
Comparison of Two Human Skin Cell Isolation Protocols and Their Influence on Keratinocyte and Fibroblast Culture

 ,
,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
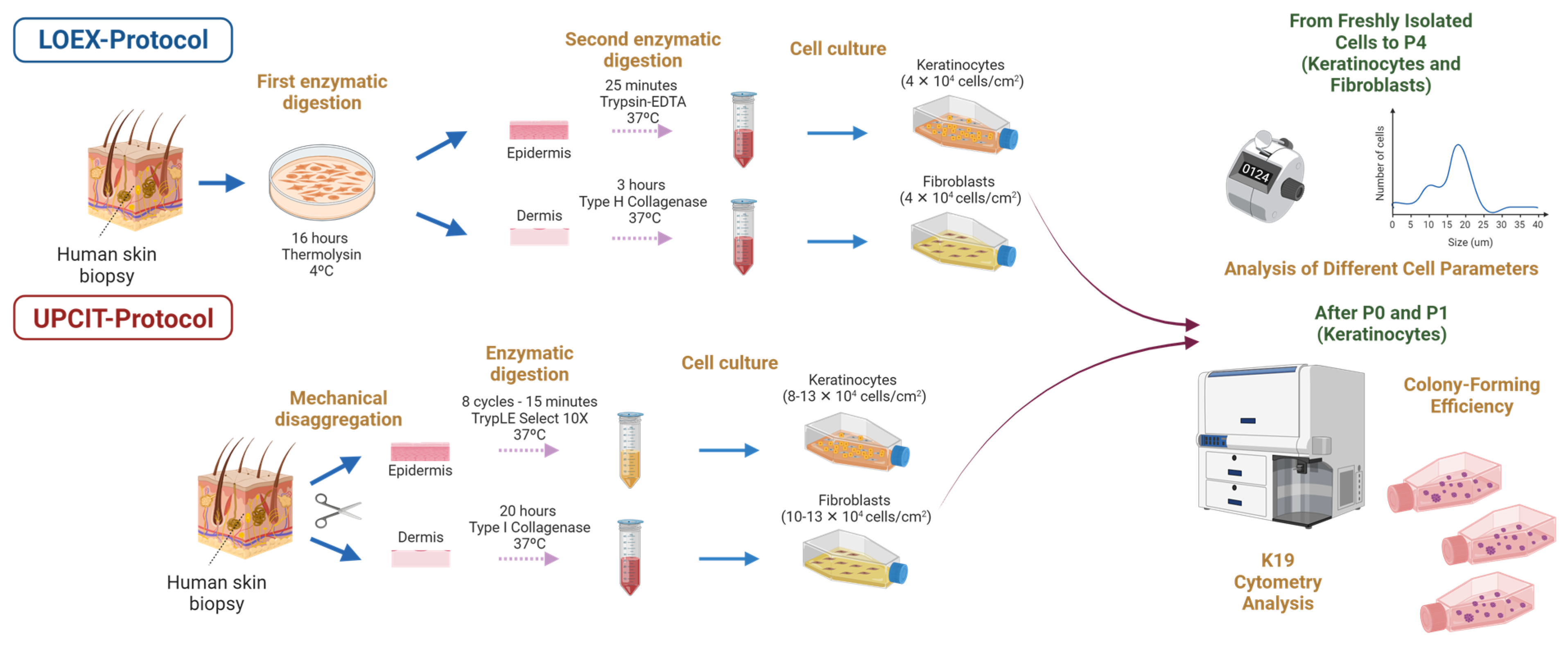
2.1. Cell Size, Viability and Yield
2.1.1. Epithelial Cells
2.1.2. Dermal Cells
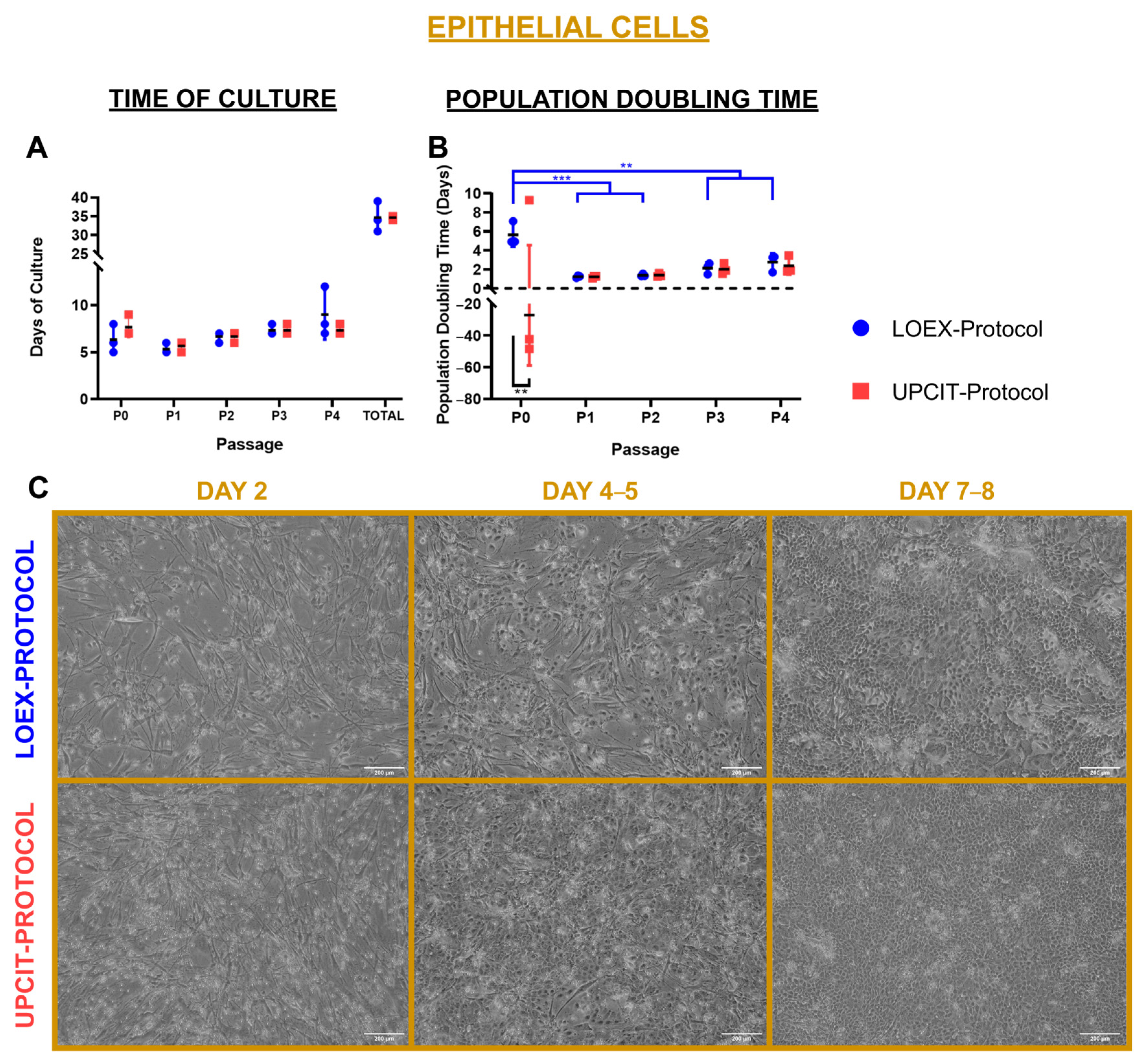
2.2. Culture and Population Doubling Time
2.2.1. Epithelial Cells
2.2.2. Dermal Cells
2.3. Clonogenicity of Epithelial Cell Cultures
2.4. Keratin 19 (K19) Analysis by Flow Cytometry
3. Discussion
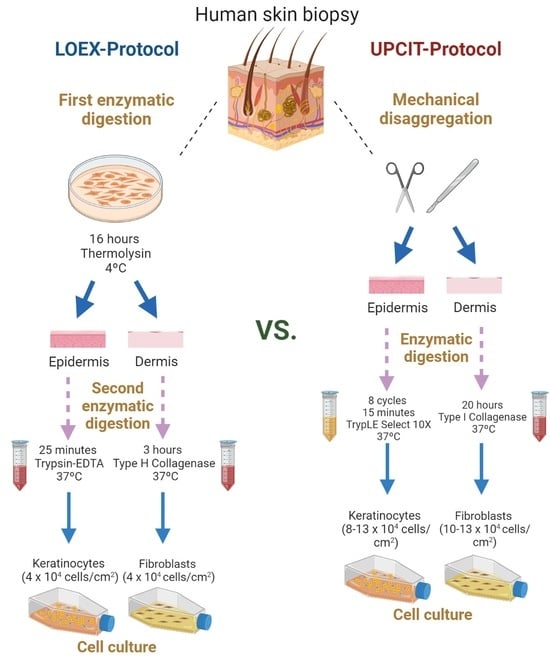
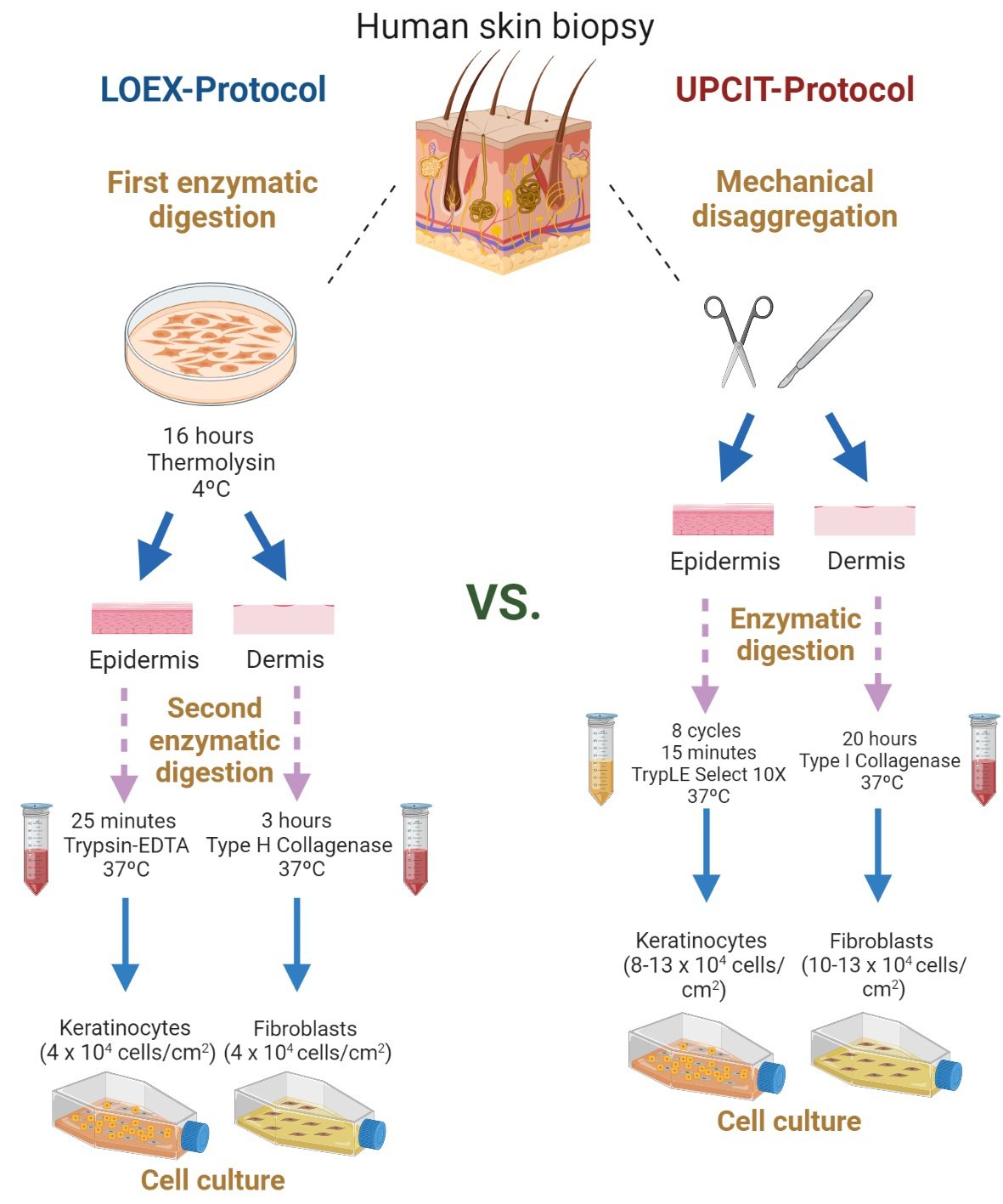
4. Materials and Methods
4.1. Cell Isolation and Culture
4.1.1. Two-Step Digestion Protocol (LOEX-Protocol)
Epithelial Cell Isolation
Dermal Cell Isolation
4.1.2. One-Step Digestion Protocol (UPCIT-Protocol)
Epithelial Cell Isolation
Dermal Cell Isolation
4.1.3. Culture Maintenance and Passages
4.2. Cell Size, Viability and Yield
4.3. Culture and Population Doubling Time
4.4. Clonogenicity of Epithelial Cell Cultures
4.5. Keratin 19 (K19) Analysis by Flow Cytometry
4.6. Statistical Analysis
4.7. Ethics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Phelan, K.; May, K.M. Mammalian Cell Tissue Culture Techniques. Curr. Protoc. Mol. Biol. 2017, 117, A.3F.1–A.3F.23. [Google Scholar] [CrossRef] [PubMed]
- Combes, R.D. The Use of Human Cells in Biomedical Research and Testing. Altern. Lab. Anim. 2004, 32 (Suppl. 1A), 43–49. [Google Scholar] [CrossRef] [PubMed]
- Advanced Therapy Medicinal Products: Overview|European Medicines Agency. Available online: https://www.ema.europa.eu/en/human-regulatory/overview/advanced-therapy-medicinal-products-overview#advanced-therapies-in-the-product-lifecycle-section (accessed on 31 March 2023).
- Philippeos, C.; Hughes, R.D.; Dhawan, A.; Mitry, R.R. Introduction to Cell Culture. Methods Mol. Biol. 2012, 806, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kino-oka, M.; Taya, M. Recent Developments in Processing Systems for Cell and Tissue Cultures toward Therapeutic Application. J. Biosci. Bioeng. 2009, 108, 267–276. [Google Scholar] [CrossRef]
- Kino-oka, M.; Mizutani, M.; Medcalf, N. Cell Manufacturability. Cell Gene Ther. Insights 2019, 5, 1347–1359. [Google Scholar] [CrossRef]
- Sierra-Sánchez, Á.; Kim, K.H.; Blasco-Morente, G.; Arias-Santiago, S. Cellular Human Tissue-Engineered Skin Substitutes Investigated for Deep and Difficult to Heal Injuries. NPJ Regen. Med. 2021, 6, 35. [Google Scholar] [CrossRef]
- Ścieżyńska, A.; Nogowska, A.; Sikorska, M.; Konys, J.; Karpińska, A.; Komorowski, M.; Ołdak, M.; Malejczyk, J. Isolation and Culture of Human Primary Keratinocytes—A Methods Review. Exp. Dermatol. 2019, 28, 107–112. [Google Scholar] [CrossRef]
- Larouche, D.; Paquet, C.; Fradette, J.; Carrier, P.; Auger, F.A.; Germain, L. Regeneration of Skin and Cornea by Tissue Engineering. Methods Mol. Biol. 2009, 482, 233–256. [Google Scholar] [CrossRef]
- Germain, L.; Rouabhia, M.; Guignard, R.; Carrier, L.; Bouvard, V.; Auger, F.A. Improvement of Human Keratinocyte Isolation and Culture Using Thermolysin. Burns 1993, 19, 99–104. [Google Scholar] [CrossRef]
- Sierra-Sánchez, Á.; Fernández-González, A.; Lizana-Moreno, A.; Espinosa-Ibáñez, O.; Martinez-Lopez, A.; Guerrero-Calvo, J.; Fernández-Porcel, N.; Ruiz-García, A.; Ordóñez-Luque, A.; Carriel, V.; et al. Hyaluronic Acid Biomaterial for Human Tissue-Engineered Skin Substitutes: Preclinical Comparative in Vivo Study of Wound Healing. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 2414–2427. [Google Scholar] [CrossRef]
- Michel, M.; Török, N.; Godbout, M.J.; Lussier, M.; Gaudreau, P.; Royal, A.; Germain, L. Keratin 19 as a Biochemical Marker of Skin Stem Cells in Vivo and in Vitro: Keratin 19 Expressing Cells Are Differentially Localized in Function of Anatomic Sites, and Their Number Varies with Donor Age and Culture Stage. J. Cell Sci. 1996, 109 Pt 5, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Llames, S.G.; Del Rio, M.; Larcher, F.; García, E.; Escamez, M.J.; Jorcano, J.L.; Holguín, P.; Meana, A. Human Plasma as a Dermal Scaffold for the Generation of a Completely Autologous Bioengineered Skin. Transplantation 2004, 77, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Dervish, S.; Jackson, C.J. Isolation of Human Skin Epidermal Stem Cells Based on the Expression of Endothelial Protein C Receptor. Methods Mol. Biol. 2019, 1879, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, F.L.; Bernstam, L.I. Isolation, Purification, and Cultivation of Murine and Human Keratinocytes. Methods Mol. Biol. 2005, 290, 187–206. [Google Scholar] [CrossRef]
- Rheinwald, J.G.; Green, H. Serial Cultivation of Strains of Human Epidermal Keratinocytes: The Formation of Keratinizing Colonies from Single Cells. Cell 1975, 6, 331–343. [Google Scholar] [CrossRef]
- Johansen, C. Generation and Culturing of Primary Human Keratinocytes from Adult Skin. J. Vis. Exp. 2017, 2017, e56863. [Google Scholar] [CrossRef]
- Daniels, J.T.; Kearney, J.N.; Ingham, E. Human Keratinocyte Isolation and Cell Culture: A Survey of Current Practices in the UK. Burns 1996, 22, 35–39. [Google Scholar] [CrossRef]
- Aasen, T.; Belmonte, J.C.I. Isolation and Cultivation of Human Keratinocytes from Skin or Plucked Hair for the Generation of Induced Pluripotent Stem Cells. Nat. Protoc. 2010, 5, 371–382. [Google Scholar] [CrossRef]
- Braziulis, E.; Diezi, M.; Biedermann, T.; Pontiggia, L.; Schmucki, M.; Hartmann-Fritsch, F.; Luginbühl, J.; Schiestl, C.; Meuli, M.; Reichmann, E. Modified Plastic Compression of Collagen Hydrogels Provides an Ideal Matrix for Clinically Applicable Skin Substitutes. Tissue Eng. Part C Methods 2012, 18, 464–474. [Google Scholar] [CrossRef]
- Korosec, A.; Frech, S.; Lichtenberger, B.M. Isolation of Papillary and Reticular Fibroblasts from Human Skin by Fluorescence-Activated Cell Sorting. J. Vis. Exp. 2019, 2019, e59372. [Google Scholar] [CrossRef]
- Supp, D.M.; Hahn, J.M.; Combs, K.A.; McFarland, K.L.; Powell, H.M. Isolation and Feeder-Free Primary Culture of Four Cell Types from a Single Human Skin Sample. STAR Protoc. 2022, 3, 101172. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Kang, H.J.; Kim, C.H.; Kim, C.H.; Han, E.S.; Han, K.; Kim, T.H.; Gin, Y.J.; Son, Y.S. Application of Physical Force Is Essential to Enrich for Epidermal Stem Cells in Primary Human Keratinocyte Isolation. Tissue Eng. 2004, 10, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Rakhorst, H.A.; Tra, W.M.W.; Posthumus-van Sluijs, S.J.; de Groot, E.; van Osch, G.J.V.M.; van Neck, J.W.; Hofer, S.O.P. Mucosal Keratinocyte Isolation: A Short Comparative Study on Thermolysin and Dispase. Int. J. Oral Maxillofac. Surg. 2006, 35, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Prost, C.; Dubertret, L.; Fosse, M.; Wechsler, J.; Touraine, R. A Routine Immuno-Electron Microscopic Technique for Localizing an Auto-Antibody on Epidermal Basement Membrane. Br. J. Dermatol. 1984, 110, 1–7. [Google Scholar] [CrossRef]
- Walzer, C.; Benathan, M.; Frenk, E. Thermolysin Treatment: A New Method for Dermo-Epidermal Separation. J. Investig. Dermatol. 1989, 92, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Visse, R.; Inouye, M.; Nagase, H.; Brodsky, B. Defining Requirements for Collagenase Cleavage in Collagen Type III Using a Bacterial Collagen System. J. Biol. Chem. 2012, 287, 22988. [Google Scholar] [CrossRef]
- Jeon, Y.R.; Kang, E.H.; Yang, C.E.; Yun, I.S.; Lee, W.J.; Lew, D.H. The Effect of Platelet-Rich Plasma on Composite Graft Survival. Plast. Reconstr. Surg. 2014, 134, 239–246. [Google Scholar] [CrossRef]
- Hosseini, M.; Shafiee, A. Engineering Bioactive Scaffolds for Skin Regeneration. Small 2021, 17, 2101384. [Google Scholar] [CrossRef]
- Tedesco, M.; Pranteda, G.; Chichierchia, G.; Paolino, G.; Latini, A.; Orsini, D.; Cristaudo, A.; Foddai, M.L.; Migliano, E.; Morrone, A. The Use of PRP (Platelet-Rich Plasma) in Patients Affected by Genital Lichen Sclerosus: Clinical Analysis and Results. J. Eur. Acad. Dermatol. Venereol. 2019, 33, e58–e59. [Google Scholar] [CrossRef]
- Hybbinette, S.; Boström, M.; Lindberg, K. Enzymatic Dissociation of Keratinocytes from Human Skin Biopsies for in Vitro Cell Propagation. Exp. Dermatol. 1999, 8, 30–38. [Google Scholar] [CrossRef]
- Raxworthy, M.J.; Cunlife, W.J.; Wood, E.J. The Influence of Proteases on the Colony-Forming Efficiency of Human Keratinocytes in Culture. Biochem. Soc. Trans. 1987, 15, 519–520. [Google Scholar] [CrossRef]
- Gragnani, A.; Sobral, C.S.; Ferreira, L.M. Thermolysin in Human Cultured Keratinocyte Isolation. Braz. J. Biol. 2007, 67, 105–109. [Google Scholar] [CrossRef]
- Bisson, F.; Rochefort, É.; Lavoie, A.; Larouche, D.; Zaniolo, K.; Simard-Bisson, C.; Damour, O.; Auger, F.A.; Guérin, S.L.; Germain, L. Irradiated Human Dermal Fibroblasts Are as Efficient as Mouse Fibroblasts as a Feeder Layer to Improve Human Epidermal Cell Culture Lifespan. Int. J. Mol. Sci. 2013, 14, 4684. [Google Scholar] [CrossRef] [PubMed]
- Barrandon, Y.; Grasset, N.; Zaffalon, A.; Gorostidi, F.; Claudinot, S.; Droz-Georget, S.L.; Nanba, D.; Rochat, A. Capturing Epidermal Stemness for Regenerative Medicine. Semin. Cell Dev. Biol. 2012, 23, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Barrandon, Y.; Green, H. Three Clonal Types of Keratinocyte with Different Capacities for Multiplication. Proc. Natl. Acad. Sci. USA 1987, 84, 2302. [Google Scholar] [CrossRef] [PubMed]
- Ghio, S.C.; Barbier, M.A.; Doucet, E.J.; Debbah, I.; Safoine, M.; Le-Bel, G.; Cartier, A.; Jolibois, E.; Morissette, A.; Larouche, D.; et al. A Newly Developed Chemically Defined Serum-Free Medium Suitable for Human Primary Keratinocyte Culture and Tissue Engineering Applications. Int. J. Mol. Sci. 2023, 24, 1821. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sierra-Sánchez, Á.; Barbier, M.A.; Magne, B.; Larouche, D.; Arias-Santiago, S.; Germain, L. Comparison of Two Human Skin Cell Isolation Protocols and Their Influence on Keratinocyte and Fibroblast Culture. Int. J. Mol. Sci. 2023, 24, 14712. https://doi.org/10.3390/ijms241914712
Sierra-Sánchez Á, Barbier MA, Magne B, Larouche D, Arias-Santiago S, Germain L. Comparison of Two Human Skin Cell Isolation Protocols and Their Influence on Keratinocyte and Fibroblast Culture. International Journal of Molecular Sciences. 2023; 24(19):14712. https://doi.org/10.3390/ijms241914712
Chicago/Turabian StyleSierra-Sánchez, Álvaro, Martin A. Barbier, Brice Magne, Danielle Larouche, Salvador Arias-Santiago, and Lucie Germain. 2023. "Comparison of Two Human Skin Cell Isolation Protocols and Their Influence on Keratinocyte and Fibroblast Culture" International Journal of Molecular Sciences 24, no. 19: 14712. https://doi.org/10.3390/ijms241914712