

Genome-Wide Identification of MADS-Box Family Genes in Safflower (Carthamus tinctorius L.) and Functional Analysis of CtMADS24 during Flowering
 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Phylogenetic Analysis of MADS-Box Family Members
2.2. Analysis of the Conserved Motifs and Gene Structure
2.3. Analysis of Cis-Regulatory Elements in the Promoters of MADS-Box Family Genes
2.4. Gene Expression Analysis by Transcriptome Data of CtMADS during Safflower Floral Development
2.5. Gene Expression Analysis of CtMADS in Different Flower Organs
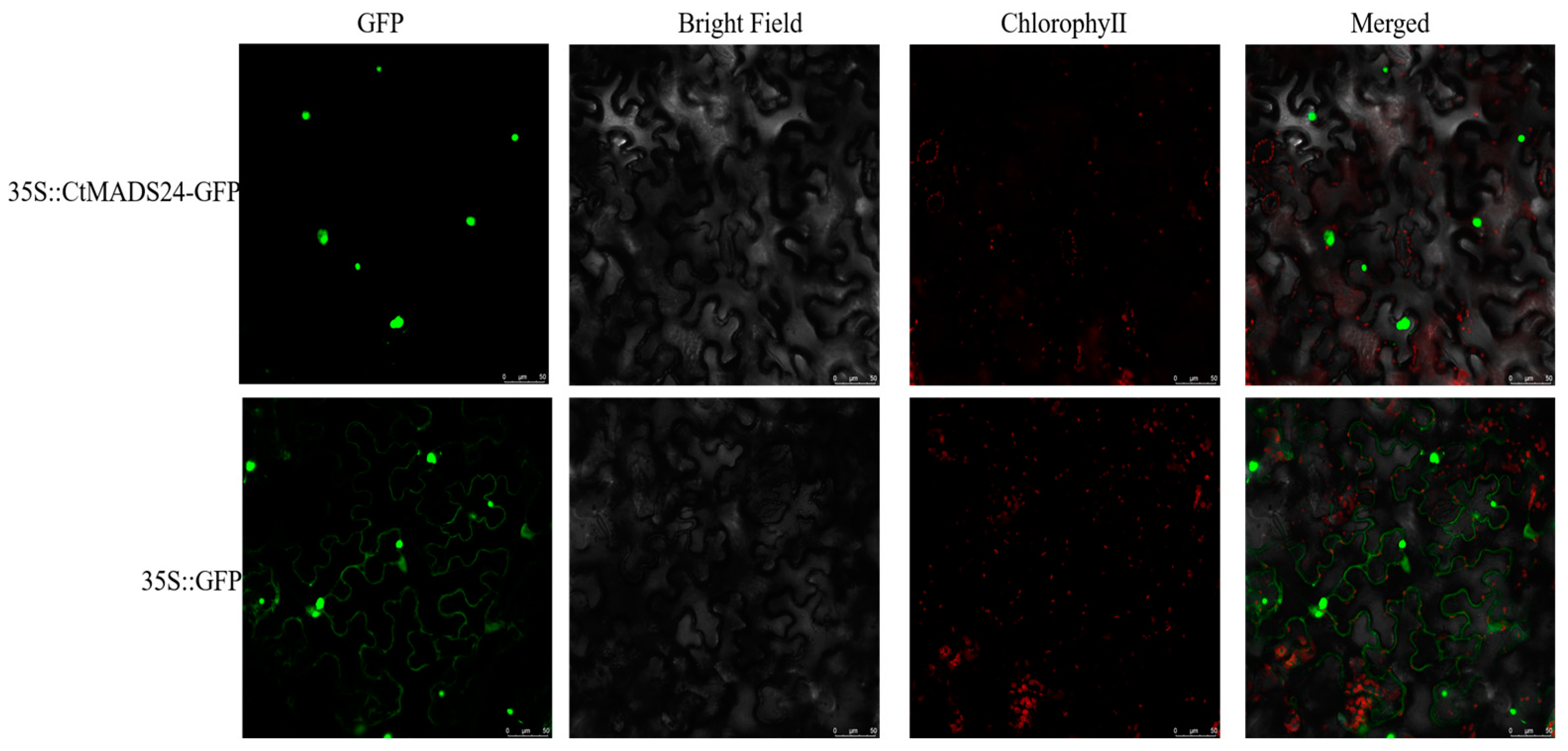
2.6. Subcellular Location of CtMADS24
2.7. Ectopic Expression of CtMADS24 and Expression Level of Flowering-Related Genes in Arabidopsis
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Identification of MADS-Box Family Genes
4.3. Phylogenetic Analysis, Conserved Motif, and Gene Structure Analysis of CtMADSs
4.4. Prediction of Cis-Acting Elements in Promoter Sequences
4.5. Safflower MADS-Box Genes Expression Pattern Analysis
4.6. RNA Isolation and Quantitative Real-Time PCR Analysis
4.7. ORF Cloning and Subcellular Localization
4.8. Generation of CtMADS24-Overexpressed Transgenic Arabidopsis Lines and Ectopic Expression Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shore, P.; Sharrocks, A.D. The MADS-box family of transcription factors. Eur. J. Biochem. 1995, 229, 1–13. [Google Scholar] [CrossRef]
- Gramzow, L.; Theissen, G. A hitchhiker’s guide to the MADS world of plants. Genome Biol. 2010, 11, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Buylla, E.R.; Pelaz, S.; Liljegren, S.J.; Gold, S.E.; Burgeff, C.; Ditta, G.S.; Ribas de Pouplana, L.; Martínez-Castilla, L.; Yanofsky, M.F. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc. Natl. Acad. Sci. USA 2000, 97, 5328–5333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Folter, S.; Angenent, G.C. trans meets cis in MADS science. Trends Plant Sci. 2006, 11, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Smaczniak, C.; Immink, R.G.; Angenent, G.C.; Kaufmann, K. Developmental and evolutionary diversity of plant MADS-domain factors: Insights from recent studies. Development 2012, 139, 3081–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramzow, L.; Weilandt, L.; Theissen, G. MADS goes genomic in conifers: Towards determining the ancestral set of MADS-box genes in seed plants. Ann. Bot. 2014, 114, 1407–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramzow, L.; Theissen, G. Phylogenomics reveals surprising sets of essential and dispensable clades of MIKC(c)-group MADS-box genes in flowering plants. J. Exp. Zool. B Mol. Dev. Evol. 2015, 324, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Pan, S.; Kennedy, A.; Melzer, R. MADS-box genes and crop domestication: The jack of all traits. J. Exp. Bot. 2018, 69, 1447–1469. [Google Scholar] [CrossRef]
- Zobell, O.; Faigl, W.; Saedler, H.; Munster, T. MIKC* MADS-box proteins: Conserved regulators of the gametophytic generation of land plants. Mol. Biol. Evol. 2010, 27, 1201–1211. [Google Scholar] [CrossRef] [Green Version]
- Masiero, S.; Colombo, L.; Grini, P.E.; Schnittger, A.; Kater, M.M. The emerging importance of type I MADS box transcription factors for plant reproduction. Plant Cell 2011, 23, 865–872. [Google Scholar] [CrossRef]
- Qiu, Y.; Köhler, C. Endosperm Evolution by Duplicated and Neofunctionalized Type I MADS-Box Transcription Factors. Mol. Biol. Evol. 2022, 39, msab355. [Google Scholar] [CrossRef] [PubMed]
- Abraham-Juarez, M.J.; Schrager-Lavelle, A.; Man, J.; Whipple, C.; Handakumbura, P.; Babbitt, C.; Bartlett, M. Evolutionary Variation in MADS Box Dimerization Affects Floral Development and Protein Abundance in Maize. Plant Cell 2020, 32, 3408–3424. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Li, F.; Shi, G.; Wang, L.; Wang, L.; Fan, L. Identification of MADS-Box Transcription Factors in Iris laevigata and Functional Assessment of IlSEP3 and IlSVP during Flowering. Int. J. Mol. Sci. 2022, 23, 9950. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Wang, F.; Geng, S.; Guan, J.; Tao, S.; Jia, M.; Sun, G.; Wang, Z.; Wang, K.; Ye, X.; et al. The wheat AGL6-like MADS-box gene is a master regulator for floral organ identity and a target for spikelet meristem development manipulation. Plant Biotechnol. J. 2022, 20, 75–88. [Google Scholar] [CrossRef]
- Theissen, G.; Melzer, R.; Rumpler, F. MADS-domain transcription factors and the floral quartet model of flower development: Linking plant development and evolution. Development 2016, 143, 3259–3271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugouvieux, V.; Silva, C.S.; Jourdain, A.; Stigliani, A.; Charras, Q.; Conn, V.; Conn, S.J.; Carles, C.C.; Parcy, F.; Zubieta, C. Tetramerization of MADS family transcription factors SEPALLATA3 and AGAMOUS is required for floral meristem determinacy in Arabidopsis. Nucleic Acids Res. 2018, 46, 4966–4977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Cai, Y.; Wang, H.; Zeng, Y.; Zhuang, X.; Li, B.; Jiang, L. Trans-Golgi network-located AP1 gamma adaptins mediate dileucine motif-directed vacuolar targeting in Arabidopsis. Plant Cell 2014, 26, 4102–4118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teo, Z.W.N.; Zhou, W.; Shen, L. Dissecting the Function of MADS-Box Transcription Factors in Orchid Reproductive Development. Front. Plant Sci. 2019, 10, 1474. [Google Scholar] [CrossRef]
- Alvarez-Buylla, E.R.; Garcia-Ponce, B.; Garay-Arroyo, A. Unique and redundant functional domains of APETALA1 and CAULIFLOWER, two recently duplicated Arabidopsis thaliana floral MADS-box genes. J. Exp. Bot. 2006, 57, 3099–3107. [Google Scholar] [CrossRef] [Green Version]
- Pabón-Mora, N.; Hidalgo, O.; Gleissberg, S.; Litt, A. Assessing duplication and loss of APETALA1/FRUITFULL homologs in Ranunculales. Front. Plant Sci. 2013, 4, 358. [Google Scholar] [CrossRef]
- Parenicová, L.; de Folter, S.; Kieffer, M.; Horner, D.S.; Favalli, C.; Busscher, J.; Cook, H.E.; Ingram, R.M.; Kater, M.M.; Davies, B.; et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: New openings to the MADS world. Plant Cell 2003, 15, 1538–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenbussche, M.; Theissen, G.; Van de Peer, Y.; Gerats, T. Structural diversification and neo-functionalization during floral MADS-box gene evolution by C-terminal frameshift mutations. Nucleic Acids Res. 2003, 31, 4401–4409. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Jang, S.; Chae, S.; Chung, K.M.; Moon, Y.H.; An, G.; Jang, S.K. Analysis of the C-terminal region of Arabidopsis thaliana APETALA1 as a transcription activation domain. Plant Mol. Biol. 1999, 40, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Yalovsky, S.; Kulukian, A.; Rodríguez-Concepción, M.; Young, C.A.; Gruissem, W. Functional requirement of plant farnesyltransferase during development in Arabidopsis. Plant Cell 2000, 12, 1267–1278. [Google Scholar] [CrossRef] [Green Version]
- Berbel, A.; Navarro, C.; Ferrándiz, C.; Cañas, L.A.; Madueño, F.; Beltrán, J.P. Analysis of PEAM4, the pea AP1 functional homologue, supports a model for AP1-like genes controlling both floral meristem and floral organ identity in different plant species. Plant J. 2001, 25, 441–451. [Google Scholar] [CrossRef]
- Benlloch, R.; d’Erfurth, I.; Ferrandiz, C.; Cosson, V.; Beltrán, J.P.; Cañas, L.A.; Kondorosi, A.; Madueño, F.; Ratet, P. Isolation of mtpim proves Tnt1 a useful reverse genetics tool in Medicago truncatula and uncovers new aspects of AP1-like functions in legumes. Plant Physiol. 2006, 142, 972–983. [Google Scholar] [CrossRef] [Green Version]
- Müller, B.M.; Saedler, H.; Zachgo, S. The MADS-box gene DEFH28 from Antirrhinum is involved in the regulation of floral meristem identity and fruit development. Plant J. 2001, 28, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Balanzà, V.; Martínez-Fernández, I.; Sato, S.; Yanofsky, M.F.; Kaufmann, K.; Angenent, G.C.; Bemer, M.; Ferrándiz, C. Genetic control of meristem arrest and life span in Arabidopsis by a FRUITFULL-APETALA2 pathway. Nat. Commun. 2018, 9, 565. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Sun, Y.; Yu, X.; Li, H.; Bao, M.; He, Y. Functional Conservation and Divergence of Five AP1/FUL-like Genes in Marigold (Tagetes erecta L.). Genes 2021, 12, 2011. [Google Scholar] [CrossRef]
- Ahmad, N.; Li, T.; Liu, Y.; Hoang, N.Q.V.; Ma, X.; Zhang, X.; Liu, J.; Yao, N.; Liu, X.; Li, H. Molecular and biochemical rhythms in dihydroflavonol 4-reductase-mediated regulation of leucoanthocyanidin biosynthesis in Carthamus tinctorius L. Ind. Crops Prod. 2020, 156, 112838. [Google Scholar] [CrossRef]
- Ahmad, N.; Jianyu, L.; Xu, T.; Noman, M.; Jameel, A.; Na, Y.; Yuanyuan, D.; Nan, W.; Xiaowei, L.; Fawei, W. Overexpression of a novel cytochrome P450 promotes flavonoid biosynthesis and osmotic stress tolerance in transgenic Arabidopsis. Genes 2019, 10, 756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Ahmad, N.; Hong, Y.; Zhu, M.; Zaman, S.; Wang, N.; Yao, N.; Liu, X. Molecular Characterization of an Isoflavone 2′-Hydroxylase Gene Revealed Positive Insights into Flavonoid Accumulation and Abiotic Stress Tolerance in Safflower. Molecules 2022, 27, 8001. [Google Scholar] [CrossRef] [PubMed]
- Hoang, N.Q.; Kong, J.; Ahmad, N.; Ma, X.; Zhang, X.; Wu, Y.; Wang, N.; Yao, N.; Liu, X.; Jin, L. Genome-wide identification and expression profiling of duplicated flavonoid 3′-hydroxylase gene family in Carthamus tinctorius L. Not. Bot. Horti Agrobot. Cluj-Napoca 2021, 49, 12509. [Google Scholar] [CrossRef]
- Sun, F.; Ahmad, N.; Jin, L.; Xinyue, Z.; Xintong, M.; Hoang, N.Q.; Mallano, A.I.; Wang, N.; Zhuoda, Y.; Liu, X. Genome-wide investigation of Hydroxycinnamoyl CoA: Shikimate Hydroxycinnamoyl Transferase (HCT) gene family in Carthamus tinctorius L. Not. Bot. Horti Agrobot. Cluj-Napoca 2021, 49, 12489. [Google Scholar]
- Lu, Y.; Chi, M.; Li, L.; Li, H.; Noman, M.; Yang, Y.; Ji, K.; Lan, X.; Qiang, W.; Du, L. Genome-wide identification, expression profiling, and functional validation of oleosin gene family in Carthamus tinctorius L. Front. Plant Sci. 2018, 9, 1393. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Ahmad, N.; Zhang, J.; Lv, Y.; Zhang, X.; Ma, X.; Liu, X.; Na, Y. Genome-wide analysis and transcriptional reprogrammings of MYB superfamily revealed positive insights into abiotic stress responses and anthocyanin accumulation in Carthamus tinctorius L. Mol. Genet. Genom. 2022, 297, 125–145. [Google Scholar] [CrossRef]
- Hong, Y.; Ahmad, N.; Tian, Y.; Liu, J.; Wang, L.; Wang, G.; Liu, X.; Dong, Y.; Wang, F.; Liu, W.; et al. Genome-Wide Identification, Expression Analysis, and Subcellular Localization of Carthamus tinctorius bHLH Transcription Factors. Int. J. Mol. Sci. 2019, 20, 3044. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Li, L.; ShangGuan, G.; Jia, C.; Deng, S.; Noman, M.; Liu, Y.; Guo, Y.; Han, L.; Zhang, X. Genome-wide identification and expression analysis of bZIP gene family in Carthamus tinctorius L. Sci. Rep. 2020, 10, 15521. [Google Scholar] [CrossRef]
- Kim, E.J.; Hong, W.J.; Kim, Y.J.; Jung, K.H. Transcriptome Analysis of Triple Mutant for OsMADS62, OsMADS63, and OsMADS68 Reveals the Downstream Regulatory Mechanism for Pollen Germination in Rice (Oryza sativa). Int. J. Mol. Sci. 2021, 23, 239. [Google Scholar] [CrossRef]
- Goloveshkina, E.N.; Shul’ga, O.A.; Shchennikova, A.V.; Kamionskaya, A.M.; Skryabin, K.G. Constitutive expression of the sunflower and chrysanthemum genes of the AP1/FUL group changes flowering timing in transgenic tobacco plants. Dokl. Biol. Sci. 2010, 434, 322–324. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, D.; Lin, X.; Ding, M.; Tong, Z. Genome-wide identification of MADS-box family genes in moso bamboo (Phyllostachys edulis) and a functional analysis of PeMADS5 in flowering. BMC Plant Biol. 2018, 18, 176. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Cao, Y.; Chen, T.; Abdullah, M.; Jin, Q.; Fan, H.; Lin, Y.; Cai, Y. Evolution and functional divergence of MADS-box genes in Pyrus. Sci. Rep. 2019, 9, 1266. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Kennedy, A.; Pan, S.; Jermiin, L.S.; Melzer, R. Genome-wide analysis of MIKC-type MADS-box genes in wheat: Pervasive duplications, functional conservation and putative neofunctionalization. New Phytol. 2020, 225, 511–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.; Li, Y.; Guan, Y.; Wang, S.; Luo, H.; Li, X.; Li, H.; Zhang, Z. Auxin-induced AUXIN RESPONSE FACTOR4 activates APETALA1 and FRUITFULL to promote flowering in woodland strawberry. Hortic. Res. 2021, 8, 115. [Google Scholar] [CrossRef]
- Zhang, C.; Wei, L.; Wang, W.; Qi, W.; Cao, Z.; Li, H.; Bao, M.; He, Y. Identification, characterization and functional analysis of AGAMOUS subfamily genes associated with floral organs and seed development in Marigold (Tagetes erecta). BMC Plant Biol. 2020, 20, 439. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Ni, Z.; Dai, Y.; Yao, Y.; Nie, X.; Sun, Q. Characterization and expression of 42 MADS-box genes in wheat (Triticum aestivum L.). Mol. Genet. Genom. 2006, 276, 334–350. [Google Scholar] [CrossRef]
- Gramzow, L.; Ritz, M.S.; Theissen, G. On the origin of MADS-domain transcription factors. Trends Genet. 2010, 26, 149–153. [Google Scholar] [CrossRef]
- Hsu, H.F.; Chen, W.H.; Shen, Y.H.; Hsu, W.H.; Mao, W.T.; Yang, C.H. Multifunctional evolution of B and AGL6 MADS box genes in orchids. Nat. Commun. 2021, 12, 902. [Google Scholar] [CrossRef]
- Li, C.; Wang, Y.; Xu, L.; Nie, S.; Chen, Y.; Liang, D.; Sun, X.; Karanja, B.K.; Luo, X.; Liu, L. Genome-Wide Characterization of the MADS-Box Gene Family in Radish (Raphanus sativus L.) and Assessment of Its Roles in Flowering and Floral Organogenesis. Front. Plant Sci. 2016, 7, 1390. [Google Scholar] [CrossRef] [Green Version]
- Kanno, A.; Saeki, H.; Kameya, T.; Saedler, H.; Theissen, G. Heterotopic expression of class B floral homeotic genes supports a modified ABC model for tulip (Tulipa gesneriana). Plant Mol. Biol. 2003, 52, 831–841. [Google Scholar] [CrossRef]
- Litt, A.; Kramer, E.M. The ABC model and the diversification of floral organ identity. Semin. Cell Dev. Biol. 2010, 21, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Q.; Yang, S.; Lin, S.; Bao, M.; Bendahmane, M.; Wu, Q.; Wang, C.; Fu, X. Identification and Characterization of the MADS-Box Genes and Their Contribution to Flower Organ in Carnation (Dianthus caryophyllus L.). Genes 2018, 9, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.J.; Zheng, B.Q.; Wang, J.Y.; Tsai, W.C.; Lu, H.C.; Zou, L.H.; Wan, X.; Zhang, D.Y.; Qiao, H.J.; Liu, Z.J.; et al. New insight into the molecular mechanism of colour differentiation among floral segments in orchids. Commun. Biol. 2020, 3, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, H.; Zhang, N.; Liu, C.; Xu, G.; Zhang, J.; Chen, Z.; Kong, H. Patterns of gene duplication and functional diversification during the evolution of the AP1/SQUA subfamily of plant MADS-box genes. Mol. Phylogenet. Evol. 2007, 44, 26–41. [Google Scholar] [CrossRef]
- Burko, Y.; Shleizer-Burko, S.; Yanai, O.; Shwartz, I.; Zelnik, I.D.; Jacob-Hirsch, J.; Kela, I.; Eshed-Williams, L.; Ori, N. A role for APETALA1/fruitfull transcription factors in tomato leaf development. Plant Cell 2013, 25, 2070–2083. [Google Scholar] [CrossRef] [Green Version]
- Pabón-Mora, N.; Sharma, B.; Holappa, L.D.; Kramer, E.M.; Litt, A. The Aquilegia FRUITFULL-like genes play key roles in leaf morphogenesis and inflorescence development. Plant J. 2013, 74, 197–212. [Google Scholar] [CrossRef]
- Ferrándiz, C.; Gu, Q.; Martienssen, R.; Yanofsky, M.F. Redundant regulation of meristem identity and plant architecture by FRUITFULL, APETALA1 and CAULIFLOWER. Development 2000, 127, 725–734. [Google Scholar] [CrossRef]
- Cheng, X.; Wang, H.; Wei, H.; Gu, L.; Hao, P.; Sun, H.; Wu, A.; Cheng, S.; Yu, S. The MADS transcription factor GhAP1.7 coordinates the flowering regulatory pathway in upland cotton (Gossypium hirsutum L.). Gene 2021, 769, 145235. [Google Scholar] [CrossRef]
- Li, C.; Chen, L.; Fan, X.; Qi, W.; Ma, J.; Tian, T.; Zhou, T.; Ma, L.; Chen, F. MawuAP1 promotes flowering and fruit development in the basal angiosperm Magnolia wufengensis (Magnoliaceae). Tree Physiol. 2020, 40, 1247–1259. [Google Scholar] [CrossRef]
- Morel, P.; Chambrier, P.; Boltz, V.; Chamot, S.; Rozier, F.; Rodrigues Bento, S.; Trehin, C.; Monniaux, M.; Zethof, J.; Vandenbussche, M. Divergent Functional Diversification Patterns in the SEP/AGL6/AP1 MADS-Box Transcription Factor Superclade. Plant Cell 2019, 31, 3033–3056. [Google Scholar] [CrossRef] [Green Version]
- Alejandra Mandel, M.; Gustafson-Brown, C.; Savidge, B.; Yanofsky, M.F. Molecular characterization of the Arabidopsis floral homeotic gene APETALA1. Nature 1992, 360, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Tang, A.; Yu, J.; Cheng, T.; Wang, J.; Yang, W.; Pan, H.; Zhang, Q. RcAP1, a Homolog of APETALA1, is Associated with Flower Bud Differentiation and Floral Organ Morphogenesis in Rosa chinensis. Int. J. Mol. Sci. 2019, 20, 3557. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Fei, Y.; Zhang, K.; Fang, Z. Ectopic Expression of a Fagopyrum esculentum APETALA1 Ortholog only Rescues Sepal Development in Arabidopsis ap1 Mutant. Int. J. Mol. Sci. 2019, 20, 2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Li, Y.; Cao, Y.; Shi, W.; Xie, E.; Mu, N.; Du, G.; Shen, Y.; Tang, D.; Cheng, Z. Nitrogen nutrition contributes to plant fertility by affecting meiosis initiation. Nat. Commun. 2022, 13, 485. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, A.; Huang, H.; Zheng, M.; Zaman, W.; Li, D.; Saqib, S.; Zhao, H.; Lü, S. Molecular Cloning and Functional Analysis of GmLACS2-3 Reveals Its Involvement in Cutin and Suberin Biosynthesis along with Abiotic Stress Tolerance. Int. J. Mol. Sci. 2021, 22, 9175. [Google Scholar] [CrossRef]
- Ayaz, A.; Saqib, S.; Huang, H.; Zaman, W.; Lü, S.; Zhao, H. Genome-wide comparative analysis of long-chain acyl-CoA synthetases (LACSs) gene family: A focus on identification, evolution and expression profiling related to lipid synthesis. Plant Physiol. Biochem. 2021, 161, 1–11. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Naeem, M.; Shahzad, K.; Saqib, S.; Shahzad, A.; Nasrullah; Younas, M.; Afridi, M.I. The Solanum melongena COP1LIKE manipulates fruit ripening and flowering time in tomato (Solanum lycopersicum). Plant Growth Regul. 2022, 96, 369–382. [Google Scholar] [CrossRef]
- Sparkes, I.A.; Runions, J.; Kearns, A.; Hawes, C. Rapid, transient expression of fluorescent fusion proteins in tobacco plants and generation of stably transformed plants. Nat. Protoc. 2006, 1, 2019–2025. [Google Scholar] [CrossRef]
- Clough, S.J.; Bent, A.F. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998, 16, 735–743. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Ge, H.; Ahmad, N.; Li, J.; Wang, Y.; Liu, X.; Liu, W.; Li, X.; Wang, N.; Wang, F.; et al. Genome-Wide Identification of MADS-Box Family Genes in Safflower (Carthamus tinctorius L.) and Functional Analysis of CtMADS24 during Flowering. Int. J. Mol. Sci. 2023, 24, 1026. https://doi.org/10.3390/ijms24021026
Wang Y, Ge H, Ahmad N, Li J, Wang Y, Liu X, Liu W, Li X, Wang N, Wang F, et al. Genome-Wide Identification of MADS-Box Family Genes in Safflower (Carthamus tinctorius L.) and Functional Analysis of CtMADS24 during Flowering. International Journal of Molecular Sciences. 2023; 24(2):1026. https://doi.org/10.3390/ijms24021026
Chicago/Turabian StyleWang, Yifei, Hengshuo Ge, Naveed Ahmad, Jia Li, Yijin Wang, Xinyi Liu, Weican Liu, Xiaowei Li, Nan Wang, Fawei Wang, and et al. 2023. "Genome-Wide Identification of MADS-Box Family Genes in Safflower (Carthamus tinctorius L.) and Functional Analysis of CtMADS24 during Flowering" International Journal of Molecular Sciences 24, no. 2: 1026. https://doi.org/10.3390/ijms24021026