
A Pathogenic Variant Reclassified to the Pseudogene PMS2P1 in a Patient with Suspected Hereditary Cancer
 ,
,
Abstract
:1. Introduction
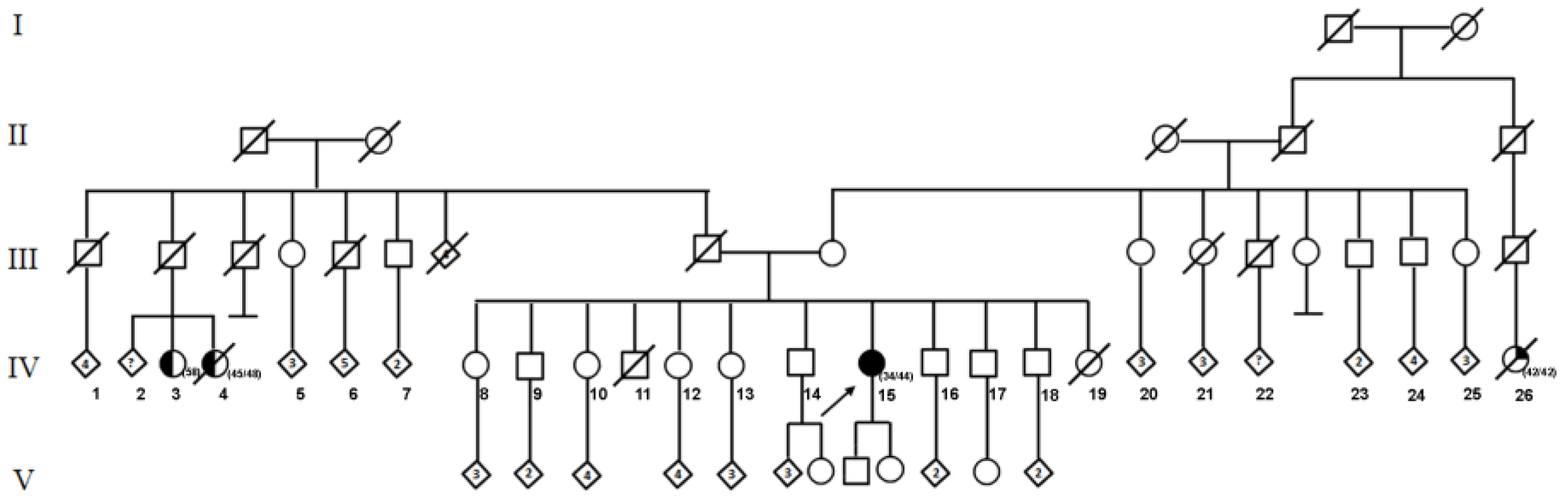
2. Case Presentation
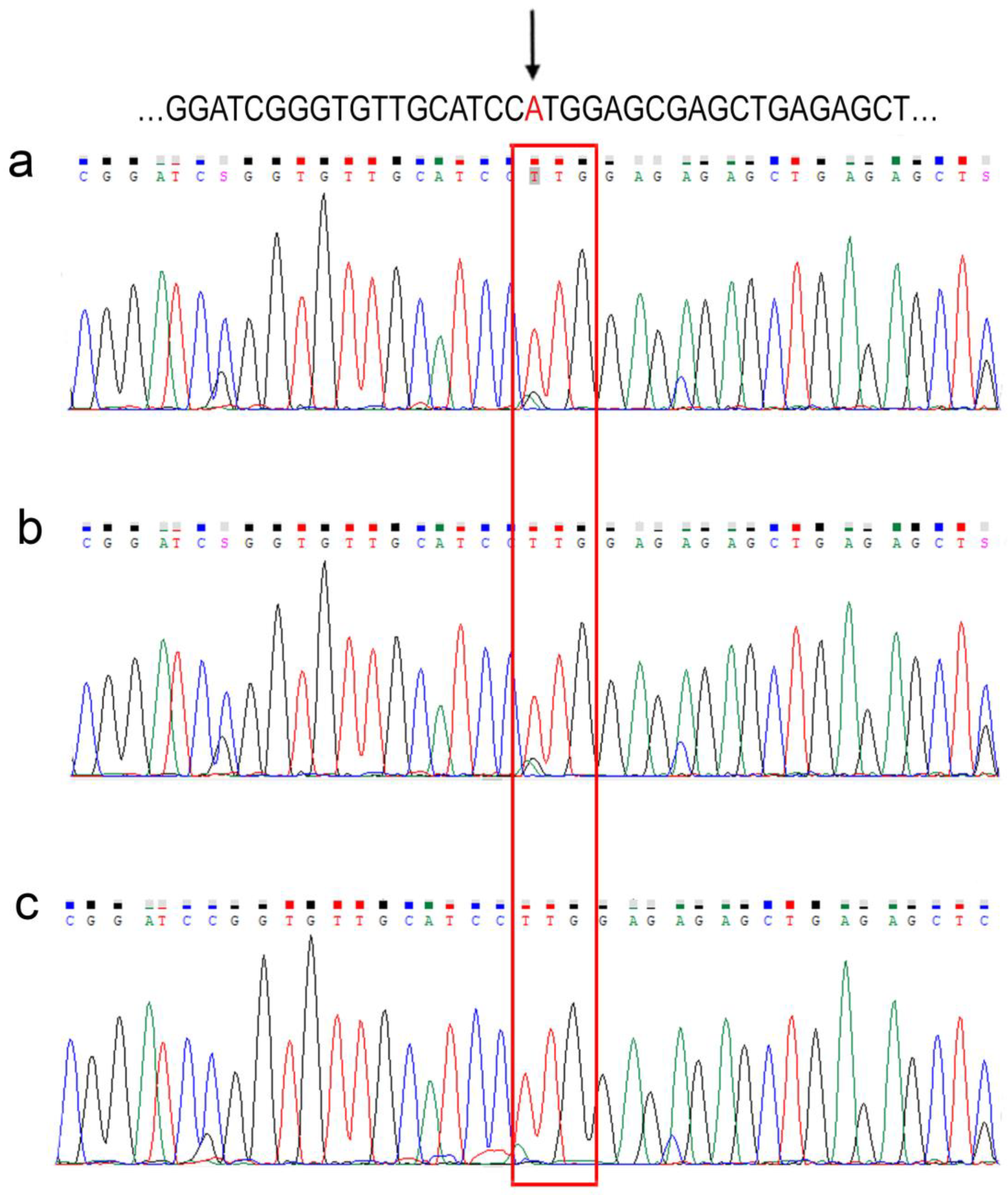
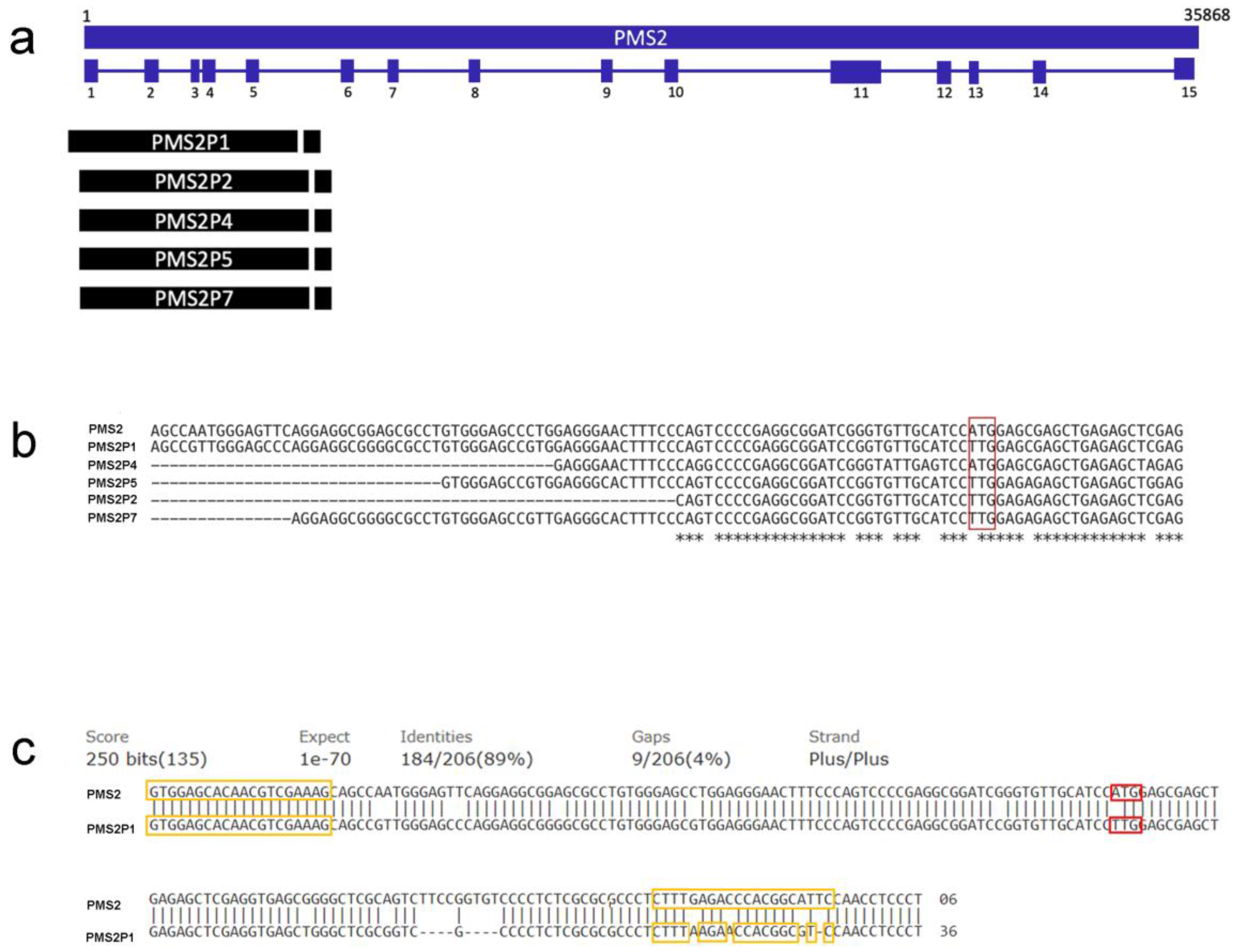
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lynch, H.T.; de la Chapelle, A. Hereditary Colorectal Cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, M.; Heald, B.; Yanda, C.; Kelly, E.D.; Grobmyer, S.; Eng, C.; Kalady, M.; Pederson, H. Investigating the Link between Lynch Syndrome and Breast Cancer. Eur. J. Breast Health 2020, 16, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.J.; da Silva, E.M.; Marra, A.; Gazzo, A.M.; Selenica, P.; Rai, V.K.; Mandelker, D.; Pareja, F.; Misyura, M.; D’Alfonso, T.M.; et al. Morphologic and Genomic Characteristics of Breast Cancers Occurring in Individuals with Lynch Syndrome. Clin. Cancer Res. 2022, 28, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Cerretelli, G.; Ager, A.; Arends, M.J.; Frayling, I.M. Molecular pathology of Lynch syndrome. J. Pathol. 2020, 250, 518–531. [Google Scholar] [CrossRef] [Green Version]
- Provenzale, D.; Gupta, S.; Ahnen, D.J.; Bray, T.; Cannon, J.A.; Cooper, G.; David, D.S.; Early, D.S.; Erwin, D.; Ford, J.M.; et al. Genetic/Familial High-Risk Assessment: Colorectal Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2016, 14, 1010–1030. [Google Scholar] [CrossRef]
- Therkildsen, C.; Jensen, L.H.; Rasmussen, M.; Bernstein, I. An Update on Immune Checkpoint Therapy for the Treatment of Lynch Syndrome. Clin. Exp. Gastroenterol. 2021, 14, 181–197. [Google Scholar] [CrossRef]
- Nicolaides, N.C.; Carter, K.C.; Shell, B.K.; Papadopoulos, N.; Vogelstein, B.; Kinzler, K.W. Genomic organization of the human PMS2 gene family. Genomics 1995, 30, 195–206. [Google Scholar] [CrossRef]
- Huang, Y.; Li, G.-M. DNA mismatch repair preferentially safeguards actively transcribed genes. DNA Repair 2018, 71, 82–86. [Google Scholar] [CrossRef]
- Espenschied, C.R.; LaDuca, H.; Li, S.; McFarland, R.; Gau, C.-L.; Hampel, H. Multigene Panel Testing Provides a New Perspective on Lynch Syndrome. J. Clin. Oncol. 2017, 35, 2568–2575. [Google Scholar] [CrossRef]
- Ten Broeke, S.W.; van der Klift, H.M.; Tops, C.M.J.; Aretz, S.; Bernstein, I.; Buchanan, D.D.; De La Chapelle, A.; Capella, G.; Clendenning, M.; Engel, C.; et al. Cancer Risks for PMS2 -Associated Lynch Syndrome. J. Clin. Oncol. 2018, 36, 2961–2968. [Google Scholar] [CrossRef] [Green Version]
- Steward, C.A.; Parker, A.P.J.; Minassian, B.A.; Sisodiya, S.M.; Frankish, A.; Harrow, J. Genome annotation for clinical genomic diagnostics: Strengths and weaknesses. Genome Med. 2017, 9, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Niessen, R.C.; Kleibeuker, J.H.; Jager, P.O.J.; Sijmons, R.H.; Hofstra, R.M.W. Getting rid of thePMS2 pseudogenes: Mission impossible? Hum Mutat. 2007, 28, 414. [Google Scholar] [CrossRef]
- Jansen, A.M.L.; Tops, C.M.J.; Ruano, D.; van Eijk, R.; Wijnen, J.T.; ten Broeke, S.; Nielsen, M.; Hes, F.J.; van Wezel, T.; Morreau, H. The complexity of screening PMS2 in DNA isolated from formalin-fixed paraffin-embedded material. Eur. J. Hum. Genet. 2020, 28, 333–338. [Google Scholar] [CrossRef]
- Blount, J.; Prakash, A. The changing landscape of Lynch syndrome due to PMS2 mutations. Clin. Genet. 2018, 94, 61–69. [Google Scholar] [CrossRef]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S.R.F.; Wilkie, A.O.M.; McVean, G.; Lunter, G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014, 46, 912–918. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Allen, B.; Kaldate, R.R.; Bowles, K.R.; Judkins, T.; Kaushik, P.; Roa, B.B.; Wenstrup, R.J.; Hartman, A.-R.; Syngal, S. Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients With Suspected Lynch Syndrome. Gastroenterology 2015, 149, 604–613.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, R.; Spier, I.; Zhao, B.; Kloth, M.; Marquez, J.; Hinrichsen, I.; Kirfel, J.; Tafazzoli, A.; Horpaopan, S.; Uhlhaas, S.; et al. Exome Sequencing Identifies Biallelic MSH3 Germline Mutations as a Recessive Subtype of Colorectal Adenomatous Polyposis. Am. J. Hum. Genet. 2016, 99, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Mighton, C.; Charames, G.S.; Wang, M.; Zakoor, K.-R.; Wong, A.; Shickh, S.; Watkins, N.; Lebo, M.S.; Bombard, Y.; Lerner-Ellis, J. Variant classification changes over time in BRCA1 and BRCA2. Genet Med. 2019, 21, 2248–2254. [Google Scholar] [CrossRef]
- Chong, A.; Chong, G.; Foulkes, W.D.; Saskin, A. Reclassification of a frequent African-origin variant from PMS2 to the pseudogene PMS2CL. Hum. Mutat. 2020, 41, 749–752. [Google Scholar] [CrossRef]
- Tsaousis, G.N.; Papadopoulou, E.; Agiannitopoulos, K.; Pepe, G.; Tsoulos, N.; Boukovinas, I.; Floros, T.; Iosifidou, R.; Katopodi, O.; Koumarianou, A.; et al. Revisiting the Implications of Positive Germline Testing Results Using Multi-gene Panels in Breast Cancer Patients. Cancer Genom. Proteom. 2022, 19, 60–78. [Google Scholar] [CrossRef]
- Sekine, M.; Enomoto, T. Precision medicine for hereditary tumors in gynecologic malignancies. J. Obstet. Gynaecol. Res. 2021, 47, 2597–2606. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ID | Gender | Age (Years) | Diagnosis | Relationship | Sample | PMS2 c.1A > T, (p.Met1Leu) |
|---|---|---|---|---|---|---|
| 1 | F | 44 | Breast cancer | Proband | PB | positive |
| 2 | M | 19 | healthy | son | PB | positive |
| 3 | M | 37 | healthy | brother | PB | positive |
| 4 | M | 40 | healthy | brother | PB | positive |
| 5 | M | 42 | healthy | brother | PB | positive |
| 6 | F | 49 | healthy | sister | PB | positive |
| 7 | F | 52 | healthy | sister | PB | positive |
| C1 | M | 29 | healthy | UC | PB | positive |
| C2 | M | 30 | healthy | UC | PB | positive |
| C3 | M | 36 | healthy | UC | PB | positive |
| C4 | M | 37 | healthy | UC | PB | positive |
| C5 | F | 35 | healthy | UC | PB | positive |
| C6 | F | 47 | healthy | UC | PB | positive |
| C7 | F | 55 | healthy | UC | PB | positive |
| Gene/Pseudogene | Description | NCBI Reference Sequence | Match Exon 1 |
|---|---|---|---|
| PMS2 | Homo sapiens PMS1 homolog 2, mismatch repair system component (PMS2), RefSeqGene (LRG_161) on chromosome 7 | NG_008466.1 | Reference |
| PMS2P1 | PMS1 homolog 2, mismatch repair system component pseudogene 1 [Homo sapiens] | NC_000007.14:c100336307-100320640 | Yes |
| PMS2P2 | PMS1 homolog 2, mismatch repair system component pseudogene 2 [Homo sapiens] | NC_000007.14:c75358997-75343937 | Yes |
| PMS2P3 | PMS1 homolog 2, mismatch repair system component pseudogene 3 [Homo sapiens] | NC_000007.14:c75528123-75507747 | No |
| PMS2P4 | PMS1 homolog 2, mismatch repair system component pseudogene 4 [Homo sapiens] | NC_000007.14:c67302442-67276131 | Yes |
| PMS2P5 | PMS1 homolog 2, mismatch repair system component pseudogene 5 [Homo sapiens] | NC_000007.14:74890768-74921138 | Yes |
| PMS2P6 | PMS1 homolog 2, mismatch repair system component pseudogene 6 [Homo sapiens] | NC_000007.14:73093644-73096950 | No |
| PMS2P7 | PMS1 homolog 2, mismatch repair system component pseudogene 7 [Homo sapiens] | NC_000007.14:c73006080-73016375 | Yes |
| PMS2P8 | PMS1 homolog 2, mismatch repair system component pseudogene 8 [Homo sapiens] | NC_000007.14:73037373-73040804 | No |
| PMS2P9 | PMS1 homolog 2, mismatch repair system component pseudogene 9 [Homo sapiens] | NC_000007.14:77039480-77053038 | No |
| PMS2P10 | PMS1 homolog 2, mismatch repair system component pseudogene 10 [Homo sapiens] | NC_000007.14:c75327776-75324481 | No |
| PMS2P11 | PMS1 homolog 2, mismatch repair system component pseudogene 11 [Homo sapiens] | NC_000007.14:77011447-77025554 | No |
| PMS2P12 | PMS1 homolog 2, mismatch repair system component pseudogene 12 [Homo sapiens] | NC_000007.14:102337315-102339139 | No |
| PMS2CL | PMS2 C-terminal like pseudogene [Homo sapiens] | NC_000007.14:c6735305-6751601 | No |
| LOC441259 | PMS1 homolog 2, mismatch repair system component pseudogene [Homo sapiens] | NC_000007.14:c75299806-75296553 | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fragoso-Ontiveros, V.; De la Fuente-Hernandez, M.A.; Gonzalez-Osnaya, V.; Gamez-Rosales, M.; Perez-Montiel, M.D.; Isla-Ortiz, D.; Cantu-De Leon, D.F.; Alvarez-Gomez, R.M. A Pathogenic Variant Reclassified to the Pseudogene PMS2P1 in a Patient with Suspected Hereditary Cancer. Int. J. Mol. Sci. 2023, 24, 1398. https://doi.org/10.3390/ijms24021398
Fragoso-Ontiveros V, De la Fuente-Hernandez MA, Gonzalez-Osnaya V, Gamez-Rosales M, Perez-Montiel MD, Isla-Ortiz D, Cantu-De Leon DF, Alvarez-Gomez RM. A Pathogenic Variant Reclassified to the Pseudogene PMS2P1 in a Patient with Suspected Hereditary Cancer. International Journal of Molecular Sciences. 2023; 24(2):1398. https://doi.org/10.3390/ijms24021398
Chicago/Turabian StyleFragoso-Ontiveros, Veronica, Marcela Angelica De la Fuente-Hernandez, Vincent Gonzalez-Osnaya, Mario Gamez-Rosales, Maria Delia Perez-Montiel, David Isla-Ortiz, David Francisco Cantu-De Leon, and Rosa Maria Alvarez-Gomez. 2023. "A Pathogenic Variant Reclassified to the Pseudogene PMS2P1 in a Patient with Suspected Hereditary Cancer" International Journal of Molecular Sciences 24, no. 2: 1398. https://doi.org/10.3390/ijms24021398