
Evaluation of Two Simultaneous Metabolomic and Proteomic Extraction Protocols Assessed by Ultra-High-Performance Liquid Chromatography Tandem Mass Spectrometry
 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Results
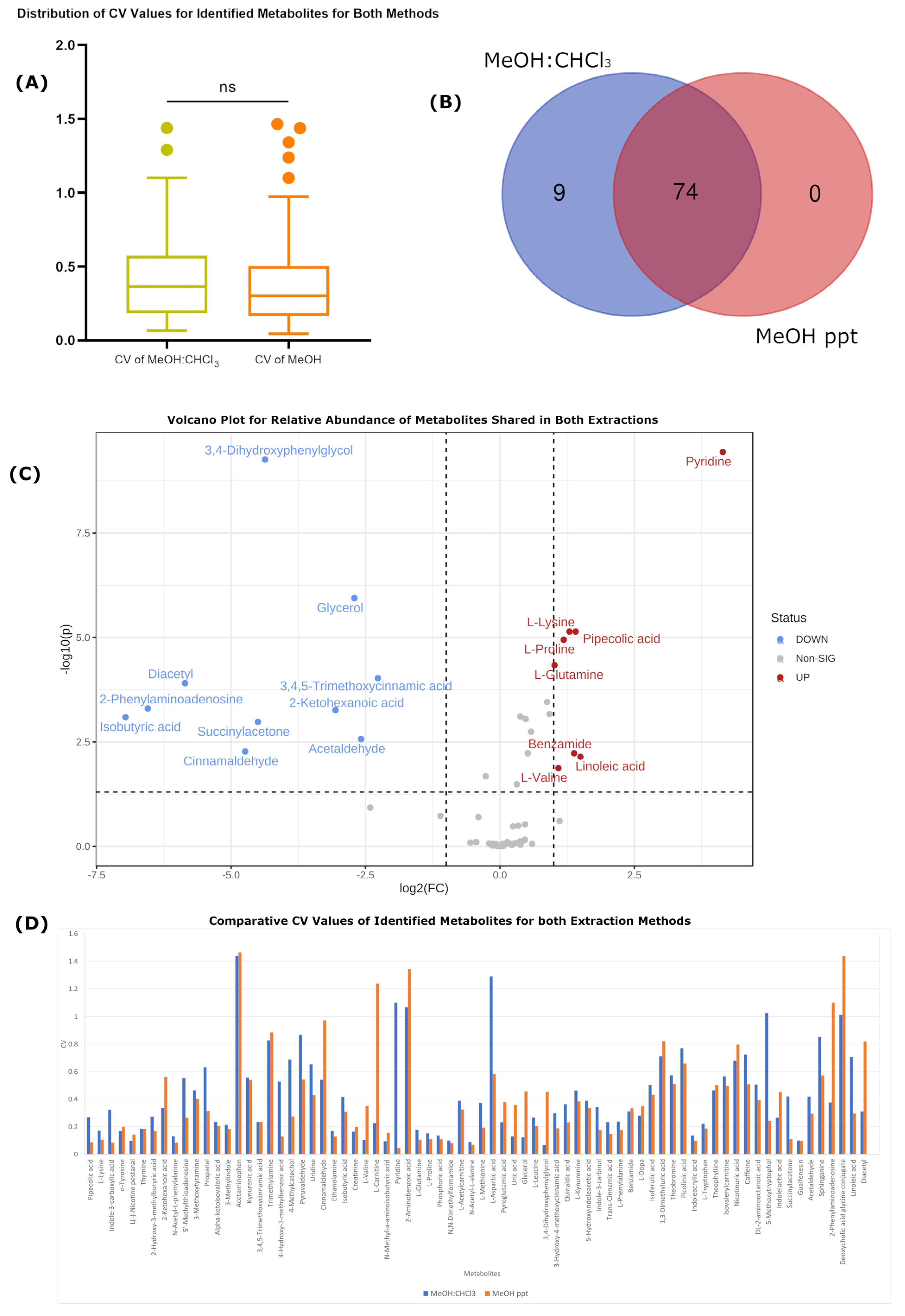
2.1. Biological Metabolomic Variation Exceeds That of Both Protocols
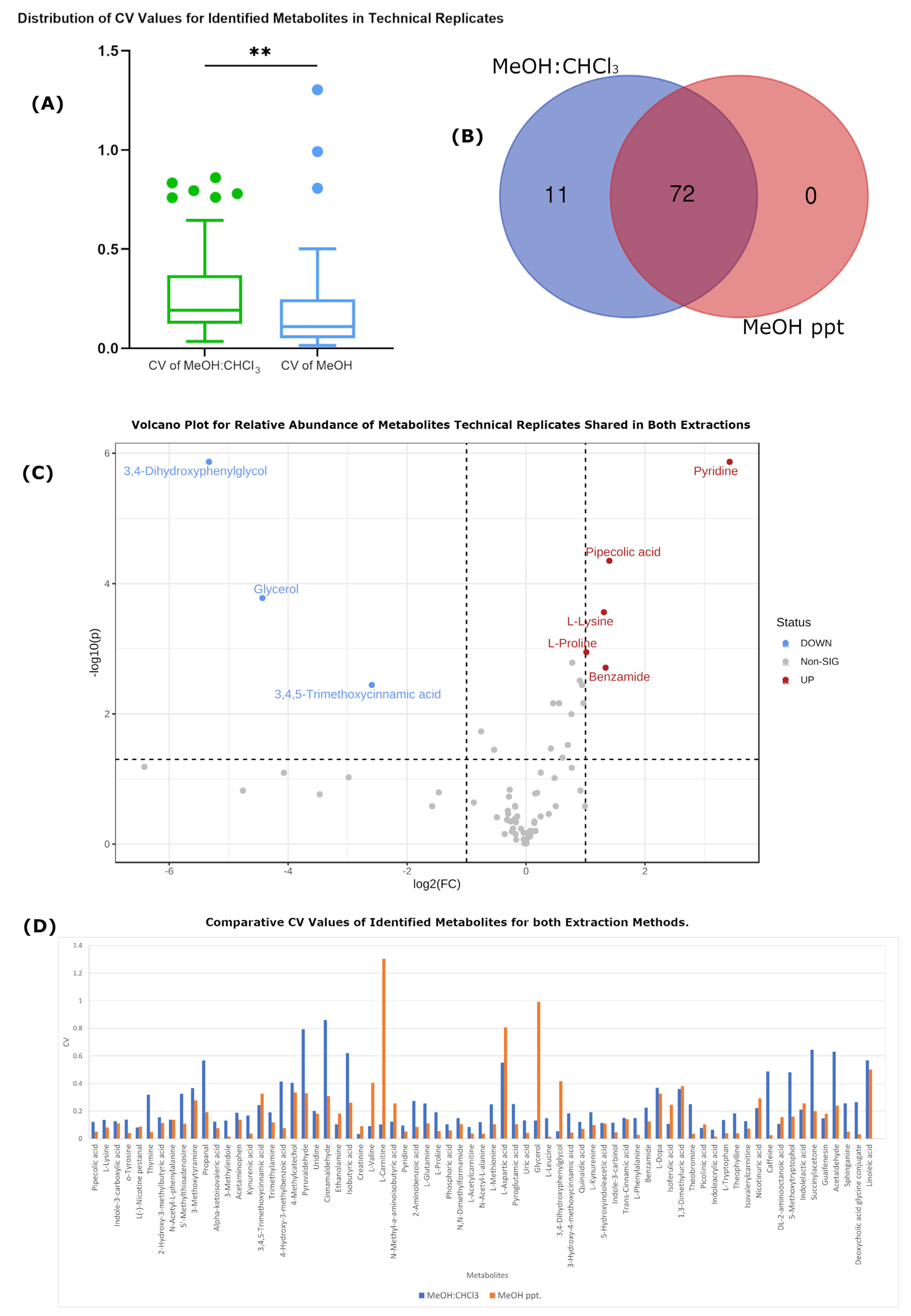
Technical Variation Is Improved in MeOH-Only Extraction
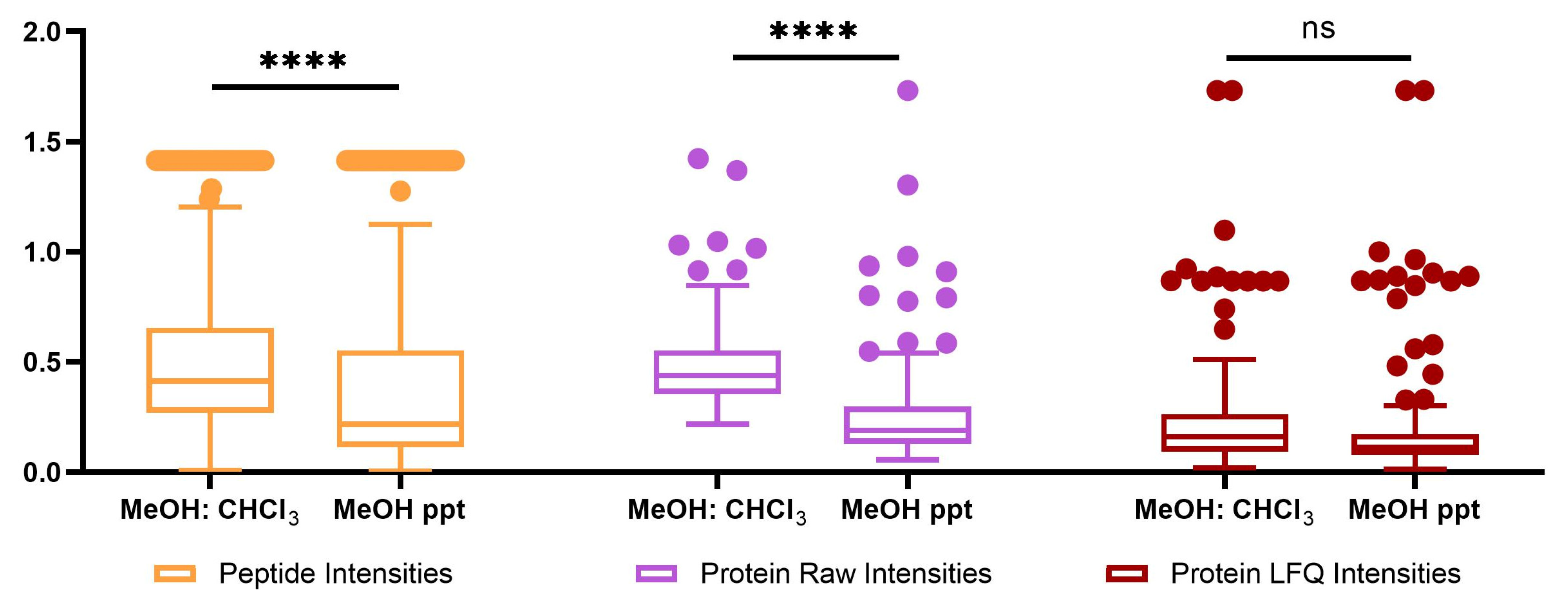
2.2. Proteomics Analysis
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Samples Collection
4.3. Sample Preparation
4.3.1. Methanol Precipitation for Metabolomic Extraction
4.3.2. Methanol: Chloroform for Metabolomic Extraction
4.3.3. Proteomics Sample Preparation
4.4. Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS)
4.5. Data Analysis and Statistical Approach
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Menyhárt, O.; Győrffy, B. Multi-omics approaches in cancer research with applications in tumor subtyping, prognosis, and diagnosis. Comput. Struct. Biotechnol. J. 2021, 19, 949–960. [Google Scholar] [CrossRef]
- Pang, Z.; Zhou, G.; Ewald, J.; Le Chang, L.; Hacariz, O.; Basu, N.; Xia, J. Using MetaboAnalyst 5.0 for LC–HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat. Protoc. 2022, 17, 1735–1761. [Google Scholar] [CrossRef]
- Kang, M.; Ko, E.; Mersha, T.B. A roadmap for multi-omics data integration using deep learning. Briefings Bioinform. 2021, 23, bbab454. [Google Scholar] [CrossRef]
- Zhou, G.; Pang, Z.; Lu, Y.; Ewald, J.; Xia, J. OmicsNet 2.0: A web-based platform for multi-omics integration and network visual analytics. Nucleic Acids Res. 2022, 50, W527–W533. [Google Scholar] [CrossRef]
- Iturria-Medina, Y.; Adewale, Q.; Khan, A.F.; Ducharme, S.; Rosa-Neto, P.; O’Donnell, K.; Petyuk, V.A.; Gauthier, S.; De Jager, P.L.; Breitner, J.; et al. Unified epigenomic, transcriptomic, proteomic, and metabolomic taxonomy of Alzheimer’s disease progression and heterogeneity. Sci. Adv. 2022, 8, eabo6764. [Google Scholar] [CrossRef]
- Zhou, D.; Long, C.; Shao, Y.; Li, F.; Sun, W.; Zheng, Z.; Wang, X.; Huang, Y.; Pan, F.; Chen, G.; et al. Integrated Metabolomics and Proteomics Analysis of Urine in a Mouse Model of Posttraumatic Stress Disorder. Front. Neurosci. 2022, 16, 828382. [Google Scholar] [CrossRef]
- Monribot-Villanueva, J.L.; Altúzar-Molina, A.; Aluja, M.; Zamora-Briseño, J.A.; Elizalde-Contreras, J.M.; Bautista-Valle, M.V.; de Los Santos, J.A.; Sánchez-Martínez, D.E.; Rivera-Reséndiz, F.J.; Vázquez-Rosas-Landa, M.; et al. Integrating proteomics and metabolomics approaches to elucidate the ripening process in white Psidium guajava. Food Chem. 2021, 367, 130656. [Google Scholar] [CrossRef]
- Lai, M.; Zhang, X.; Zhou, D.; Zhu, M.; Liu, Q.; Zhang, Y.; Wang, D. Integrating serum proteomics and metabolomics to compare the common and distinct features between acute aggressive ischemic stroke (APIS) and acute non-aggressive ischemic stroke (ANPIS). J. Proteom. 2022, 261, 104581. [Google Scholar] [CrossRef]
- Guan, F.; Xiang, X.; Xie, Y.; Li, H.; Zhang, W.; Shu, Y.; Wang, J.; Qin, W. Simultaneous metabolomics and proteomics analysis of plasma-derived extracellular vesicles. Anal. Methods 2021, 13, 1930–1938. [Google Scholar] [CrossRef]
- Maghuly, F.; Marzban, G.J. Omics Technologies Toward Systems Biology. Front. Genet. 2021, 12, 756847. [Google Scholar] [CrossRef]
- Díaz-Beltrán, L.; González-Olmedo, C.; Luque-Caro, N.; Díaz, C.; Martín-Blázquez, A.; Fernández-Navarro, M.; Ortega-Granados, A.; Gálvez-Montosa, F.; Vicente, F.; del Palacio, J.P.; et al. Human Plasma Metabolomics for Biomarker Discovery: Targeting the Molecular Subtypes in Breast Cancer. Cancers 2021, 13, 147. [Google Scholar] [CrossRef]
- Mielko, K.A.; Pudełko-Malik, N.; Tarczewska, A.; Młynarz, P. NMR spectroscopy as a “green analytical method” in metabolomics and proteomics studies. Sustain. Chem. Pharm. 2021, 22, 100474. [Google Scholar] [CrossRef]
- Sarkar, A.A. Advances in Omics Protocols. Genet. Eng. Biotechnol. News 2021, 41, P30–P31. [Google Scholar] [CrossRef]
- Martias, C.; Baroukh, N.; Mavel, S.; Blasco, H.; Lefèvre, A.; Roch, L.; Montigny, F.; Gatien, J.; Schibler, L.; Dufour-Rainfray, D.; et al. Optimization of Sample Preparation for Metabolomics Exploration of Urine, Feces, Blood and Saliva in Humans Using Combined NMR and UHPLC-HRMS Platforms. Molecules 2021, 26, 4111. [Google Scholar] [CrossRef]
- Iverson, S.J.; Lang, S.L.C.; Cooper, M.H. Comparison of the bligh and dyer and folch methods for total lipid determination in a broad range of marine tissue. Lipids 2001, 36, 1283–1287. [Google Scholar] [CrossRef]
- Löfgren, L.; Ståhlman, M.; Forsberg, G.-B.; Saarinen, S.; Nilsson, R.; Hansson, G.I. The BUME method: A novel automated chloroform-free 96-well total lipid extraction method for blood plasma [S]. J. Lipid Res. 2012, 53, 1690–1700. [Google Scholar] [CrossRef] [Green Version]
- Abbott, S.K.; Jenner, A.M.; Mitchell, T.W.; Brown, S.H.J.; Halliday, G.M.; Garner, B. An Improved High-Throughput Lipid Extraction Method for the Analysis of Human Brain Lipids. Lipids 2013, 48, 307–318. [Google Scholar] [CrossRef] [Green Version]
- Sarafian, M.H.; Gaudin, M.; Lewis, M.R.; Martin, F.-P.; Holmes, E.; Nicholson, J.K.; Dumas, M.-E. Objective Set of Criteria for Optimization of Sample Preparation Procedures for Ultra-High Throughput Untargeted Blood Plasma Lipid Profiling by Ultra Performance Liquid Chromatography–Mass Spectrometry. Anal. Chem. 2014, 86, 5766–5774. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. J. Lipid Res. 1959, 37, 911–917. [Google Scholar]
- Hammad, S.M.; Pierce, J.S.; Soodavar, F.; Smith, K.J.; Al Gadban, M.M.; Rembiesa, B.; Klein, R.L.; Hannun, Y.A.; Bielawski, J.; Bielawska, A. Blood sphingolipidomics in healthy humans: Impact of sample collection methodology. J. Lipid Res. 2010, 51, 3074–3087. [Google Scholar] [CrossRef] [Green Version]
- Gika, H.; Theodoridis, G. Sample preparation prior to the LC–MS-based metabolomics/metabonomics of blood-derived samples. Bioanalysis 2011, 3, 1647–1661. [Google Scholar] [CrossRef]
- Gowda, G.N.; Raftery, D. Quantitating metabolites in protein precipitated serum using NMR spectroscopy. Anal. Chem. 2014, 86, 5433–5440. [Google Scholar] [CrossRef]
- Al-Salhi, R.; Monfort, C.; Bonvallot, N.; David, A. Analytical strategies to profile the internal chemical exposome and the metabolome of human placenta. Anal. Chim. Acta 2022, 1219, 339983. [Google Scholar] [CrossRef]
- Alsoud, L.O.; Soares, N.C.; Al-Hroub, H.M.; Mousa, M.; Kasabri, V.; Bulatova, N.; Suyagh, M.; Alzoubi, K.H.; El-Huneidi, W.; Abu-Irmaileh, B.; et al. Identification of Insulin Resistance Biomarkers in Metabolic Syndrome Detected by UHPLC-ESI-QTOF-MS. Metab. 2022, 12, 508. [Google Scholar] [CrossRef]
- Kiseleva, O.; Kurbatov, I.; Ilgisonis, E.; Poverennaya, E. Defining Blood Plasma and Serum Metabolome by GC-MS. Metabolites 2021, 12, 15. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, J.; Zhang, R.; Abliz, Z. Methods used to increase the comprehensive coverage of urinary and plasma metabolomes by MS. Bioanalysis 2016, 8, 981–997. [Google Scholar] [CrossRef]
- Coman, C.; Solari, F.A.; Hentschel, A.; Sickmann, A.; Zahedi, R.P.; Ahrends, R. Simultaneous Metabolite, Protein, Lipid Extraction (SIMPLEX): A Combinatorial Multimolecular Omics Approach for Systems Biology. Mol. Cell. Proteom. 2016, 15, 1435–1466. [Google Scholar] [CrossRef] [Green Version]
- Sharaf, B.M.; Giddey, A.D.; Al-Hroub, H.M.; Menon, V.; Okendo, J.; El-Awady, R.; Mousa, M.; Almehdi, A.; Semreen, M.H.; Soares, N.C. Mass spectroscopy-based proteomics and metabolomics analysis of triple-positive breast cancer cells treated with tamoxifen and/or trastuzumab. Cancer Chemother. Pharmacol. 2022, 90, 467–488. [Google Scholar] [CrossRef]
- Weckwerth, W.; Wenzel, K.; Fiehn, O.J.P. Process for the integrated extraction, identification and quantification of metabolites, proteins and RNA to reveal their co-regulation in biochemical networks. Proteomics. 2004, 4, 78–83. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Blankenburg, M.; Haberland, L.; Elvers, H.-D.; Tannert, C.; Jandrig, B. High-Throughput Omics Technologies: Potential Tools for the Investigation of Influences of EMF on Biological Systems. Curr. Genom. 2009, 10, 86–92. [Google Scholar] [CrossRef]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics* s⃞. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Sawada, T.; Hatayama, K.; Ohyagi, A.; Tsukuda, Y.; Namekawa, K.; Ito, R.; Saito, K.; Nakazawa, H. Separation Technique for the Determination of Highly Polar Metabolites in Biological Samples. Metabolites 2012, 2, 496–515. [Google Scholar] [CrossRef]
- Chou, S.; Spoo, W. Toxicological Profile for Chloroform; US Department of Health and Human Services: Atlanta, GA, USA, 1997; pp. 1–291. [Google Scholar]
- Sekar, A.; Varghese, G.K.; Varma, M. Chloroform—An Emerging Pollutant in the Air. In New Trends in Emerging Environmental Contaminants; Springer: Berlin/Heidelberg, Germany, 2022; pp. 101–129. [Google Scholar]
- Wong, M.W.K.; Braidy, N.; Pickford, R.; Sachdev, P.; Poljak, A. Comparison of Single Phase and Biphasic Extraction Protocols for Lipidomic Studies Using Human Plasma. Front. Neurol. 2019, 10, 879. [Google Scholar] [CrossRef]
- Alshehry, Z.H.; Barlow, C.K.; Weir, J.M.; Zhou, Y.; McConville, M.J.; Meikle, P.J. An Efficient Single Phase Method for the Extraction of Plasma Lipids. Metabolites 2015, 5, 389–403. [Google Scholar] [CrossRef]
- Gil, A.; Zhang, W.; Wolters, J.C.; Permentier, H.; Boer, T.; Horvatovich, P.; Heiner-Fokkema, M.R.; Reijngoud, D.-J.; Bischoff, R. One- vs two-phase extraction: Re-evaluation of sample preparation procedures for untargeted lipidomics in plasma samples. Anal. Bioanal. Chem. 2018, 410, 5859–5870. [Google Scholar] [CrossRef] [Green Version]
- Southam, A.D.; Haglington, L.D.; Najdekr, L.; Jankevics, A.; Weber, R.J.M.; Dunn, W.B. Assessment of human plasma and urine sample preparation for reproducible and high-throughput UHPLC-MS clinical metabolic phenotyping. Analyst 2020, 145, 6511–6523. [Google Scholar] [CrossRef]
- Couvillion, S.P.; Zhu, Y.; Nagy, G.; Adkins, J.N.; Ansong, C.; Renslow, R.S.; Piehowski, P.; Ibrahim, Y.M.; Kelly, R.T.; Metz, T.O. New mass spectrometry technologies contributing towards comprehensive and high throughput omics analyses of single cells. Analyst 2019, 144, 794–807. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef]
- Ramagli, L.; Rodriguez, L.J.E. Quantitation of microgram amounts of protein in SDS-mercaptoethanol-tris electrophoresis sample buffer. Anal. Biochem. 1985, 6, 559–563. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| MeOH: CHCl3 | MeOH ppt | |||
|---|---|---|---|---|
| Different Biological Samples (n = 6) | Technical Replicates (n = 4) | Different Biological Samples (n = 6) | Technical Replicates (n = 4) | |
| Number of detected features | 5556 | 5702 | 3708 | 3584 |
| Number of identified metabolites | 83 | 83 | 74 | 72 |
| Average RSD | 0.427405 | 0.27521863 | 0.389594 | 0.179235 |
| MeOH: CHCl3 | MeOH ppt | |||||
|---|---|---|---|---|---|---|
| Protein LFQ Intensities | Raw Protein Intensities | Peptides | Protein LFQ Intensities | Raw Protein Intensities | Peptides | |
| Number of quantifiable features | 104 | 114 | 1243 | 103 | 115 | 1254 |
| Average RSD of quantitation | 0.264476 | 0.482346 | 0.483063 | 0.232188 | 0.26544306 | 0.369035 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zenati, R.A.; Giddey, A.D.; Al-Hroub, H.M.; Hagyousif, Y.A.; El-Huneidi, W.; Bustanji, Y.; Abu-Gharbieh, E.; Alqudah, M.A.Y.; Shara, M.; Abuhelwa, A.Y.; et al. Evaluation of Two Simultaneous Metabolomic and Proteomic Extraction Protocols Assessed by Ultra-High-Performance Liquid Chromatography Tandem Mass Spectrometry. Int. J. Mol. Sci. 2023, 24, 1354. https://doi.org/10.3390/ijms24021354
Zenati RA, Giddey AD, Al-Hroub HM, Hagyousif YA, El-Huneidi W, Bustanji Y, Abu-Gharbieh E, Alqudah MAY, Shara M, Abuhelwa AY, et al. Evaluation of Two Simultaneous Metabolomic and Proteomic Extraction Protocols Assessed by Ultra-High-Performance Liquid Chromatography Tandem Mass Spectrometry. International Journal of Molecular Sciences. 2023; 24(2):1354. https://doi.org/10.3390/ijms24021354
Chicago/Turabian StyleZenati, Ruba A., Alexander D. Giddey, Hamza M. Al-Hroub, Yousra A. Hagyousif, Waseem El-Huneidi, Yasser Bustanji, Eman Abu-Gharbieh, Mohammad A. Y. Alqudah, Mohd Shara, Ahmad Y. Abuhelwa, and et al. 2023. "Evaluation of Two Simultaneous Metabolomic and Proteomic Extraction Protocols Assessed by Ultra-High-Performance Liquid Chromatography Tandem Mass Spectrometry" International Journal of Molecular Sciences 24, no. 2: 1354. https://doi.org/10.3390/ijms24021354