS: A user-friendly web-based tool for molecule library selection and structure-based virtual screening. Bioinformatics 2020, 36, 4200–4202. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Bauer, M.R.; Mackey, M.D. Electrostatic complementarity as a fast and effective tool to optimize binding and selectivity of protein–ligand complexes. J. Med. Chem. 2019, 62, 3036–3050. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucl. Acids. Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 2019, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Turq, P.; Lantelme, F.; Friedman, H.L. Brownian dynamics: Its application to ionic solutions. J. Chem. Phys. 1977, 66, 3039–3044. [Google Scholar] [CrossRef]
- Åqvist, J.; Wennerström, P.; Nervall, M.; Bjelic, S.; Brandsdal, B.O. Molecular dynamics simulations of water and biomolecules with a Monte Carlo constant pressure algorithm. Chem. Phys. Lett. 2004, 384, 288–294. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arantes, P.R.; Polêto, M.D.; Pedebos, C.; Ligabue-Braun, R. Making it rain: Cloud-based molecular simulations for everyone. J. Chem. Inf. Model. 2021, 61, 4852–4856. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins Struct. Funct. Genet. 2004, 55, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Tuccinardi, T. What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert Opin. Drug Discov. 2021, 16, 1233–1237. [Google Scholar] [CrossRef]
- Forouzesh, N.; Mishra, N. An effective MM/GBSA protocol for absolute binding free energy calculations: A case study on SARS-CoV-2 spike protein and the human ACE2 receptor. Molecules 2021, 26, 2383. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Coordinate of Docking Box | Structure | Amino Acid Residues |
|---|---|---|---|
| Aquaporin | X: 4.7541; Y: 0.6196; Z: 0.8248 |  | Phe22, Gly23, Val45, Val49, Glu138, Leu145, Gly198, Ala199, Phe200, Asn201, Pro202, Gly203, Ile204 |
| CTP synthase | X: −11.6484; Y: −1.5365; Z: 7.2675 |  | Ala59, Glu64, Ile65, Ile292, Thr293, Arg294, Tyr295, Val301, Tyr302, Leu305, Cys347, Pro348, Gly349, Gly350, Phe351, Gly352, Thr354, Lys359, Ile376, Cys377, Leu378, Arg453, His499, Glu502, Leu503 |
| Thymidine kinase | X: 6.300; Y: −0.1442; Z: −3.7617 |  | Val11, Ser12, Cys13, Gly14, Lys15, Thr16, Ilc17, Lys39, Asp43, Arg45, Tyr46, Ser50, Ile51, Lys52, Ser53, Ala54, Asp85, Glu86, Gln88, Phe89, Gly113, Leu114, Lys116, Asp117, Phe118, Phe123, Ser161, Lys180, Cys183, Gly184, Gly185, Ile186, Tyr189 |
| Dihydrofolate reductase | X: 3.5346; Y: 3.8013;Z: 4.8650 |  | Leu27, Val28, Ala29, Ile37, Ser38, Gly40, Glu41, Lys42, Met43, Trp45, Arg47, Leu48, Ser4, Asp51, Phe52, Ala53, Met55, Lys56, Met59, Gly70, Arg71, Lys72, Thr73, Glu75, Val76, Ala77, Lys78, Tyr79, Thr80, Asn81, Tyr82, Leu87, Ser88, Arg89, Lys102, ser103, Phe104, Ala119, Gly120, Thr138, Arg150 |
| Methionine Aminopeptidase 2 |  | Asn75, Asn76, Gly77, Ile78, Gly79, Phe80, Pro81, Gly83, Ser85, Ala90, Ala91, His92, Lys111, Asp113, Asp124, Leu191, His194, Ile201, His202, Glu226, Phe228, His224, Phe245, Met246, Pro275, Pro303, Try304, Pro305, Pro306, Leu307, Gln317, Glu319 |
| Protein Name | E. hepatopenaei | L. vannamei |
|---|---|---|
| Aquaporin | Phe22, Gly23, Val45, Val49, Glu138, Leu145, Gly198, Ala199, Phe200, Asn201, Pro202, Gly203, Ile204 | A0A193KUU7: Leu36, Val37, Ile59, Phe63, Glu149, Pro156, Pro200, Ala201, Arg 202 |
| A0A3R7N2N5: Glu60, Leu67, Phe131, Cys232, Phe233, Pro234, Pro265, Asn226, Pro267 | ||
| A0A3R7PEU6: Met26, Gly30, Tyr112,Trp119 | ||
| A0A3R7Q089: Phe25, Gly26, Trp49, Gln156, Leu163, Ala284, Ser285, Leu286, Gly287 | ||
| CTP synthase | Ala59, Glu64, Ile65, Ile292, Thr293, Arg294, Tyr295, Val301, Tyr302, Leu305, Cys347, Pro348, Gly349, Gly350, Phe351, Gly352, Thr354, Lys359, Ile376, Cys377, Leu378, Arg453, His499, Glu502, Leu503 | Phe64, Gly69, Val70, Val317, Gly318, Lys319, Tyr320, Ser326, Tyr327, Val330, Val382, Pro383, Gly384, Ile385, Gly386, Gly387, Arg389, Lys394, Val411, Cys412, Leu413, Gly492, Val530, Tyr 532,Val534 |
| Dihydrofolate reductase | Leu27, Val28, Ala29, Ile37, Ser38, Gly40, Glu41, Lys42, Met43, Trp45, Arg47, Leu48, Ser4, Asp51, Phe52, Ala53, Met55, Lys56, Met59, Gly70, Arg71, Lys72, Thr73, Glu75, Val76, Ala77, Lys78, Tyr79, Thr80, Asn81, Tyr82, Leu87, Ser88, Arg89, Lys102, Ser103, Phe104, Ala119, Gly120, Thr138, Arg150 | Val2, Tyr3, Ile15, Ala16, Asn19, Asn20, Glu21, Leu22, Trp24, His26, Glu27, Gly33, Asp35, Phe36, Gly37, Ser39, Ala40, Gln43, Gly51, Arg52, Lys53, Thr54, Asp56, Val58, Ala59, Gly60, Phe61, Asp62, Pro66, Tyr67, Phe72, Val73, Leu74, Lys88, Val89, Phe90, Ala101, Gly107, Try108, Asn109, Glu110, Leu111, Tyr112, Asp114, Pro145 |
| Methionine aminopeptidase | Asn75, Asn76, Gly77, Ile78, Gly79, Phe80, Pro81, Gly83, Ser85, Ala90, Ala91, His92, Lys111, Asp113, Asp124, Leu191, His194, Ile201, His202, Glu226, Phe228, His224, Phe245, Met246, Pro275, Pro303, Try304, Pro305, Pro306, Leu307, Gln317, Glu319 | A0A423SS39: Lys201, Ala202, Gly203, Leu204, Ala205, Phe206, Pro207, Gly209, Ser211, Ala216, Ala217, H218, Lys238, Asp240, Asp251, Leu315, His318, Ile325, His326, Glu351, Tyr370, Met371, Ala401, Pro430, Tyr431, Pro432, Pro433, Leu434, Gln317, Glu446 |
| A0A423SV11: His3, Ile10, His11, Glu36, Tyr55, Met56, Ala73, P102, Tyr103, Pro104, Pro105, Leu106, Gln116, Glu118 | ||
| A0A423SSE3: Asn176, Try177, His178, Gly179, Phe180, Phe181, Ser183, Ser187, Ile192, Cys193, His194, Asn209, Asp211, Phe219, His220, Trp331, Pro332, Gln347, Glu349 | ||
| A0A423TUC3: Leu79, Tyr80, His81, Gly82, Phe83, Pro84, Ser86, Ser90, Ile95, Cys96, His97, Asn112, Asp114, Phe122, His123, Gln196, Leu198 | ||
| A0A423SU95: Leu148, Asn149, Tyr150, His151, Gly152, Phe153, Pro154, Ser156, Ser160, Ala165, Cys166, His167, Asn182, Asp184, Tyr192, His193, Gly203, Glu222, Ala223, Tyr271, Gly284, Thr286 | ||
| Thymidine kinases | Val11, Ser12, Cys13, Gly14, Lys15, Thr16, Ilc17, Lys39, Asp43, Arg45, Tyr46, Ser50, Ile51, Lys52, Ser53, Ala54, Asp85, Glu86, Gln88, Phe89, Gly113, Leu114, Lys116, Asp117, Phe118, Phe123, Ser161, Lys180, Cys183, Gly184, Gly185, Ile186, Tyr189 | Gly4, Lys5, Thr6, Thr7, Asp31, Arg33, Tyr34, Gly38, Ile39, Ala40, Thr41, His42, Asp78, Thr79, Glu81, Pro107, Arg146, Phe159, Glu161, Vol162, Gly164, Ser166, Tyr169 |
| Easy Vs | Flare | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Molecular ID | Affinity Score | Total Number of Bonds | PatchDock Score | Electrostatic Complementarity Score | Electrostatic Complementarity (Pearson R) | Electrostatic Complementarity (Spearman’s Rho) | Number of Hydrogen Bonds | Amino Acids Involved in Hydrogen Bonds (Bond Distance Å) | Number of π–π Bonds | Amino Acids in π–π Bond(s) (Bond Distance Å) |
| Aquaporin | ||||||||||
| CHEMBL3703838 | −8 | 9 | 0.286 | 0.337 | 0.335 | 5 | Gly197 (1.8), Gly198 (2.9), Ala 199 (1.8), Lys34 (2.7) (2.7) | 1 | Thr121 (2.6) | |
| ZINC000002243083 | −7.6 | 9 | 0.302 | 0.451 | 0.325 | 6 | Gly198 (2.4) (1.7), Thr33 (1.8), Ala123 (2.5) (2.6), Ser196 (2.2) | |||
| CHEMBL133039 | −7.6 | 9 | 0.337 | 0.448 | 0.549 | 5 | Gly198 (2.1 Å), Gly197 (2.1 Å), Leu119 (2.5 Å), Thr33 (1.8 Å), Ile120 (1.7 Å) | 1 | Lys34 (3.7) | |
| CHEMBL3140193 | −7.5 | 9 | 0.295 | 0.361 | 0.341 | 6 | Gly189 (1.9), Gly46 (3.0), Ser193 (1.7) (2.7), Lys34 (2.5), Thr33 (2.2) | 1 | Thr33 (3.9) | |
| CHEMBL2132563 | −7.5 | 9 | 0.289 | 0.303 | 0.311 | 5 | Gly197 (2.6), Ser193 (3.0), Lys34 (2.6), Thr33 (1.9) (2.4) | |||
| CTPsynthase | ||||||||||
| CHEMBL48494 | −9.6 | 11 | 5011 | 0.331 | 0.249 | 0.284 | 7 | Arg453 (2.2), Ile65 (2.0), Tyr295 (2.2), Gly349 (2.6), Glu501 (2.0) (1.7), Gly349 (2.3) | 3 | Phe351 (3.1) (3.1), Arg453 (3.1) |
| CHEMBL1162979 | −8.6 | 16 | 5250 | 0.301 | 0.467 | 0.459 | 13 | Gly55 (1.7), Ala59 (2.6), Ile65 (2.5) (2.9), Glu64 (2.3), Glu501 (1.8), Gly349 (2.3), Arg453 (2.8) (3.0) (2.3) (2.1), His499 (2.8), Gly350 (2.5) | 2 | Arg453 (4.1), Phe351 (3.9) |
| CHEMBL133039 | −8.3 | 12 | 5496 | 0.296 | 0.409 | 0.421 | 8 | Ile65 (2.1), Phe351 (1.9) (2.7), Tyr295 (2.4), Val301 (3.4), Glu64 (2.1), Gly349 (2.0) (2.4) | 3 | Phe351 (3.0), Arg453 (4.3), Tyr302 (3.3) |
| CHEMBL525202 | −8.1 | 11 | 5494 | 0.291 | 0.367 | 0.429 | 10 | Phe351 (2.6), Vol301 (2.9), Ile65 (2.6) (1.9) (2.6), Arg453 (2.3) (2.6) (2.2) (2.4), Tyr295 (1.9) | ||
| CHEMBL1091856 | −7.6 | 17 | 5128 | 0.297 | 0.408 | 0.364 | 15 | Ala59 (2.3), Tyr295 (2.2), Arg453 (2.9) (2.2) (2.1) (2.5), Phe351 (2.6) (2.4), Ile65 (1.8) (1.8), Glu64 (2.4) (1.9), Glu501 (1.8), Gly349 (2.8), His499 (2.9) | 1 | Phe351 (2.9) |
| Thymidine kinase | ||||||||||
| CHEMBL3674540 | −9.5 | 11 | 5118 | 0.286 | 0.389 | 0.433 | 7 | Glu86 (2.5) (2.7) (2.1), Lys15 (2.5), Gly14 (2.3), Ala54 (2.0), Ile186 (2.2), | ||
| CHEMBL1683320 | −8.6 | 15 | 4888 | 0.267 | 0.352 | 0.381 | 13 | Ser12 (1.9) (2.7), Gly14 (3.0), Arg45 (2.4) (2.0), Glu86 (2.8) (1.9) (1.9), Thr16 (2.8) (2.1) (2.2), Ile17 (2.2), Ala54 (2.3) | ||
| CHEMBL391279 | −8.3 | 16 | 4702 | 0.329 | 0.461 | 0.521 | 12 | Ile186 (2.7), Tyr46 (3.0) (2.4), Arg45 (3.0), Glu86 (2.1) (2.9), Thr16 (1.8), Glu86 (2.1) (2.9), Lys15 (2.1) (2.4), Pro10 (2.9), Cys13 (2.8) | 1 | Arg45 (4.9) |
| ZINC000031750813 | −8.2 | 14 | 3210 | 0.308 | 0.531 | 0.449 | 10 | Gly14 (2.6), Ile17 (3.0), Ala54 (2.5), Thr16 (2.4) (2.3), Ser12 (2.1) (2.3), Gly86 (2.0) (2.3) (2.0) | 1 | Arg45 (3.4) |
| CHEMBL4078273 | −8.1 | 12 | 5638 | 0.312 | 0.37 | 0.352 | 8 | Glu86 (1.7) (2.9) (2.9) (2.0), Arg45 (2.4), Ser12 (2.3) (2.8), Thr16 (1.9) | 1 | Tyr46 (4.5) |
| Dihydrofolate reductase | ||||||||||
| CHEMBL1966988 | −8.7 | 11 | 6342 | 0.287 | 0.421 | 0.477 | 7 | Ile37 (1.9) (2.6), Gly40 (2.2), Lys72 (2.5), Gly121 (1.8), Arg150 (1.8) (2.8) | ||
| CHEMBL340488 | −8.1 | 9 | 6330 | 0.311 | 0.353 | 0.343 | 7 | Lys87 (1.9), Lys72 (2.1), Glu123 (2.8), Arg150 (2.9) (2.2) (2.9), Ser38 (2.2) | ||
| ZINC000016682862 | −8.0 | 11 | 7034 | 0.287 | 0.408 | 0.399 | 6 | Arg150 (2.4), Ile37 (2.3), Gly121 (2.2), Tyr79 (2.8), Lys78 (2.1) | 2 | Phe52 (2.9), Tyr79 (2.7) |
| ZINC000828645375 | −7.7 | 14 | 6246 | 0.351 | 0.518 | 0.521 | 8 | Thr73 (2.1) (2.6), Gly121 (3.0) (2.2), Tyr125 (2.0), Ile37 (1.7), Ala29 (2.2), Tyr125 (2.0) | 3 | Met55 (3.0), Phe52 (2.7), Tyr79 (2.6) |
| CHEMBL3901573 | −7.5 | 11 | 4999 | 0.306 | 0.411 | 0.369 | 8 | Arg150 (2.5) (2.5) Ser38 (2.2), Gly121 (1.9), Ala199 (2.0), Tyr125 (2.4), Leu27 (2.1), Ala29 (2.1) | 1 | Met55 (3.8) |
| Methionine aminopeptidase 2 | ||||||||||
| CHEMBL3913373 | −9 | 11 | 5428 | 0.299 | 0.311 | 0.371 | 9 | Asn192 (2.1) (2.1), Ser206 (2.1), Glu226 (2.0) (2.9) (2.8), His194 (2.7), His202 (2.2), His244 (2.8) | 1 | His92 (2.6) |
| CHEMBL1962731 | −8.9 | 15 | 5458 | 0.282 | 0.426 | 0.399 | 6 | Asn192 (2.1), Glu226 (2.0), His202 (2.1), Asp113 (2.9) (2.4), Pro81 (2.6) | 2 | His202 (4.6), His92 (2.3) |
| CHEMBL3142997 | −8.1 | 12 | 5264 | 0.323 | 0.436 | 0.536 | 8 | Ser206 (1.8), Asn192 (1.8) (1.8) (2.2), Glu226 (2.6) (2.0), His202 (2.0) (2.3) | ||
| ZINC000199197855 | −8.0 | 13 | 5160 | 0.359 | 0.412 | 0.542 | 9 | Ser206 (2.3), Gly193 (2.9), Asn192 (1.8) (1.9) (2.0) (2.0), Asp124 (2.8), Asp113 (2.1), His92 (2.4), His202 (2.9) (2.0) | 2 | His (4.5), Tyr304 (3.1) |
| ZINC000016682972 | −7.9 | 15 | 5676 | 0.328 | 0.309 | 0.492 | 8 | His202 (2.4), Glu226 (1.9), Gln317 (2.9), His92 (2.7), Asp113 (2.4) (1.9) (1.7) | 2 | His92 (4.7), Phe80 (5.0) |
| Molecular ID | Chemical Name | Chemical Structure | Binding Energy |
|---|---|---|---|
| Aquaporin | |||
| CHEMBL3703838 | (2S,3R,4R,5S,6R)-2-[4-chloro-3-[(4,4-dioxo-2,3-dihydro-1,4lambda6-benzoxathiin-6-yl)methyl]phenyl]-6-(hydroxymethyl)oxane-3,4,5-triol |  | −36.0654 ± 2.6122 |
| ZINC000002243083 | 2-Naphthol-3,6,8-trisulfonic acid |  | −2.8214 ± 4.9570 |
| CHEMBL133039 | Sodium;2-[3,5-dihydroxy-4-[3-(3-hydroxy-4-methoxyphenyl)propanimidoyl]phenoxy]acetate |  | −30.5427 ± 3.1371 |
| CHEMBL3140193 | [(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl] (1Z)-2,3-dichloro-N-sulfooxybenzenecarboximidothioate |  | −21.3990 ± 6.5936 |
| CHEMBL2132563 | (2R,3R,4S,5R,6R)-2-[4-[3-(4-fluorophenyl)-1,2,4-oxadiazol-5-yl]-1,3-thiazol-2-yl]-6-(hydroxymethyl)oxane-3,4,5-triol |  | −32.1805 ± 4.0288 |
| CTPsynthase | |||
| CHEMBL48494 | 2-[(3-Amino-4,5,6-trihydroxyoxan-2-yl)oxymethyl]anthracene-9,10-dione |  | −37.1662 ± 3.5275 |
| CHEMBL1162979 | sodium;[(2S,3S,4R,5R,6R)-3,5-dihydroxy-2-[[3-hydroxy-5-[(Z)-2-(4-hydroxyphenyl)ethenyl]phenyl]methyl]-6-(hydroxymethyl)oxan-4-yl] sulfate |  | −45.4045 ± 2.6946 |
| CHEMBL133039 | Sodium;2-[3,5-dihydroxy-4-[3-(3-hydroxy-4-methoxyphenyl)propanimidoyl]phenoxy]acetate |  | −38.1244 ± 2.4261 |
| CHEMBL1091856 | (5-{[4-(Benzenesulfonyl)-1,2,5-oxadiazol-3-yl]oxy}-1-hydroxy-1-phosphonopentyl)phosphonic acid |  | −56.5194 ± 5.0206 |
| CHEMBL525202 | (2S,3R,4R,5R,6R)-2-[[2-(benzylamino)-4-methyl-1,3-thiazol-5-yl]methyl]-6-(hydroxymethyl)oxane-3,4,5-triol |  | −44.3102 ± 6.8784 |
| Thymidine kinase | |||
| CHEMBL3674540 | (E)-2-(Amino(1-(quinolin-6-ylmethyl)-1H-[1–3]triazolo[4,5-b]pyrazin-6-yl)methylene)hydrazinecarboxamide |  | −25.6970 ± 4.4414 |
| CHEMBL1683320 | [(2R,3S,4R,5R)-5-[4-(4-fluorophenyl)triazol-1-yl]-3,4-dihydroxyoxolan-2-yl]methyl dihydrogen phosphate |  | −26.6667 ± 4.9470 |
| CHEMBL391279 | (1S,3R,4R,7S)-3-(6-aminopurin-9-yl)-7-hydroxy-N-methyl-2-oxa-5-azabicyclo[2.2.1]heptane-1-carboxamide |  | −9.1083 ± 4.8425 |
| CHEMBL4078273 | [(2R,3S,4S,5R,6S)-6-(4-hexylphenoxy)-3,4,5-trihydroxyoxan-2-yl]methyl hydrogen sulfate |  | −33.5752 ± 4.6211 |
| ZINC000031750813 | 3-(2,3-dihydro-1H-tetrazol-5-yl)-6-(2H-tetrazol-5-yl)-1,2,4,5-tetrazine |  | −20.8656 ± 3.4276 |
| Dihydrofolate reductase | |||
| CHEMBL1966988 | 2-[4-[(2Z)-2-(2-oxooxolan-3-ylidene)hydrazinyl]phenyl]sulfonylguanidine |  | −28.0600 ± 3.0231 |
| CHEMBL340488 | ((S)-2-Amino-propionyl)-sulfamic acid (2R,3S,4R,5R)-3,4-dihydroxy-5-(4-phenyl-thiazol-2-yl)-tetrahydro-furan-2-ylmethyl ester |  | −35.9711 ± 3.1254 |
| ZINC000016682862 | ethyl 2-[[5-(benzoylcarbamothioylamino)-3,4-dicarbamoyl-1H-pyrrol-2-yl]sulfanyl]acetate |  | −23.2559 ± 3.125 |
| ZINC000828645375 | 2-(4-chlorophenyl)-5-[methyl(2,3,4,5,6-pentahydroxyhexyl)amino]-1,3-oxazole-4-carbonitrile |  | −28.9491 ± 6.23366 |
| CHEMBL3901573 | [(1R,2R,3S,4R)-2,3-dihydroxy-4-(pyridine-2-carbonylamino)cyclopentyl]methyl sulfamate |  | −13.5906 ± 6.5678 |
| Methionine aminopeptidase 2 | |||
| CHEMBL3913373 | 1-(4-hydroxy-3-nitrophenyl)-3-[(2,4,6-trioxo-1-phenyl-1,3-diazinane-5-carbonyl)amino]urea |  | −43.5796 ± 5.1314 |
| CHEMBL1962731 | 3-amino-5-(2-benzylsulfonylethylamino)-6-chloro-N-(diaminomethylidene)pyrazine-2-carboxamide |  | −29.9671 ± 9.8188 |
| CHEMBL3142997 | ((5-(2,4-Dioxo-5-vinyl-3,4-dihydro-2H-pyrimidin-1-yl)-3-hydroxy-tetrahydro-furan-2-ylmethoxy)-hydroxy-phosphoryl)acetic acid |  | −30.0314 ± 5.2458 |
| ZINC000199197855 | [(3S,4R,5R)-3,4,5,6-tetrahydroxy-2-oxohexyl] (2S)-2-amino-3-phenylpropanoate |  | −28.6175 ± 6.0841 |
| ZINC000016682972 | ethyl (2S,3R,4R,5S)-5-(1,2,4-benzotriazin-3-yl)-2,3,4,5-tetrahydroxypentanoate |  | −32.6542 ± 6.1144 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paria, P.; Tassanakajon, A. Identification of Potential Druggable Targets and Structure-Based Virtual Screening for Drug-like Molecules against the Shrimp Pathogen Enterocytozoon hepatopenaei. Int. J. Mol. Sci. 2023, 24, 1412. https://doi.org/10.3390/ijms24021412
Paria P, Tassanakajon A. Identification of Potential Druggable Targets and Structure-Based Virtual Screening for Drug-like Molecules against the Shrimp Pathogen Enterocytozoon hepatopenaei. International Journal of Molecular Sciences. 2023; 24(2):1412. https://doi.org/10.3390/ijms24021412
Chicago/Turabian StyleParia, Prasenjit, and Anchalee Tassanakajon. 2023. "Identification of Potential Druggable Targets and Structure-Based Virtual Screening for Drug-like Molecules against the Shrimp Pathogen Enterocytozoon hepatopenaei" International Journal of Molecular Sciences 24, no. 2: 1412. https://doi.org/10.3390/ijms24021412