
The Abundant and Unique Transcripts and Alternative Splicing of the Artificially Autododecaploid London Plane (Platanus × acerifolia)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
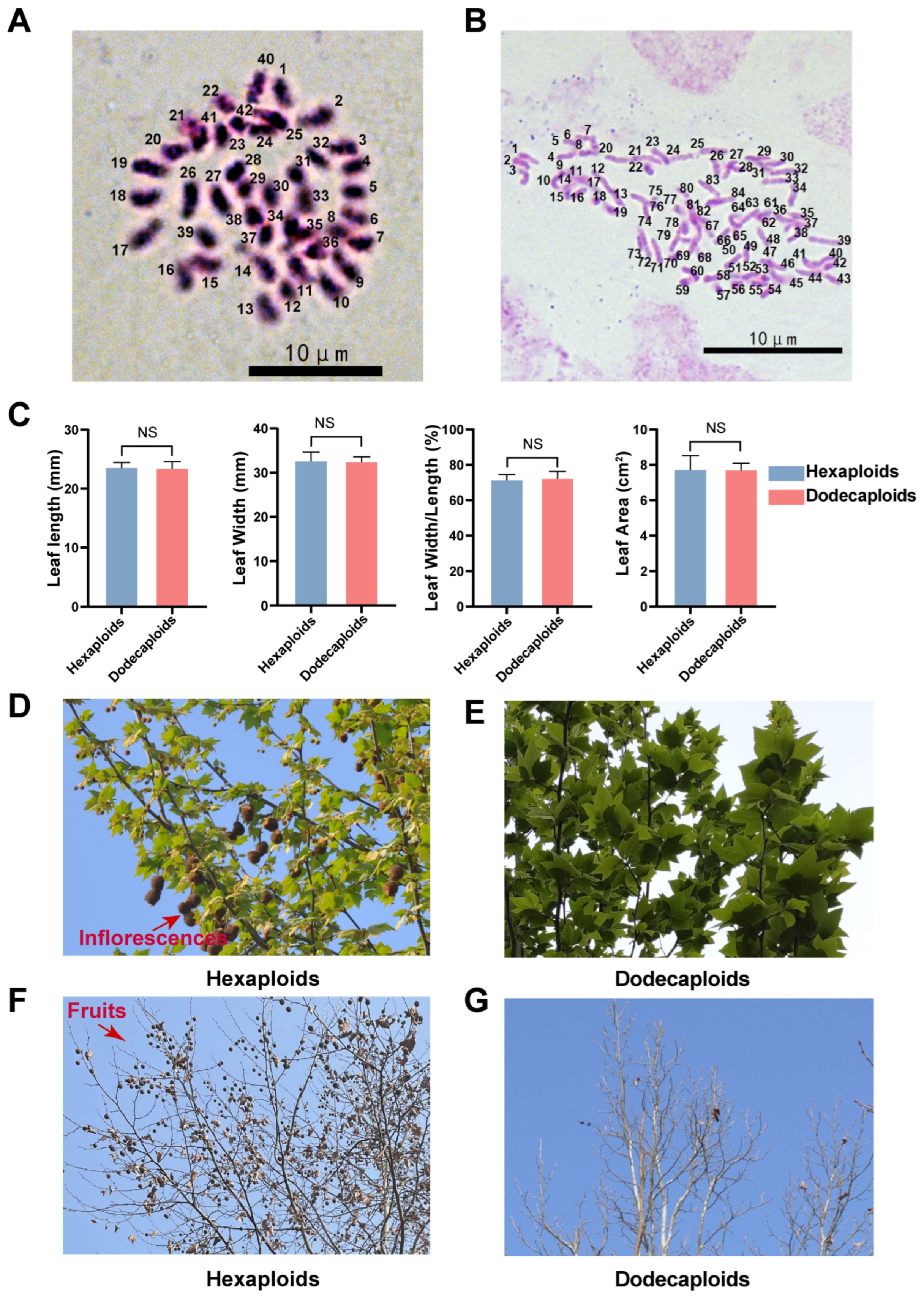
2.1. Dodecaploid Plants Exhibited a Distinct Morphology
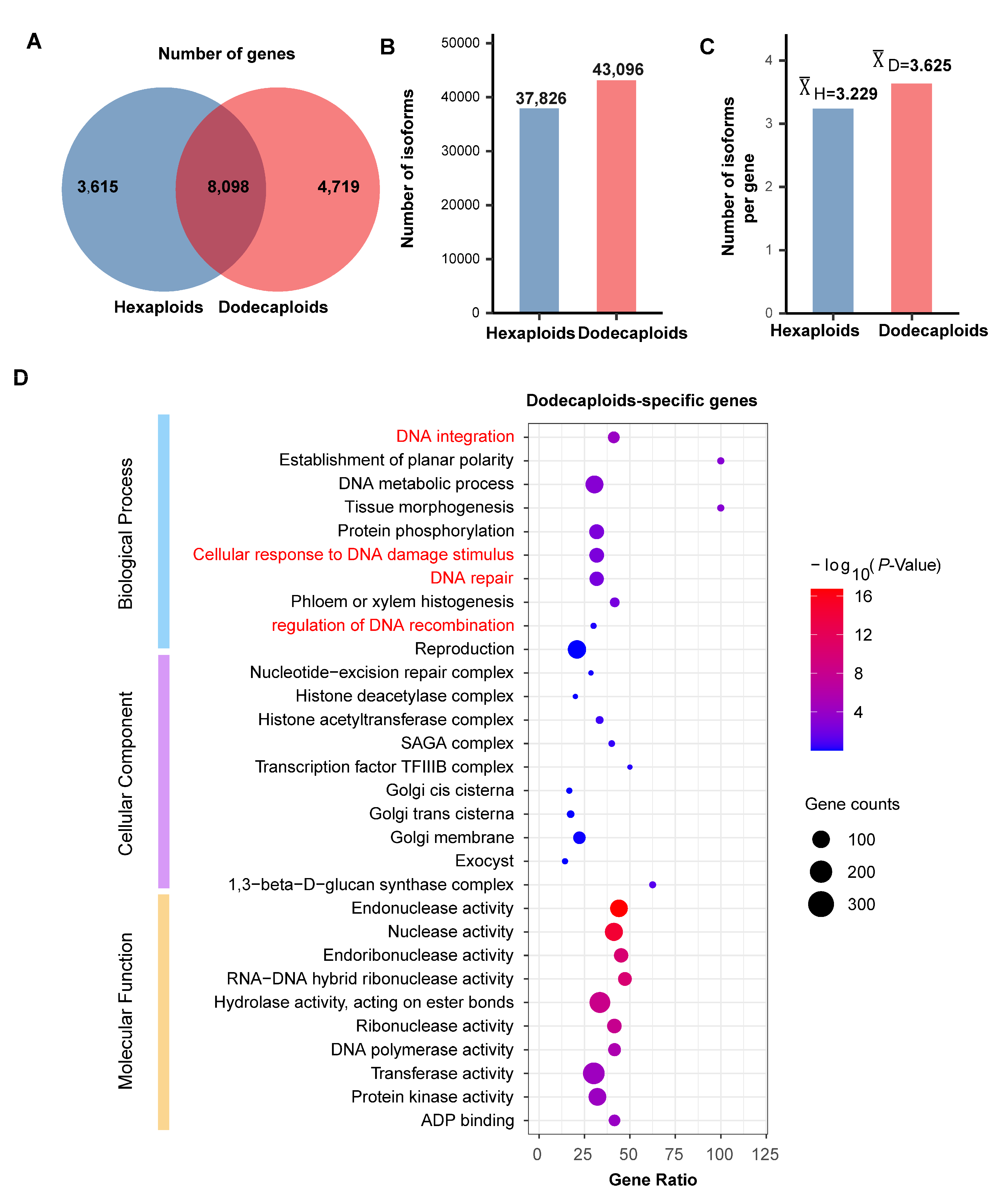
2.2. Dodecaploids Produce More Genes and Isoforms in P. × acerifolia
2.3. Dodecaploid-Specifically Transcribed Genes Associated with Chromosome Repair in P. × acerifolia
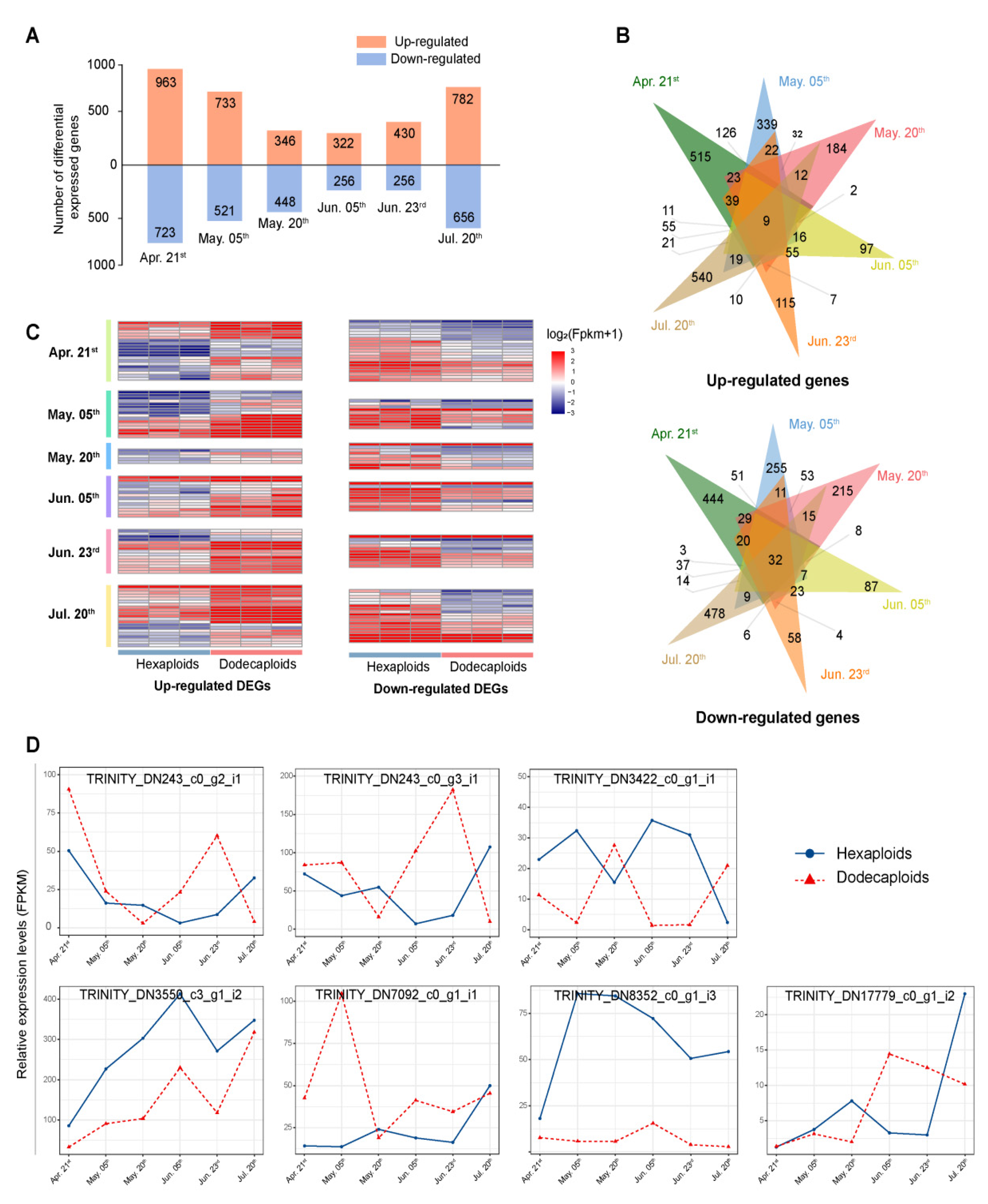
2.4. Differentially Expressed Genes between Hexaploids and Dodecaploids
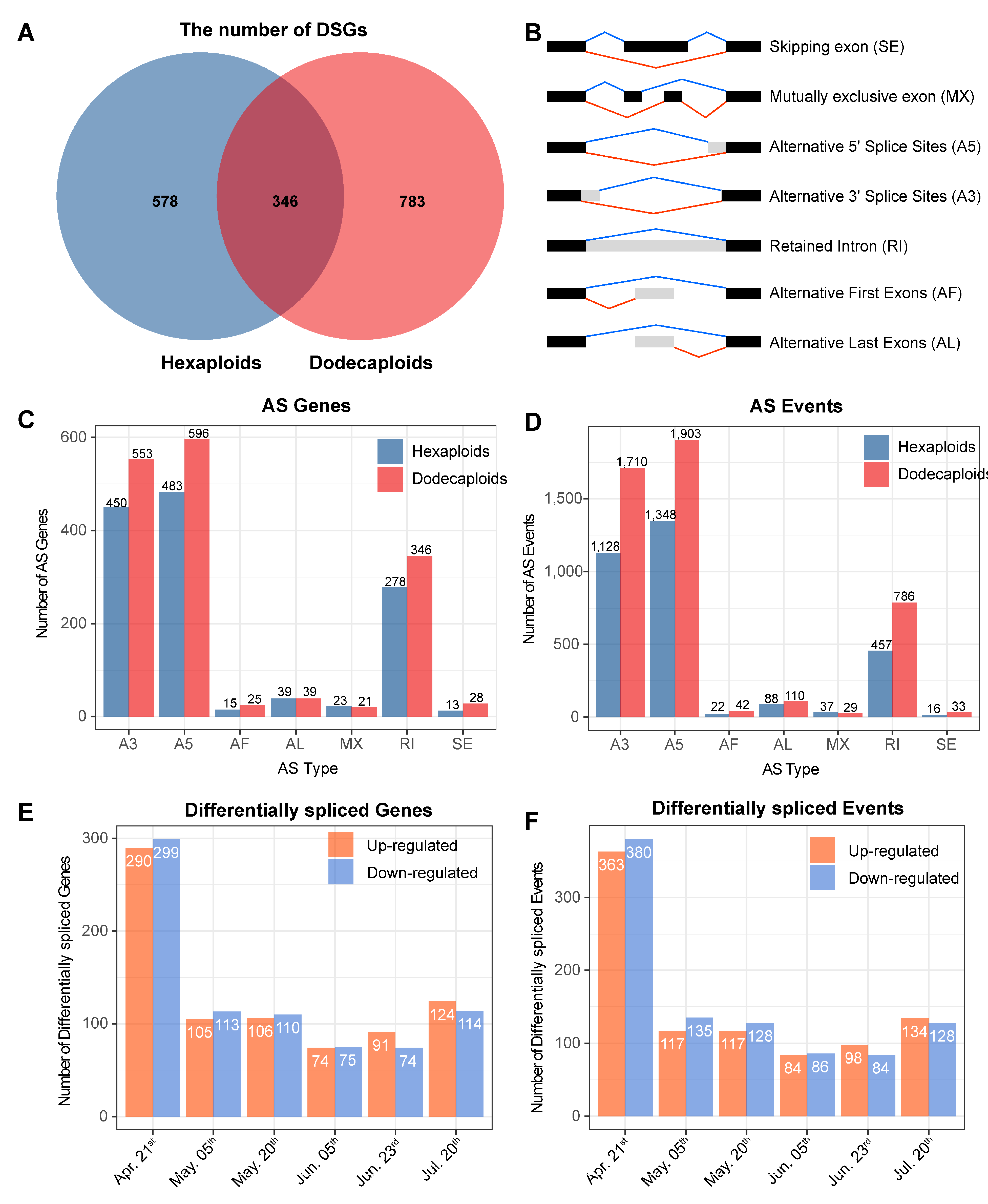
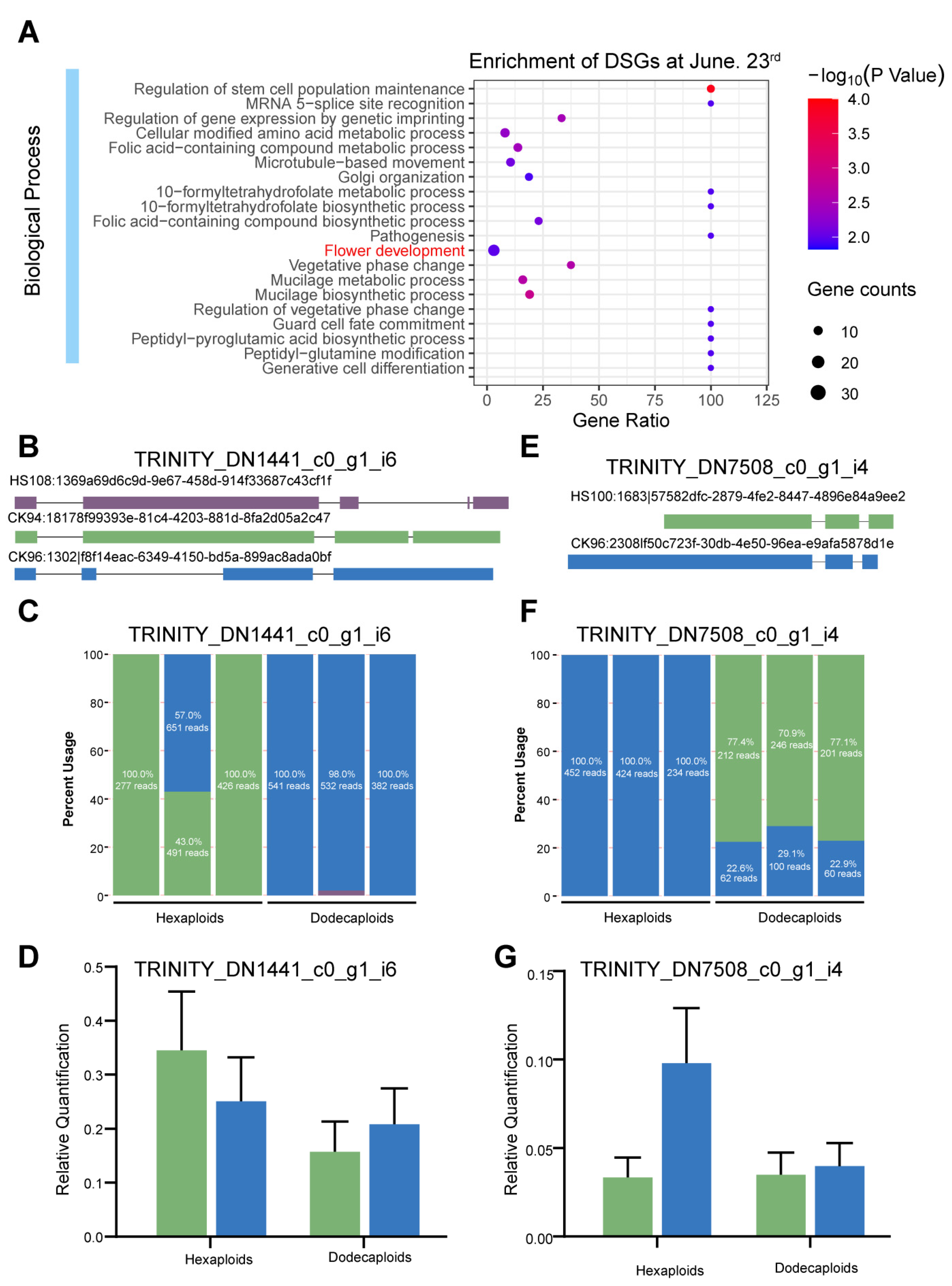
2.5. Differentially Alternative Splicing Events between Hexaploids and Dodecaploids in P. × acerifolia
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Ploidy Identification and Leaf Morphology Statistics
4.3. Sequencing of Transcriptomes
4.4. Assembling Contigs from NGS Reads
4.5. Collection of Full-Length Corrected Transcripts
4.6. Mapping Finding between Full-Length Reads and Contigs
4.7. Differential Gene Expression Analysis and Isoform Comparison between Hexaploids and Dodecaploids
4.8. Alternative Splicing Analysis
4.9. qRT-PCR Validation of AS Events
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henry, A.; Flood, M.G. The history of the London plane, Platanus acerifolia, with notes on the genus Platanus. In Proceedings of the Royal Irish Academy. Section B: Biological, Geological, and Chemical Science, 2nd ed.; JSTOR2: Dublin, Ireland, 1919; pp. 9–28. [Google Scholar]
- Ward, L.F. The paleontologic history of the genus Platanus. In Proceedings of the United States National Museum, 2nd ed.; Government Printing Office: Washington, DC, USA, 1888; pp. 39–42. [Google Scholar]
- Zonneveld, B.J. The DNA weights per nucleus (genome size) of more than 2350 species of the Flora of The Netherlands, of which 1370 are new to science, including the pattern of their DNA peaks. Forum Geobot. 2019, 8, 22. [Google Scholar]
- Bedi, Y.S. Chromosomal Conspectus and Evolutionary Status of lndian Commercial Timbers (Hardwoods). Cytologia 1991, 56, 665–672. [Google Scholar] [CrossRef]
- Oginuma, K.; Tobe, H. Karyomorphology and evolution in some Hamamelidaceae and Platanaceae (Hamamelididae; Hamamelidales). Bot. Mag. Shokubutsu-Gaku-Zasshi 1991, 104, 115–135. [Google Scholar] [CrossRef]
- Pauleit, S.; Jones, N.; Garcia-Martin, G.; Garcia-Valdecantos, J.L.; Rivière, L.M.; Vidal-Beaudet, L.; Bodson, M.; Randrup, T.B. Tree establishment practice in towns and cities--Results from a European survey. Urban For. Urban Green. 2002, 1, 83–96. [Google Scholar] [CrossRef]
- Gratani, L.; Vasheka, O.; Bigaran, F. Phenotypic plasticity of Platanus acerifolia (Platanaceae): Morphological and ana-tomical trait variations in response to different pollution levels in Rome. Mod. Phytol. 2020, 14, 55–63. [Google Scholar]
- Willis, K.J.; Petrokofsky, G. The natural capital of city trees. Science 2017, 356, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Carinanos, P.; Grilo, F.; Pinho, P.; Casares-Porcel, M.; Branquinho, C.; Acil, N.; Andreucci, M.B.; Anjos, A.; Bianco, P.M.; Brini, S.; et al. Estimation of the allergenic potential of urban trees and urban parks: Towards the healthy design of urban green spaces of the future. Int. J. Environ. Res. Public Health 2019, 16, 1357. [Google Scholar] [CrossRef] [PubMed]
- Sattler, M.C.; Carvalho, C.R.; Clarindo, W.R. The polyploidy and its key role in plant breeding. Planta 2016, 243, 281–296. [Google Scholar] [CrossRef]
- Farhadi, N.; Panahandeh, J.; Motallebi-Azar, A.; Mokhtarzadeh, S. Production of autotetraploid plants by in vitro chromosome engineering in Allium hirtifolium. Hortic. Plant J. 2022; in press. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, X.; Cheng, F. Plant polyploidy: Origin, evolution, and its influence on crop domestication. Hortic. Plant J. 2019, 5, 231–239. [Google Scholar] [CrossRef]
- Gaeta, R.T.; Chris, P.J. Homoeologous recombination in allopolyploids: The polyploid ratchet. New Phytol. 2010, 186, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Soltis, P.S.; Soltis, D.E. Ancient WGD events as drivers of key innovations in angiosperms. Curr. Opin. Plant Biol. 2016, 30, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Soltis, D.E.; Visger, C.J.; Marchant, D.B.; Soltis, P.S. Polyploidy: Pitfalls and paths to a paradigm. Am. J. Bot. 2016, 103, 1146–1166. [Google Scholar] [CrossRef] [PubMed]
- Sankoff, D.; Zheng, C. Whole Genome Duplication in Plants: Implications for Evolutionary Analysis. Methods Mol. Biol. 2018, 1704, 291–315. [Google Scholar] [PubMed]
- Zhang, X.; Chen, K.; Wang, W.; Liu, G.; Yang, C.; Jiang, J. Differences in Leaf Morphology and Related Gene Expression between Diploid and Tetraploid Birch (Betula pendula). Int. J. Mol. Sci. 2022, 23, 12966. [Google Scholar] [CrossRef]
- Soltis, D.E.; Visger, C.J.; Soltis, P.S. The polyploidy revolution then…and now: Stebbins revisited. Am. J. Bot. 2014, 101, 1057–1078. [Google Scholar] [CrossRef]
- Liu, G.; Li, Z.; Bao, M. Colchicine-induced chromosome doubling in Platanus acerifolia and its effect on plant morphology. Euphytica 2007, 157, 145–154. [Google Scholar] [CrossRef]
- Soltis, D.E.; Soltis, P.S.; Tate, J.A. Advances in the study of polyploidy since plant speciation. New Phytol. 2004, 161, 173–191. [Google Scholar] [CrossRef]
- Van Hieu, P. Polyploid gene expression and regulation in polysomic polyploids. Am. J. Plant Sci. 2019, 10, 1409–1443. [Google Scholar] [CrossRef]
- Gonzalo, A. All Ways Lead to Rome-Meiotic Stabilization Can Take Many Routes in Nascent Polyploid Plants. Genes 2022, 13, 147. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Li, B.; Tan, X.; Zhu, C.; Wu, T.; Feng, S.; Yang, Q.; Shen, S.; Yu, T.; et al. Polyploidy events shaped the expansion of transcription factors in Cucurbitaceae and exploitation of genes for tendril development. Hortic. Plant J. 2022, 8, 562–574. [Google Scholar] [CrossRef]
- Reddy, A.S. Alternative splicing of pre-messenger RNAs in plants in the genomic era. Annu. Rev. Plant Biol. 2007, 58, 267–294. [Google Scholar] [CrossRef] [PubMed]
- Wachter, A.; Tunc-Ozdemir, M.; Grove, B.C.; Green, P.J.; Shintani, D.K.; Breaker, R.R. Riboswitch control of gene expression in plants by splicing and alternative 3’ end processing of mRNAs. Plant Cell 2007, 19, 3437–3450. [Google Scholar] [CrossRef]
- Barbazuk, W.B.; Fu, Y.; McGinnis, K.M. Genome-wide analyses of alternative splicing in plants: Opportunities and challenges. Genome Res. 2008, 18, 1381–1392. [Google Scholar] [CrossRef]
- Terashima, A.; Takumi, S. Allopolyploidization reduces alternative splicing efficiency for transcripts of the wheat DREB2 homolog, WDREB2. Genome 2009, 52, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, P.; Liang, F.; Ye, Z.; Li, J.; Shen, C.; Pei, L.; Wang, F.; Hu, J.; Tu, L.; et al. A global survey of alternative splicing in allopolyploid cotton: Landscape, complexity and regulation. New Phytol. 2018, 217, 163–178. [Google Scholar] [CrossRef]
- Zheng, J.; Wen, S.; Yu, Z.; Luo, K.; Rong, J.; Ding, M. Alternative Splicing during Fiber Development in G. hirsutum. Int. J. Mol. Sci. 2023, 24, 11812. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Moshgabadi, N.; Adams, K.L. Extensive changes to alternative splicing patterns following allopolyploidy in natural and resynthesized polyploids. Proc. Natl. Acad. Sci. USA 2011, 108, 16122–16127. [Google Scholar] [CrossRef]
- Seki, M.; Oka, M.; Xu, L.; Suzuki, A.; Suzuki, Y. Transcript Identification Through Long-Read Sequencing. Methods Mol. Biol. 2021, 2284, 531–541. [Google Scholar] [CrossRef]
- Guan, D.; Halstead, M.M.; Islas-Trejo, A.D.; Goszczynski, D.E.; Cheng, H.H.; Ross, P.J.; Zhou, H. Prediction of transcript isoforms in 19 chicken tissues by Oxford Nanopore long-read sequencing. Front. Genet. 2022, 13, 997460. [Google Scholar] [CrossRef]
- Zmienko, A.; Satyr, A. Nanopore Sequencing and its Application in Biology. Postep. Biochem. 2020, 66, 193–204. [Google Scholar] [CrossRef]
- Shu, Z.; Wang, L.; Wang, J.; Zhang, L.; Hou, X.; Yan, H.; Wang, L. Integrative Analysis of Nanopore and Illumina Sequencing Reveals Alternative Splicing Complexity in Pig Longissimus Dorsi Muscle. Front. Genet. 2022, 13, 877646. [Google Scholar] [CrossRef]
- Kirov, I.; Omarov, M.; Merkulov, P.; Dudnikov, M.; Gvaramiya, S.; Kolganova, E.; Komakhin, R.; Karlov, G.; Soloviev, A. Genomic and Transcriptomic Survey Provides New Insight into the Organization and Transposition Activity of Highly Expressed LTR Retrotransposons of Sunflower (Helianthus annuus L.). Int. J. Mol. Sci. 2020, 21, 9331. [Google Scholar] [CrossRef] [PubMed]
- Landis, J.B.; Kurti, A.; Lawhorn, A.J.; Litt, A.; McCarthy, E.W. Differential gene expression with an emphasis on floral organ size differences in natural and synthetic polyploids of Nicotiana tabacum (Solanaceae). Genes 2020, 11, 1097. [Google Scholar] [CrossRef]
- Li, M.; Hu, M.; Xiao, Y.; Wu, X.; Wang, J. The activation of gene expression and alternative splicing in the formation and evolution of allopolyploid Brassica napus. Hortic. Res. 2022, 9, uhab075. [Google Scholar] [CrossRef] [PubMed]
- Ning, G.; Cheng, X.; Luo, P.; Liang, F.; Wang, Z.; Yu, G.; Li, X.; Wang, D.; Bao, M. Hybrid sequencing and map finding (HySeMaFi): Optional strategies for extensively deciphering gene splicing and expression in organisms without reference genome. Sci. Rep. 2017, 7, 43793. [Google Scholar] [CrossRef]
- Li, X.; Ma, D.; Lu, S.X.; Hu, X.; Huang, R.; Liang, T.; Xu, T.; Tobin, E.M.; Liu, H. Blue Light- and Low Temperature-Regulated COR27 and COR28 Play Roles in the Arabidopsis Circadian Clock. Plant Cell 2016, 28, 2755–2769. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Cui, X.; Zhao, C.; Shi, L.; Zhang, G.; Sun, F.; Cao, X.; Yuan, L.; Xie, Q.; Xu, X. COR27 and COR28 encode nighttime repressors integrating Arabidopsis circadian clock and cold response. J. Integr. Plant Biol. 2017, 59, 78–85. [Google Scholar] [CrossRef]
- Sablowski, R.W.; Meyerowitz, E.M. A homolog of NO APICAL MERISTEM is an immediate target of the floral homeotic genes APETALA3/PISTILLATA. Cell 1998, 92, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Gan, S. AtNAP, a NAC family transcription factor, has an important role in leaf senescence. Plant J. 2006, 46, 601–612. [Google Scholar] [CrossRef]
- Gregis, V.; Sessa, A.; Dorca-Fornell, C.; Kater, M.M. The Arabidopsis floral meristem identity genes AP1, AGL24 and SVP directly repress class B and C floral homeotic genes. Plant J. 2009, 60, 626–637. [Google Scholar] [CrossRef]
- Hassidim, M.; Harir, Y.; Yakir, E.; Kron, I.; Green, R.M. Over-expression of CONSTANS-LIKE 5 can induce flowering in short-day grown Arabidopsis. Planta 2009, 230, 481–491. [Google Scholar] [CrossRef]
- Lu, S.X.; Knowles, S.M.; Andronis, C.; Ong, M.S.; Tobin, E.M. CIRCADIAN CLOCK ASSOCIATED1 and LATE ELONGATED HYPOCOTYL function synergistically in the circadian clock of Arabidopsis. Plant Physiol. 2009, 150, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.A.; Farre, E.M.; Thomashow, M.F. Circadian clock-associated 1 and late elongated hypocotyl regulate expression of the C-repeat binding factor (CBF) pathway in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 7241–7246. [Google Scholar] [CrossRef]
- Holtan, H.E.; Bandong, S.; Marion, C.M.; Adam, L.; Tiwari, S.; Shen, Y.; Maloof, J.N.; Maszle, D.R.; Ohto, M.A.; Preuss, S.; et al. BBX32, an Arabidopsis B-Box protein, functions in light signaling by suppressing HY5-regulated gene expression and interacting with STH2/BBX21. Plant Physiol. 2011, 156, 2109–2123. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, C.; Yang, H.; Jiao, Y. Cytokinin pathway mediates APETALA1 function in the establishment of determinate floral meristems in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, 6840–6845. [Google Scholar] [CrossRef]
- Ravindran, N.; Ramachandran, H.; Job, N.; Yadav, A.; Vaishak, K.P.; Datta, S. B-box protein BBX32 integrates light and brassinosteroid signals to inhibit cotyledon opening. Plant Physiol. 2021, 187, 446–461. [Google Scholar] [CrossRef]
- Sitaraman, J.; Bui, M.; Liu, Z. LEUNIG_HOMOLOG and LEUNIG perform partially redundant functions during Arabidopsis embryo and floral development. Plant Physiol. 2008, 147, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, M.; Babolin, N.; Viana, V.E.; de Oliveira, A.C.; Gugi, B.; Caporali, E.; Herrera-Ubaldo, H.; Martinez-Estrada, E.; Driouich, A.; de Folter, S.; et al. The Genetic Control of SEEDSTICK and LEUNIG-HOMOLOG in Seed and Fruit Development: New Insights into Cell Wall Control. Plants 2022, 11, 3146. [Google Scholar] [CrossRef]
- Long, J.A.; Ohno, C.; Smith, Z.R.; Meyerowitz, E.M. TOPLESS regulates apical embryonic fate in Arabidopsis. Science 2006, 312, 1520–1523. [Google Scholar] [CrossRef]
- Mao, Y.; Pavangadkar, K.A.; Thomashow, M.F.; Triezenberg, S.J. Physical and functional interactions of Arabidopsis ADA2 transcriptional coactivator proteins with the acetyltransferase GCN5 and with the cold-induced transcription factor CBF1. Biochim. Biophys. Acta 2006, 1759, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feng, S.; Chen, F.; Chen, H.; Wang, J.; McCall, C.; Xiong, Y.; Deng, X.W. Arabidopsis DDB1-CUL4 ASSOCIATED FACTOR1 forms a nuclear E3 ubiquitin ligase with DDB1 and CUL4 that is involved in multiple plant developmental processes. Plant Cell 2008, 20, 1437–1455. [Google Scholar] [CrossRef]
- Eriksson, S.; Stransfeld, L.; Adamski, N.M.; Breuninger, H.; Lenhard, M. KLUH/CYP78A5-dependent growth signaling coordinates floral organ growth in Arabidopsis. Curr. Biol. 2010, 20, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Aki, S.; Nakai, H.; Aoyama, T.; Oka, A.; Tsuge, T. AtSAP130/AtSF3b-3 function is required for reproduction in Arabidopsis thaliana. Plant Cell Physiol. 2011, 52, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Kim, J.; Hwang, H.J.; Kim, S.; Park, C.; Kim, S.Y.; Lee, I. The FRIGIDA complex activates transcription of FLC, a strong flowering repressor in Arabidopsis, by recruiting chromatin modification factors. Plant Cell 2011, 23, 289–303. [Google Scholar] [CrossRef]
- Amasino, R.M. Vernalization and flowering time. Curr. Opin. Biotechnol. 2005, 16, 154–158. [Google Scholar] [CrossRef]
- Grasser, M.; Kane, C.M.; Merkle, T.; Melzer, M.; Emmersen, J.; Grasser, K.D. Transcript elongation factor TFIIS is involved in Arabidopsis seed dormancy. J. Mol. Biol. 2009, 386, 598–611. [Google Scholar] [CrossRef]
- Yu, T.S.; Lue, W.L.; Wang, S.M.; Chen, J. Mutation of Arabidopsis plastid phosphoglucose isomerase affects leaf starch synthesis and floral initiation. Plant Physiol. 2000, 123, 319–326. [Google Scholar] [CrossRef]
- Ding, L.; Kim, S.Y.; Michaels, S.D. FLOWERING LOCUS C EXPRESSOR family proteins regulate FLOWERING LOCUS C expression in both winter-annual and rapid-cycling Arabidopsis. Plant Physiol. 2013, 163, 243–252. [Google Scholar] [CrossRef]
- Renny-Byfield, S.; Wendel, J.F. Doubling down on genomes: Polyploidy and crop plants. Am. J. Bot. 2014, 101, 1711–1725. [Google Scholar] [CrossRef]
- Soltis, D.E.; Segovia Salcedo, M.C.; Jordon Thaden, I.; Majure, L.; Miles, N.M.; Mavrodiev, E.V.; Mei, W.; Cortez, M.B.; Soltis, P.S.; Gitzendanner, M.A. Are polyploids really evolutionary dead-ends (again)? A critical reappraisal of Mayrose et al. (2011). New Phytol. 2014, 202, 1105–1117. [Google Scholar] [CrossRef]
- Tsukaya, H. Does ploidy level directly control cell size? Counterevidence from Arabidopsis genetics. PLoS ONE 2013, 8, e83729. [Google Scholar] [CrossRef]
- Kellogg, E.A. Has the connection between polyploidy and diversification actually been tested? Curr. Opin. Plant Biol. 2016, 30, 25–32. [Google Scholar] [CrossRef]
- Chansler, M.T.; Ferguson, C.J.; Fehlberg, S.D.; Prather, L.A. The role of polyploidy in shaping morphological diversity in natural populations of Phlox amabilis. Am. J. Bot. 2016, 103, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, Y.; Hasegawa, J.; Fujikura, U.; Hoshino, R.; Matsunaga, S.; Tsukaya, H. The coordination of ploidy and cell size differs between cell layers in leaves. Development 2016, 143, 1120–1125. [Google Scholar] [CrossRef]
- Stebbins, G.L. Variation and evolution in plants. In Variation and Evolution in Plants; Columbia University Press: New York, NY, USA, 1950. [Google Scholar]
- Grant, V. Plant speciation. In Plant Speciation; Columbia University Press: New York, NY, USA, 1981; pp. 489–540. [Google Scholar]
- Grant, V. Chromosome number patterns in primitive angiosperms. Bot. Gaz. 1982, 143, 390–394. [Google Scholar] [CrossRef]
- Gaut, B.S.; Wright, S.I.; Rizzon, C.; Dvorak, J.; Anderson, L.K. Recombination: An underappreciated factor in the evolution of plant genomes. Nat. Rev. Genet. 2007, 8, 77–84. [Google Scholar] [CrossRef]
- Tayale, A.; Parisod, C. Natural pathways to polyploidy in plants and consequences for genome reorganization. Cytogenet. Genome Res. 2013, 140, 79–96. [Google Scholar] [CrossRef]
- Mandakova, T.; Lysak, M.A. Post-polyploid diploidization and diversification through dysploid changes. Curr. Opin. Plant Biol. 2018, 42, 55–65. [Google Scholar] [CrossRef]
- Li, Z.; McKibben, M.; Finch, G.S.; Blischak, P.D.; Sutherland, B.L.; Barker, M.S. Patterns and Processes of Diploidization in Land Plants. Annu. Rev. Plant Biol. 2021, 72, 387–410. [Google Scholar] [CrossRef]
- Wang, Y.; Zuo, L.; Wei, T.; Zhang, Y.; Zhang, Y.; Ming, R.; Bachar, D.; Xiao, W.; Madiha, K.; Chen, C.; et al. CHH methylation of genes associated with fatty acid and jasmonate biosynthesis contributes to cold tolerance in autotetraploids of Poncirus trifoliata. J. Integr. Plant Biol. 2022, 64, 2327–2343. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, H.; Jin, J.; Ma, X.; Li, K. Physiological and Transcriptome Analysis on Diploid and Polyploid Populus ussuriensis Kom. under Salt Stress. Int. J. Mol. Sci. 2022, 23, 7529. [Google Scholar] [CrossRef] [PubMed]
- Syed, N.H.; Kalyna, M.; Marquez, Y.; Barta, A.; Brown, J.W. Alternative splicing in plants--coming of age. Trends Plant Sci. 2012, 17, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the alternative splicing landscape in plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Jiao, Z.; Xu, M.; Wang, Y.; Li, R.; Cui, X.; Gu, L.; Zhang, S. Landscape and fruit developmental regulation of alternative splicing in tomato by genome-wide analysis. Hortic. Plant J. 2016, 2, 338–350. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, W.; Zhao, X.; Song, A.; Guo, K.; Liu, Z.; Zhang, L. Transcriptomic analysis of differentially expressed genes and alternative splicing events associated with crassulacean acid metabolism in orchids. Hortic. Plant J. 2019, 5, 268–280. [Google Scholar] [CrossRef]
- Andres, F.; Coupland, G. The genetic basis of flowering responses to seasonal cues. Nat. Rev. Genet. 2012, 13, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Damerval, C.; Becker, A. Genetics of flower development in Ranunculales-a new, basal eudicot model order for studying flower evolution. New Phytol. 2017, 216, 361–366. [Google Scholar] [CrossRef]
- Michaels, S.D.; Bezerra, I.C.; Amasino, R.M. FRIGIDA-related genes are required for the winter-annual habit in Arabidopsis. Proc. Natl. Acad. Sci. USA 2004, 101, 3281–3285. [Google Scholar] [CrossRef]
- Schmitz, R.J.; Hong, L.; Michaels, S.; Amasino, R.M. FRIGIDA-ESSENTIAL 1 interacts genetically with FRIGIDA and FRIGIDA-LIKE 1 to promote the winter-annual habit of Arabidopsis thaliana. Development 2005, 132, 5471–5478. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Sack, L.; Buckley, T.N. The Developmental Basis of Stomatal Density and Flux. Plant Physiol. 2016, 171, 2358–2363. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT--the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef]
- Langmead, B.; Wilks, C.; Antonescu, V.; Charles, R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 2019, 35, 421–432. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Malcolm, P. Heatmaps: Flexible Heatmaps for Functional Genomics and Sequence Features; R Package Version 1.6.0.; 2018. Available online: https://bioconductor.org/packages/3.17/bioc/html/heatmaps.html (accessed on 23 July 2023).
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Research 2020, 9, 304. [Google Scholar] [CrossRef]
- Trincado, J.L.; Entizne, J.C.; Hysenaj, G.; Singh, B.; Skalic, M.; Elliott, D.J.; Eyras, E. SUPPA2: Fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018, 19, 40. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, H.; Shao, C.; Cai, F.; Zhang, J.; Bao, M. PaMYB82 from Platanus acerifolia regulates trichome development in transgenic Arabidopsis. Plant Sci. 2019, 287, 110177. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, X.; Chen, X.; Li, Y.; Li, Y.; Wang, F.; Zhang, J.; Ning, G.; Bao, M. The Abundant and Unique Transcripts and Alternative Splicing of the Artificially Autododecaploid London Plane (Platanus × acerifolia). Int. J. Mol. Sci. 2023, 24, 14486. https://doi.org/10.3390/ijms241914486
Yan X, Chen X, Li Y, Li Y, Wang F, Zhang J, Ning G, Bao M. The Abundant and Unique Transcripts and Alternative Splicing of the Artificially Autododecaploid London Plane (Platanus × acerifolia). International Journal of Molecular Sciences. 2023; 24(19):14486. https://doi.org/10.3390/ijms241914486
Chicago/Turabian StyleYan, Xu, Xiyan Chen, Yangyang Li, Yuhan Li, Fei Wang, Jiaqi Zhang, Guogui Ning, and Manzhu Bao. 2023. "The Abundant and Unique Transcripts and Alternative Splicing of the Artificially Autododecaploid London Plane (Platanus × acerifolia)" International Journal of Molecular Sciences 24, no. 19: 14486. https://doi.org/10.3390/ijms241914486