
Exploring the Role of Different Cell-Death-Related Genes in Sepsis Diagnosis Using a Machine Learning Algorithm


Abstract
:1. Introduction
2. Results
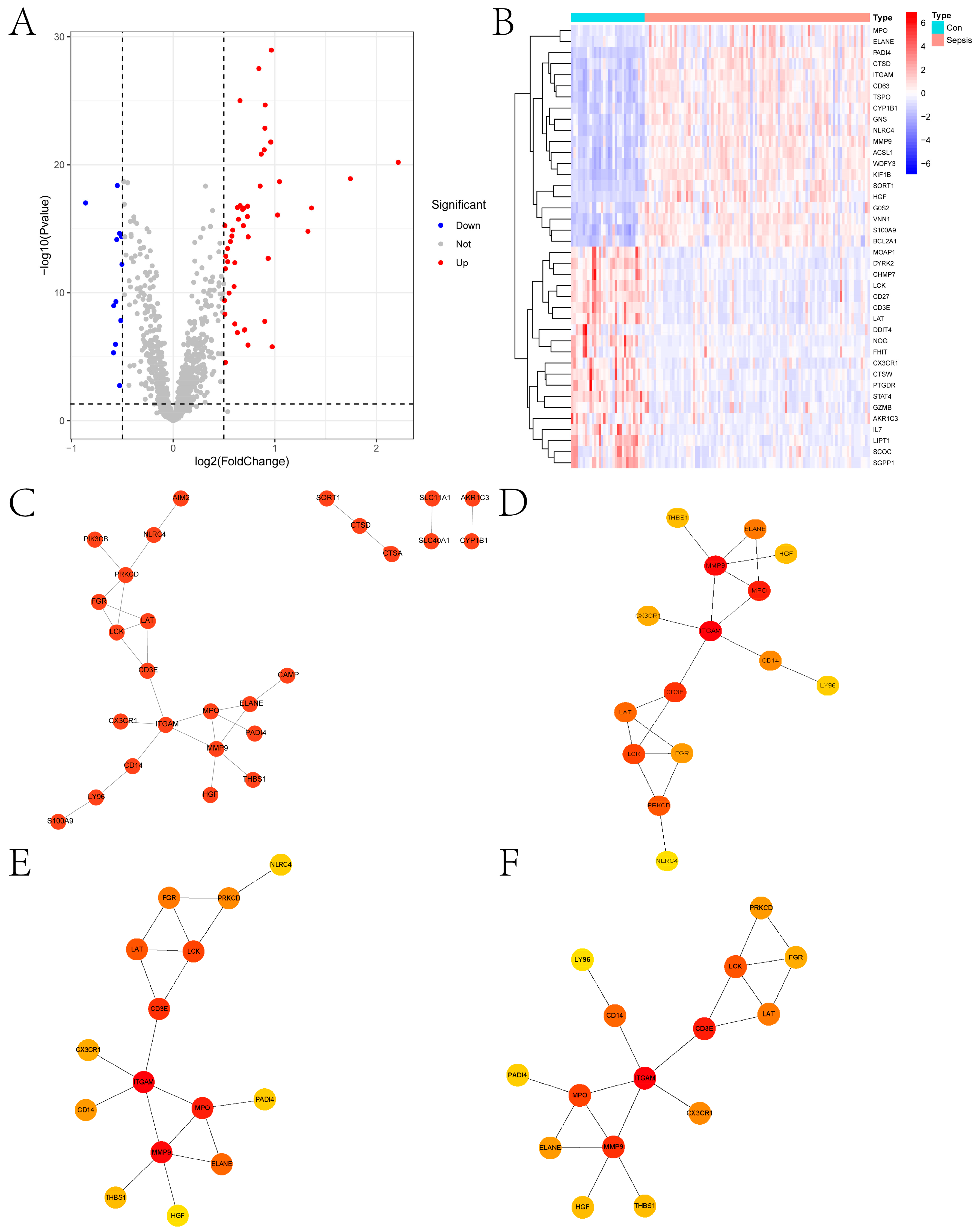
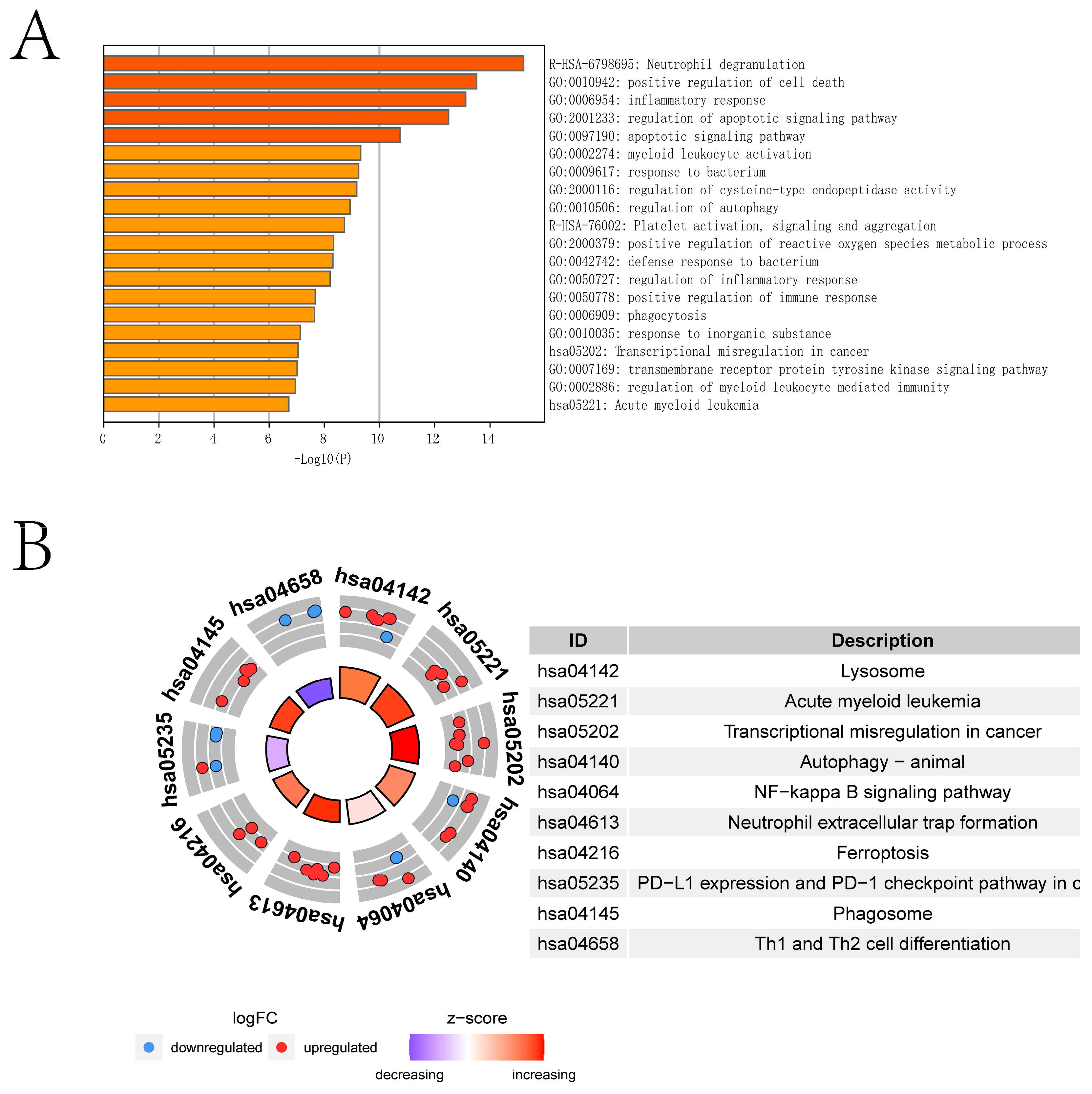
2.1. Differential Expression Analysis and Enrichment Analysis
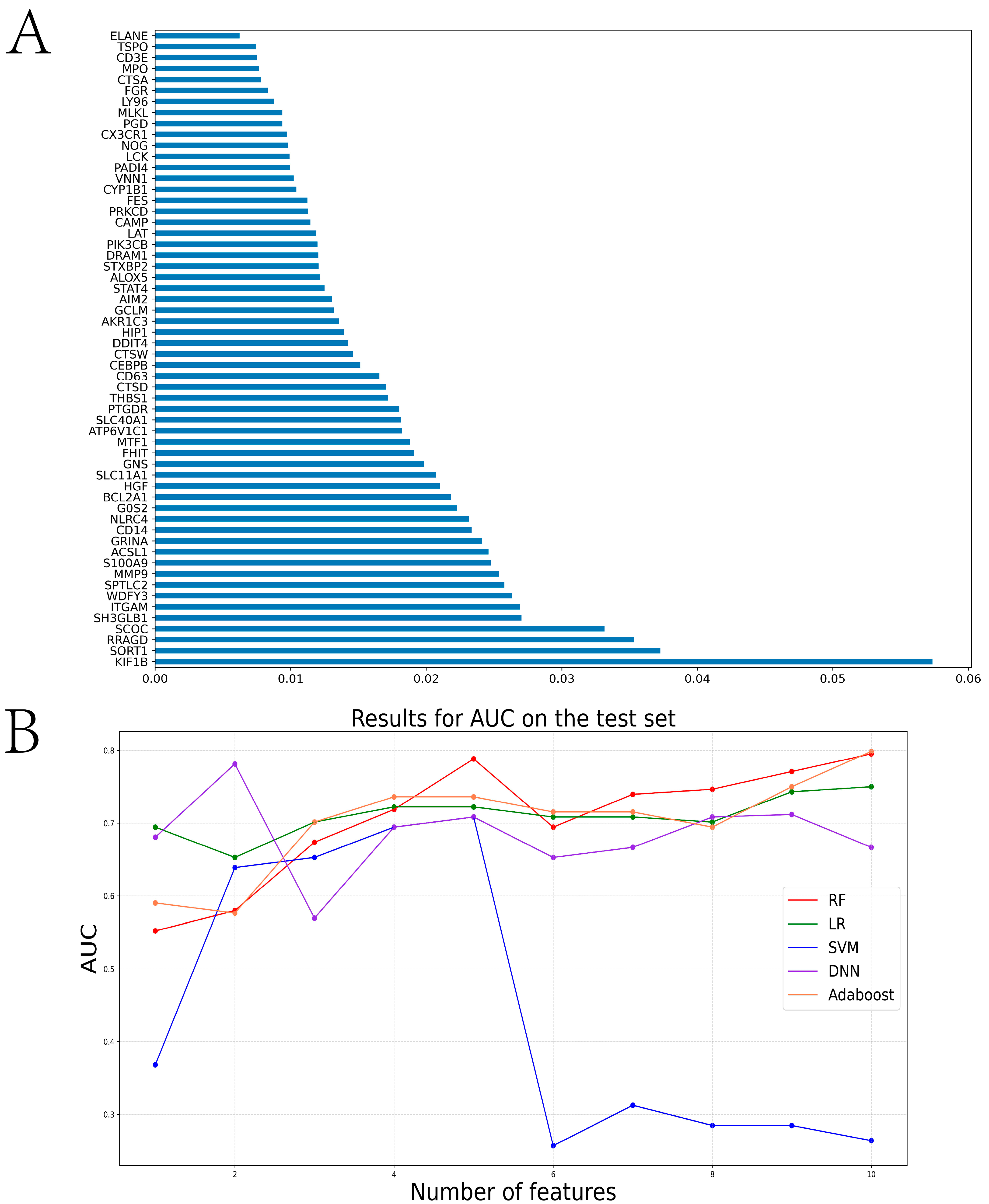
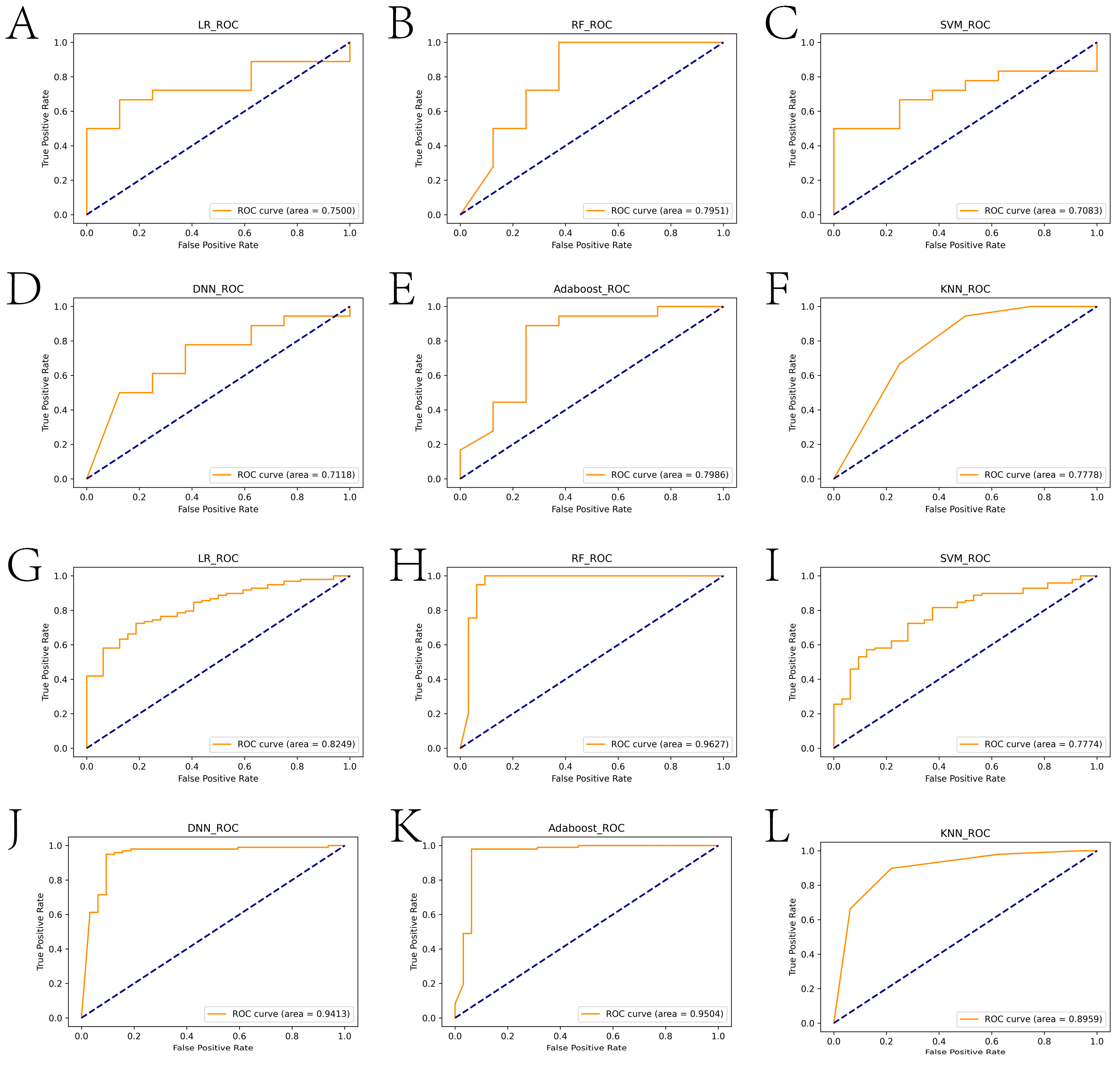
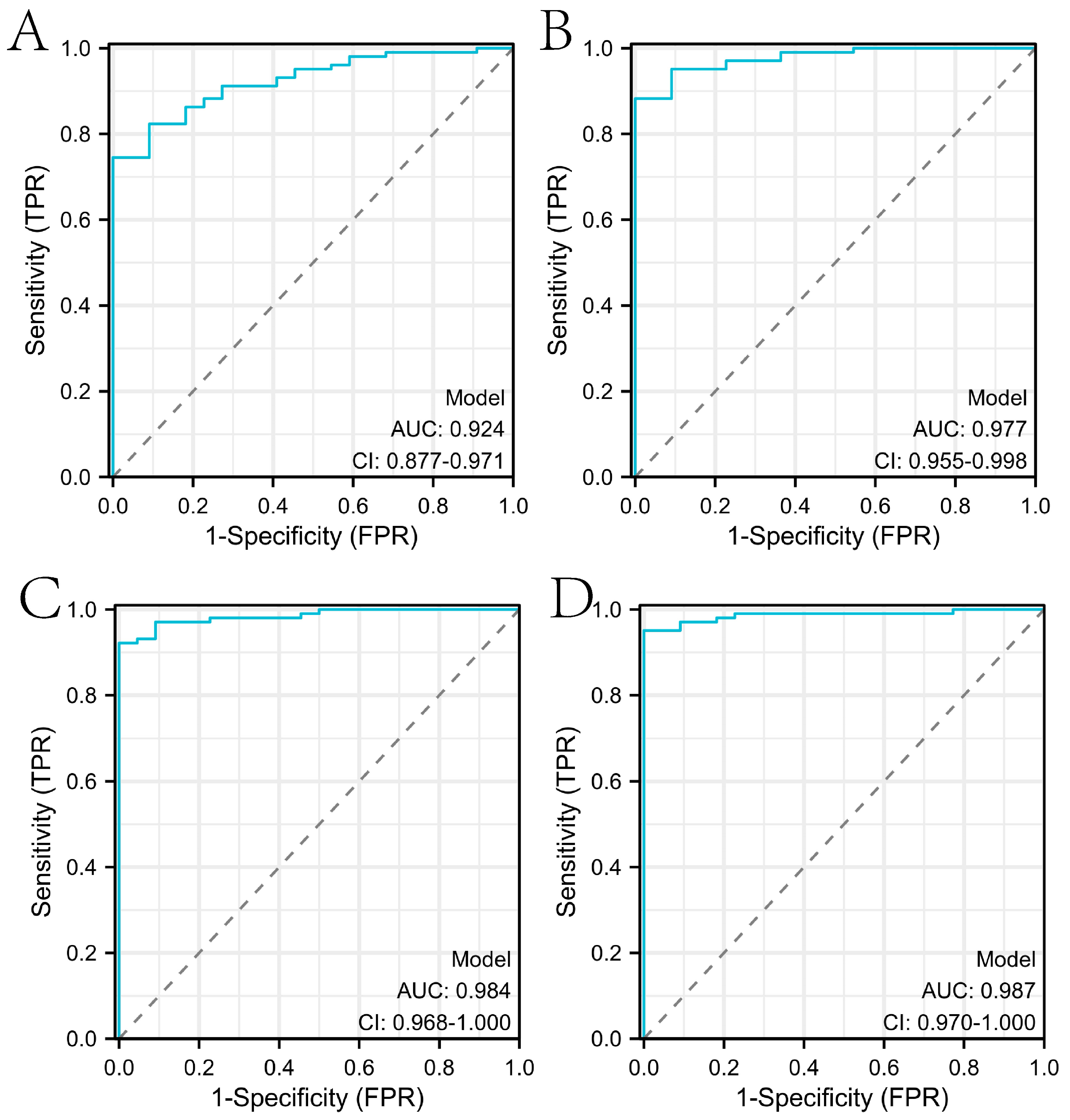
2.2. Construction of the Diagnosis-Related Model Based on Machine Learning Algorithms
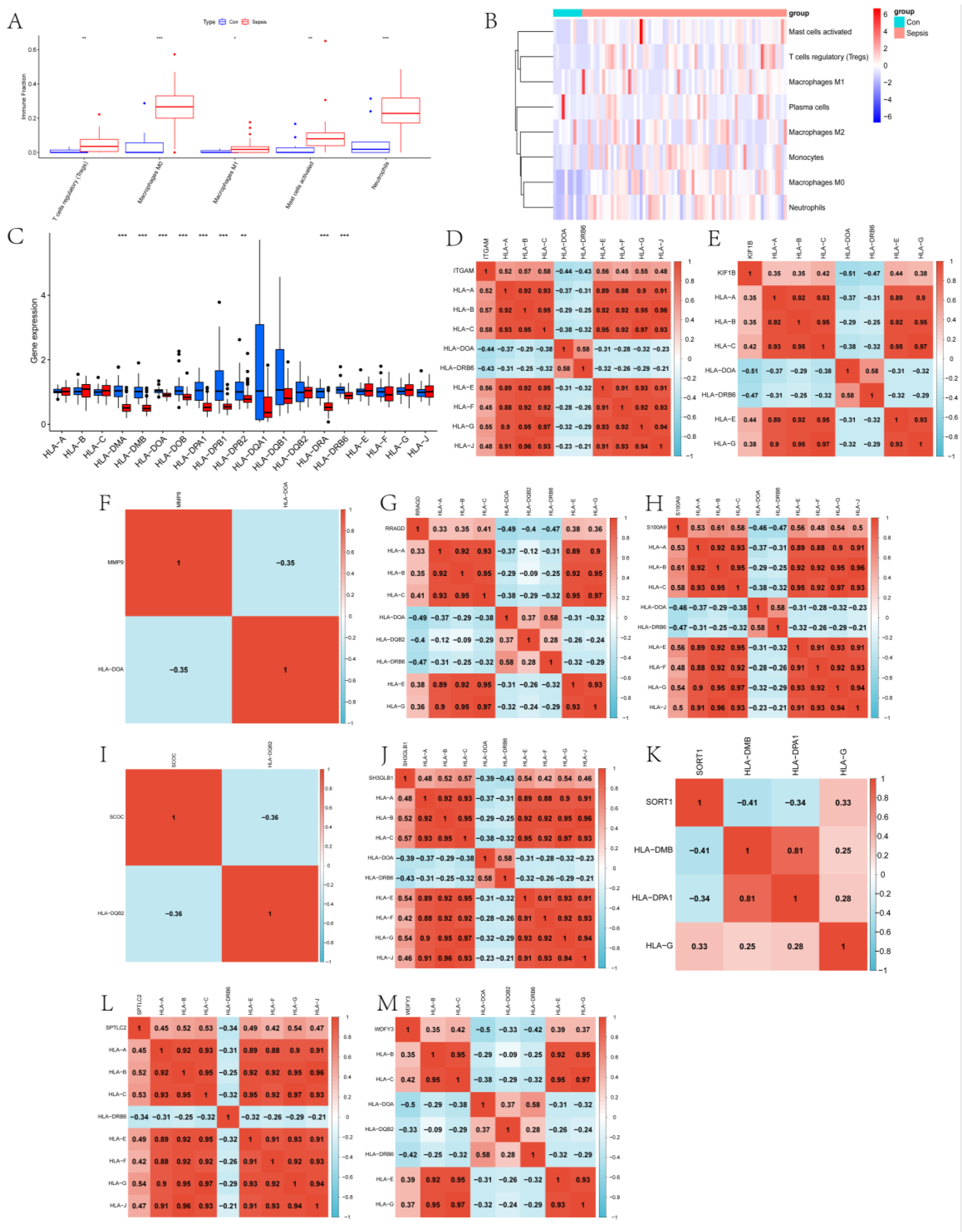
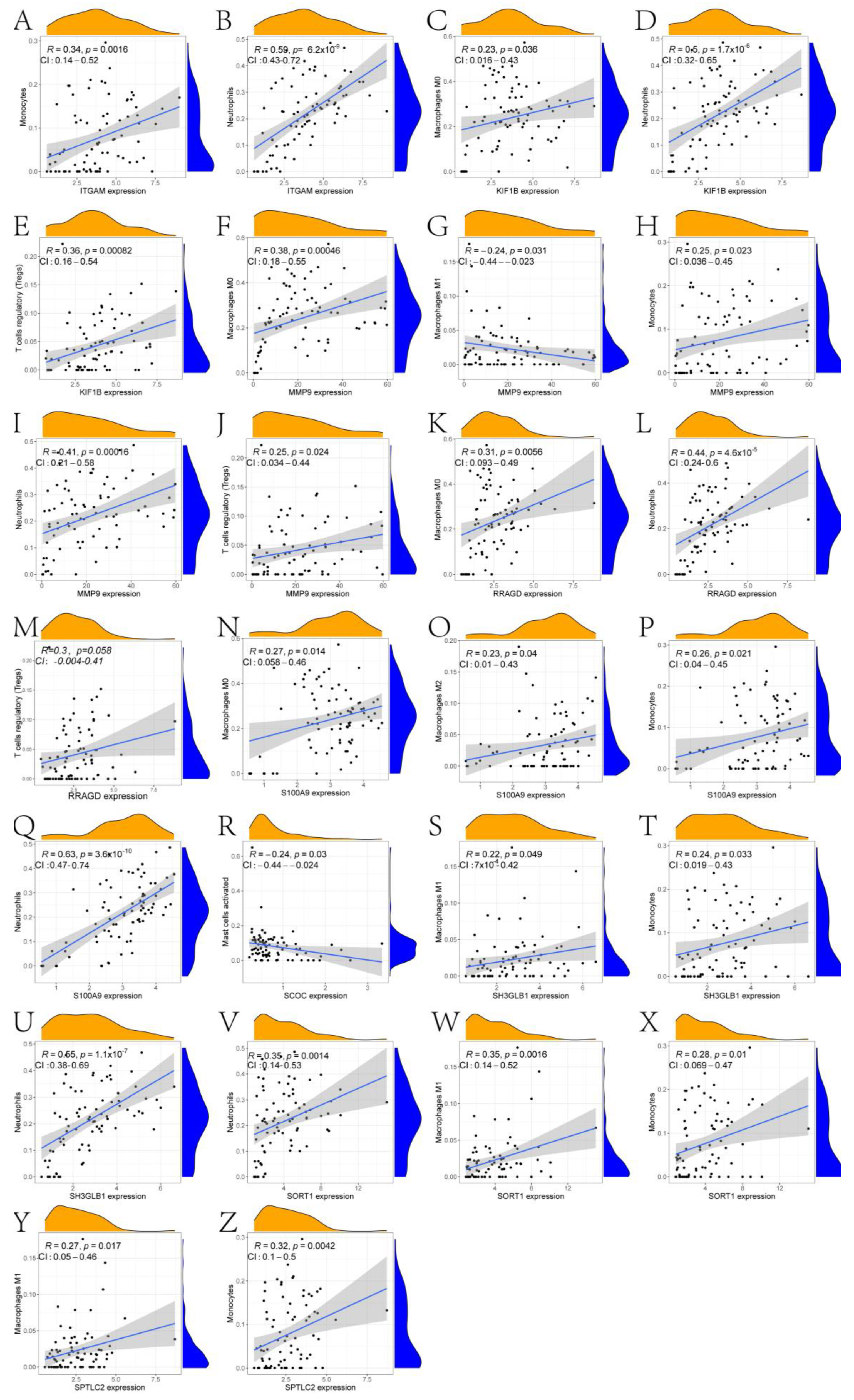
2.3. Exploration of the Immune Landscape
2.4. Comparison with Diagnostic Models Constructed in Other Studies
3. Discussion
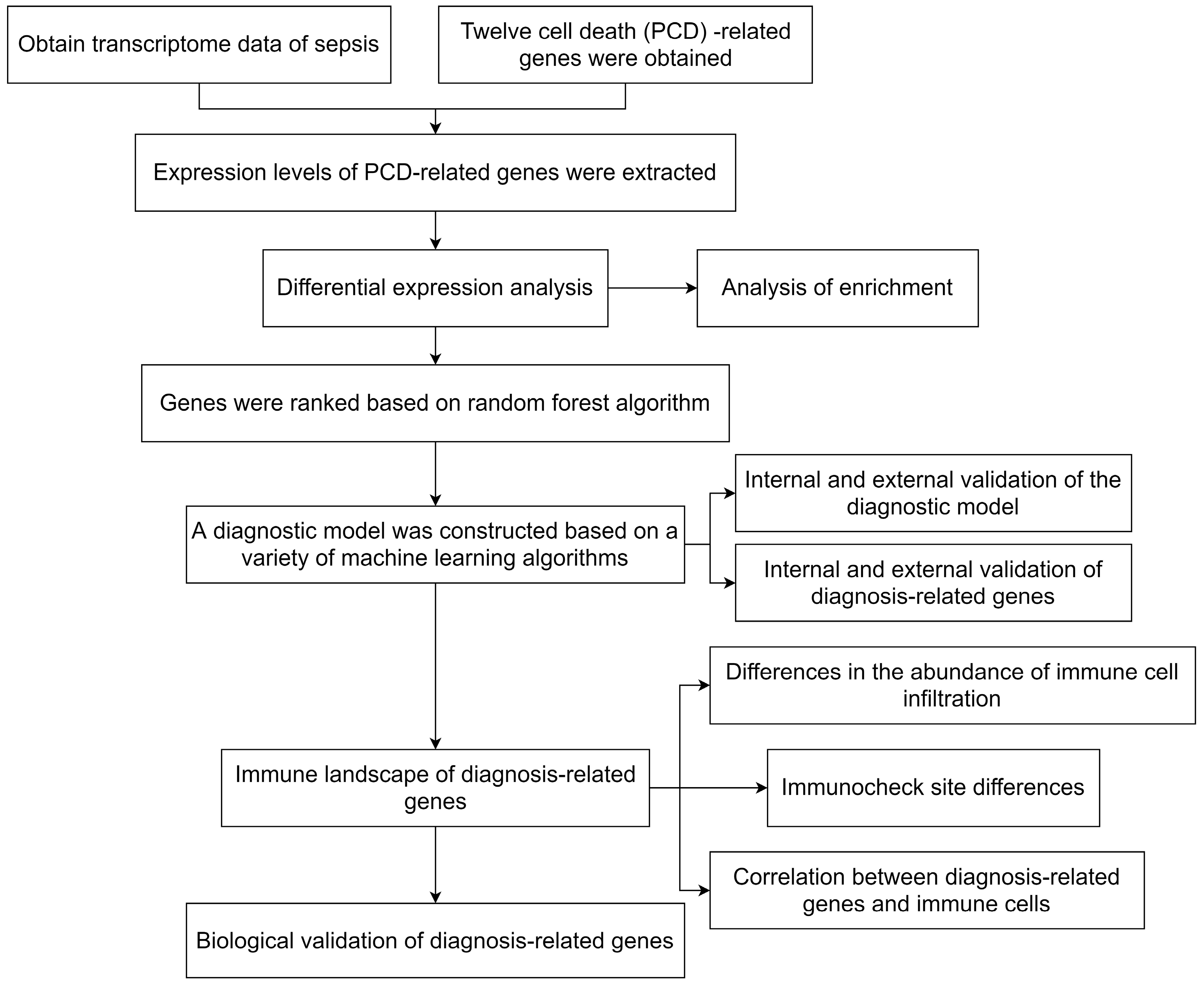
4. Materials and Methods
4.1. Data Acquisition
4.2. Differential Expression Analysis
4.3. Protein Interaction Network Construction
4.4. Enrichment Analysis
4.5. Selection of Diagnostic-Related Genes and Construction of Diagnostic Models
4.6. Immune-Related Analysis
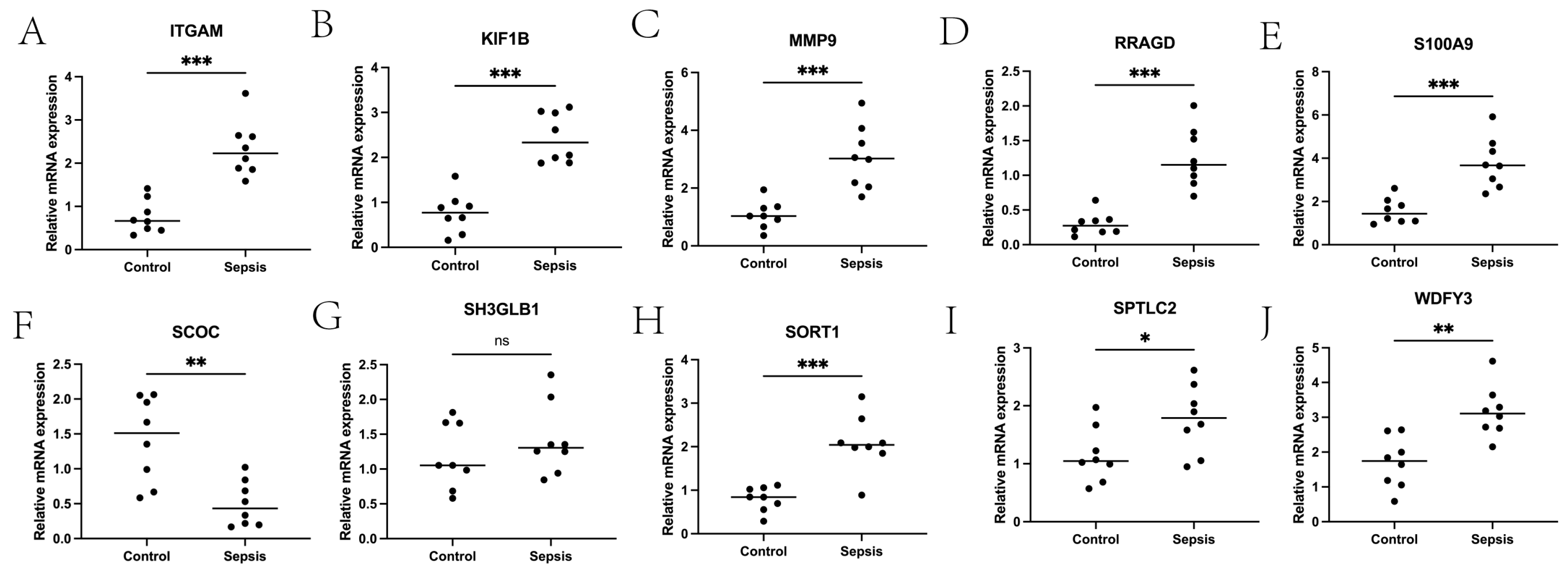
4.7. qPCR Experimental Verification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Evans, T. Diagnosis and management of sepsis. Clin. Med. 2018, 18, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Atreya, M.R.; Wong, H.R. Precision medicine in pediatric sepsis. Curr. Opin. Pediatr. 2019, 31, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Guo, Z.; Chai, Y.; Wang, Z.; Liao, H.; Wang, Z.; Wang, Z. Application Prospect of the SOFA Score and Related Modification Research Progress in Sepsis. J. Clin. Med. 2023, 12, 3493. [Google Scholar] [CrossRef]
- Reinhart, K.; Bauer, M.; Riedemann, N.C.; Hartog, C.S. New approaches to sepsis: Molecular diagnostics and biomarkers. Clin. Microbiol. Rev. 2012, 25, 609–634. [Google Scholar] [CrossRef]
- Mirijello, A.; Tosoni, A.; On Behalf of The Internal Medicine Sepsis Study, G. New Strategies for Treatment of Sepsis. Medicina 2020, 56, 527. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, C.; Liu, Y.; Wang, F.; Zhao, B.; Yang, J.; Zhao, Y.; Zhao, H.; Wang, G. Diagnostic and Predictive Values of Ferroptosis-Related Genes in Child Sepsis. Front. Immunol. 2022, 13, 881914. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Pan, Q.; Ge, H.; Xing, L.; Hong, Y.; Chen, P. Deep learning-based clustering robustly identified two classes of sepsis with both prognostic and predictive values. EBioMedicine 2020, 62, 103081. [Google Scholar] [CrossRef]
- Zou, Y.; Xie, J.; Zheng, S.; Liu, W.; Tang, Y.; Tian, W.; Deng, X.; Wu, L.; Zhang, Y.; Wong, C.W.; et al. Leveraging diverse cell-death patterns to predict the prognosis and drug sensitivity of triple-negative breast cancer patients after surgery. Int. J. Surg. 2022, 107, 106936. [Google Scholar] [CrossRef]
- Xu, X.; Lai, Y.; Hua, Z.C. Apoptosis and apoptotic body: Disease message and therapeutic target potentials. Biosci. Rep. 2019, 39, BSR20180992. [Google Scholar] [CrossRef]
- Yan, J.; Wan, P.; Choksi, S.; Liu, Z.G. Necroptosis and tumor progression. Trends Cancer 2022, 8, 21–27. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Hamann, J.C.; Kim, S.E.; Overholtzer, M. Methods for the Study of Entotic Cell Death. Methods Mol. Biol. 2019, 1880, 447–454. [Google Scholar] [PubMed]
- Berg, A.L.; Rowson-Hodel, A.; Wheeler, M.R.; Hu, M.; Free, S.R.; Carraway, K.L., III. Breast Cancer; Mayrovitz, H.N., Ed.; Exon Publications: Brisbane, Australia, 2022. [Google Scholar]
- Wang, X.; Ge, P. Parthanatos in the pathogenesis of nervous system diseases. Neuroscience 2020, 449, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef]
- Scaturro, P.; Pichlmair, A. Oxeiptosis: A discreet way to respond to radicals. Curr. Opin. Immunol. 2019, 56, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kuang, F.; Kang, R.; Tang, D. Alkaliptosis: A new weapon for cancer therapy. Cancer Gene Ther. 2020, 27, 267–269. [Google Scholar] [CrossRef]
- Wang, X.; Guo, Z.; Wang, Z.; Liao, H.; Wang, Z.; Chen, F.; Wang, Z. Diagnostic and predictive values of pyroptosis-related genes in sepsis. Front. Immunol. 2023, 14, 1105399. [Google Scholar] [CrossRef]
- Liang, S.; Xing, M.; Chen, X.; Peng, J.; Song, Z.; Zou, W. Predicting the prognosis in patients with sepsis by a pyroptosis-related gene signature. Front. Immunol. 2022, 13, 1110602. [Google Scholar] [CrossRef]
- Hao, S.; Huang, M.; Xu, X.; Wang, X.; Song, Y.; Jiang, W.; Huo, L.; Gu, J. Identification and validation of a novel mitochondrion-related gene signature for diagnosis and immune infiltration in sepsis. Front. Immunol. 2023, 14, 1196306. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, L.; Ni, S. Screening of ferroptosis-related genes in sepsis-induced liver failure and analysis of immune correlation. PeerJ 2022, 10, e13757. [Google Scholar] [CrossRef]
- Lai, Y.; Lin, C.; Lin, X.; Wu, L.; Zhao, Y.; Shao, T.; Lin, F. Comprehensive Analysis of Molecular Subtypes and Hub Genes of Sepsis by Gene Expression Profiles. Front. Genet. 2022, 13, 884762. [Google Scholar] [CrossRef]
- Lin, S.; Luo, B.; Ma, J. Multiple datasets to explore the molecular mechanism of sepsis. BMC Genom. Data 2022, 23, 66. [Google Scholar] [CrossRef]
- Martín-Fernández, M.; Tamayo-Velasco, Á.; Aller, R.; Gonzalo-Benito, H.; Martínez-Paz, P.; Tamayo, E. Endothelial Dysfunction and Neutrophil Degranulation as Central Events in Sepsis Physiopathology. Int. J. Mol. Sci. 2021, 22, 6272. [Google Scholar] [CrossRef]
- Bosmann, M.; Ward, P.A. The inflammatory response in sepsis. Trends Immunol. 2013, 34, 129–136. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [CrossRef]
- Cao, C.; Yu, M.; Chai, Y. Pathological alteration and therapeutic implications of sepsis-induced immune cell apoptosis. Cell Death Dis. 2019, 10, 782. [Google Scholar] [CrossRef]
- Wang, H.; Bai, G.; Chen, J.; Han, W.; Guo, R.; Cui, N. mTOR deletion ameliorates CD4 + T cell apoptosis during sepsis by improving autophagosome-lysosome fusion. Apoptosis 2022, 27, 401–408. [Google Scholar] [CrossRef]
- Malik, I.A.; Cardenas-Turanzas, M.; Gaeta, S.; Borthakur, G.; Price, K.; Cortes, J.; Nates, J.L. Sepsis and Acute Myeloid Leukemia: A Population-Level Study of Comparative Outcomes of Patients Discharged From Texas Hospitals. Clin. Lymphoma Myeloma Leuk. 2017, 17, e27–e32. [Google Scholar] [CrossRef]
- Zhou, H.; Li, Y.; Gui, H.; Zhao, H.; Wu, M.; Li, G.; Li, Y.; Bai, Z.; Yin, Z.; Redmond, H.P.; et al. Antagonism of Integrin CD11b Affords Protection against Endotoxin Shock and Polymicrobial Sepsis via Attenuation of HMGB1 Nucleocytoplasmic Translocation and Extracellular Release. J. Immunol. 2018, 200, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Liu, L.P.; Gui, R.; Dong, H.; Su, Y.R.; Zhou, X.H.; Liu, F.X. Discovering common pathogenetic processes between COVID-19 and sepsis by bioinformatics and system biology approach. Front. Immunol. 2022, 13, 975848. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cui, Y.; Ding, X.; Liu, S.; Han, B.; Duan, X.; Zhang, H.; Sun, T. Analysis of mRNA-lncRNA and mRNA-lncRNA-pathway co-expression networks based on WGCNA in developing pediatric sepsis. Bioengineered 2021, 12, 1457–1470. [Google Scholar] [CrossRef]
- Nangaku, M.; Sato-Yoshitake, R.; Okada, Y.; Noda, Y.; Takemura, R.; Yamazaki, H.; Hirokawa, N. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell 1994, 79, 1209–1220. [Google Scholar] [CrossRef]
- Goodwin, J.K.; Schaer, M. Septic shock. Vet. Clin. N. Am. Small Anim. Pract. 1989, 19, 1239–1258. [Google Scholar] [CrossRef]
- Fan, J.; Shi, S.; Qiu, Y.; Liu, M.; Shu, Q. Analysis of signature genes and association with immune cells infiltration in pediatric septic shock. Front. Immunol. 2022, 13, 1056750. [Google Scholar] [CrossRef]
- Hong, Y.; Chen, L.; Sun, J.; Xing, L.; Yang, Y.; Jin, X.; Cai, H.; Dong, L.; Zhou, L.; Zhang, Z. Single-cell transcriptome profiling reveals heterogeneous neutrophils with prognostic values in sepsis. iScience 2022, 25, 105301. [Google Scholar] [CrossRef]
- Tang, H.; Ivanciu, L.; Popescu, N.; Peer, G.; Hack, E.; Lupu, C.; Taylor, F.B., Jr.; Lupu, F. Sepsis-induced coagulation in the baboon lung is associated with decreased tissue factor pathway inhibitor. Am. J. Pathol. 2007, 171, 1066–1077. [Google Scholar] [CrossRef]
- Belaaouaj, A.A.; Li, A.; Wun, T.C.; Welgus, H.G.; Shapiro, S.D. Matrix metalloproteinases cleave tissue factor pathway inhibitor. Effects on coagulation. J. Biol. Chem. 2000, 275, 27123–27128. [Google Scholar] [CrossRef]
- Ryckman, C.; Vandal, K.; Rouleau, P.; Talbot, M.; Tessier, P.A. Proinflammatory activities of S100: Proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J. Immunol. 2003, 170, 3233–3242. [Google Scholar] [CrossRef]
- Ding, Z.; Du, F.; Averitt, V.R.; Jakobsson, G.; Rönnow, C.F.; Rahman, M.; Schiopu, A.; Thorlacius, H. Targeting S100A9 Reduces Neutrophil Recruitment, Inflammation and Lung Damage in Abdominal Sepsis. Int. J. Mol. Sci. 2021, 22, 12923. [Google Scholar] [CrossRef]
- Dai, J.; Kumbhare, A.; Youssef, D.; McCall, C.E.; El Gazzar, M. Intracellular S100A9 Promotes Myeloid-Derived Suppressor Cells during Late Sepsis. Front. Immunol. 2017, 8, 1565. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, F.; Teng, F.; Guo, S.; Li, H. Deficiency of S100A9 Alleviates Sepsis-Induced Acute Liver Injury through Regulating AKT-AMPK-Dependent Mitochondrial Energy Metabolism. Int. J. Mol. Sci. 2023, 24, 2112. [Google Scholar] [CrossRef] [PubMed]
- Touyama, K.; Khan, M.; Aoki, K.; Matsuda, M.; Hiura, F.; Takakura, N.; Matsubara, T.; Harada, Y.; Hirohashi, Y.; Tamura, Y.; et al. Bif-1/Endophilin B1/SH3GLB1 regulates bone homeostasis. J. Cell Biochem. 2019, 120, 18793–18804. [Google Scholar] [CrossRef]
- Huang, X.; Tan, J.; Chen, X.; Zhao, L. Identifying Potential Effective Diagnostic and Prognostic Biomarkers in Sepsis by Bioinformatics Analysis and Validation. Int. J. Gen. Med. 2022, 15, 6055–6071. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; He, Y.; Deng, Y.; Li, X.; Wang, W.; Chen, J. Ciclopirox mitigates inflammatory response in LPS-induced septic shock via inactivation of SORT1-mediated wnt/β-Catenin signaling pathway. Immunopharmacol. Immunotoxicol. 2023, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Di, C.; Du, Y.; Zhang, R.; Zhang, L.; Wang, S. Identification of autophagy-related genes and immune cell infiltration characteristics in sepsis via bioinformatic analysis. J. Thorac. Dis. 2023, 15, 1770–1784. [Google Scholar] [CrossRef]
- Monneret, G.; Debard, A.L.; Venet, F.; Bohe, J.; Hequet, O.; Bienvenu, J.; Lepape, A. Marked elevation of human circulating CD4+CD25+ regulatory T cells in sepsis-induced immunoparalysis. Crit. Care Med. 2003, 31, 2068–2071. [Google Scholar] [CrossRef]
- Venet, F.; Chung, C.S.; Kherouf, H.; Geeraert, A.; Malcus, C.; Poitevin, F.; Bohé, J.; Lepape, A.; Ayala, A.; Monneret, G. Increased circulating regulatory T cells (CD4(+)CD25 (+)CD127 (-)) contribute to lymphocyte anergy in septic shock patients. Intensive Care Med. 2009, 35, 678–686. [Google Scholar] [CrossRef]
- Schultze, J.L.; Schmidt, S.V. Molecular features of macrophage activation. Semin. Immunol. 2015, 27, 416–423. [Google Scholar] [CrossRef]
- Ip, W.K.E.; Hoshi, N.; Shouval, D.S.; Snapper, S.; Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 2017, 356, 513–519. [Google Scholar] [CrossRef]
- Qiu, P.; Liu, Y.; Zhang, J. Review: The Role and Mechanisms of Macrophage Autophagy in Sepsis. Inflammation 2019, 42, 6–19. [Google Scholar] [CrossRef]
- Zhang, S.; Wu, Z.; Chang, W.; Liu, F.; Xie, J.; Yang, Y.; Qiu, H. Classification of Patients With Sepsis According to Immune Cell Characteristics: A Bioinformatic Analysis of Two Cohort Studies. Front. Med. 2020, 7, 598652. [Google Scholar] [CrossRef]
- Yue, J.; Tan, Y.; Huan, R.; Guo, J.; Yang, S.; Deng, M.; Xiong, Y.; Han, G.; Liu, L.; Liu, J.; et al. Mast cell activation mediates blood-brain barrier impairment and cognitive dysfunction in septic mice in a histamine-dependent pathway. Front. Immunol. 2023, 14, 1090288. [Google Scholar] [CrossRef] [PubMed]
- Kovach, M.A.; Standiford, T.J. The function of neutrophils in sepsis. Curr. Opin. Infect. Dis. 2012, 25, 321–327. [Google Scholar] [CrossRef]
- Lelubre, C.; Medfai, H.; Akl, I.; Leentjens, J.; Kox, M.; Pickkers, P.; Rousseau, A.; Biston, P.; Piagnerelli, M.; Vanhaeverbeek, M.; et al. Leukocyte phosphodiesterase expression after lipopolysaccharide and during sepsis and its relationship with HLA-DR expression. J. Leukoc. Biol. 2017, 101, 1419–1426. [Google Scholar] [CrossRef]
- Mohsin, M.; Singh, P.; Khan, S.; Verma, A.K.; Jha, R.; Alsahli, M.A.; Rahmani, A.H.; Almatroodi, S.A.; Alrumaihi, F.; Kaprwan, N.; et al. Integrated transcriptomic and regulatory network analyses uncovers the role of let-7b-5p, SPIB, and HLA-DPB1 in sepsis. Sci. Rep. 2022, 12, 11963. [Google Scholar] [CrossRef] [PubMed]
- Cajander, S.; Tina, E.; Bäckman, A.; Magnuson, A.; Strålin, K.; Söderquist, B.; Källman, J. Quantitative Real-Time Polymerase Chain Reaction Measurement of HLA-DRA Gene Expression in Whole Blood Is Highly Reproducible and Shows Changes That Reflect Dynamic Shifts in Monocyte Surface HLA-DR Expression during the Course of Sepsis. PLoS ONE 2016, 11, e0154690. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Silver, J.; Oshlack, A.; Holmes, M.; Diyagama, D.; Holloway, A.; Smyth, G.K. A comparison of background correction methods for two-colour microarrays. Bioinformatics 2007, 23, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Xu, Y.; Wang, S.; Wei, K.; Wen, G.; Yu, Y.; Zhu, Y. A Novel Longitudinal Phenotype-Genotype Association Study Based on Deep Feature Extraction and Hypergraph Models for Alzheimer’s Disease. Biomolecules 2023, 13, 728. [Google Scholar] [CrossRef] [PubMed]
- Sivamurugan, J.; Sureshkumar, G. Applying dual models on optimized LSTM with U-net segmentation for breast cancer diagnosis using mammogram images. Artif. Intell. Med. 2023, 143, 102626. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Wang, Z.; Guo, Z.; Wang, Z.; Chen, F.; Wang, Z. Exploring the Role of Different Cell-Death-Related Genes in Sepsis Diagnosis Using a Machine Learning Algorithm. Int. J. Mol. Sci. 2023, 24, 14720. https://doi.org/10.3390/ijms241914720
Wang X, Wang Z, Guo Z, Wang Z, Chen F, Wang Z. Exploring the Role of Different Cell-Death-Related Genes in Sepsis Diagnosis Using a Machine Learning Algorithm. International Journal of Molecular Sciences. 2023; 24(19):14720. https://doi.org/10.3390/ijms241914720
Chicago/Turabian StyleWang, Xuesong, Ziyi Wang, Zhe Guo, Ziwen Wang, Feng Chen, and Zhong Wang. 2023. "Exploring the Role of Different Cell-Death-Related Genes in Sepsis Diagnosis Using a Machine Learning Algorithm" International Journal of Molecular Sciences 24, no. 19: 14720. https://doi.org/10.3390/ijms241914720