
The Illustration of Altered Glucose Dependency in Drug-Resistant Cancer Cells


Abstract
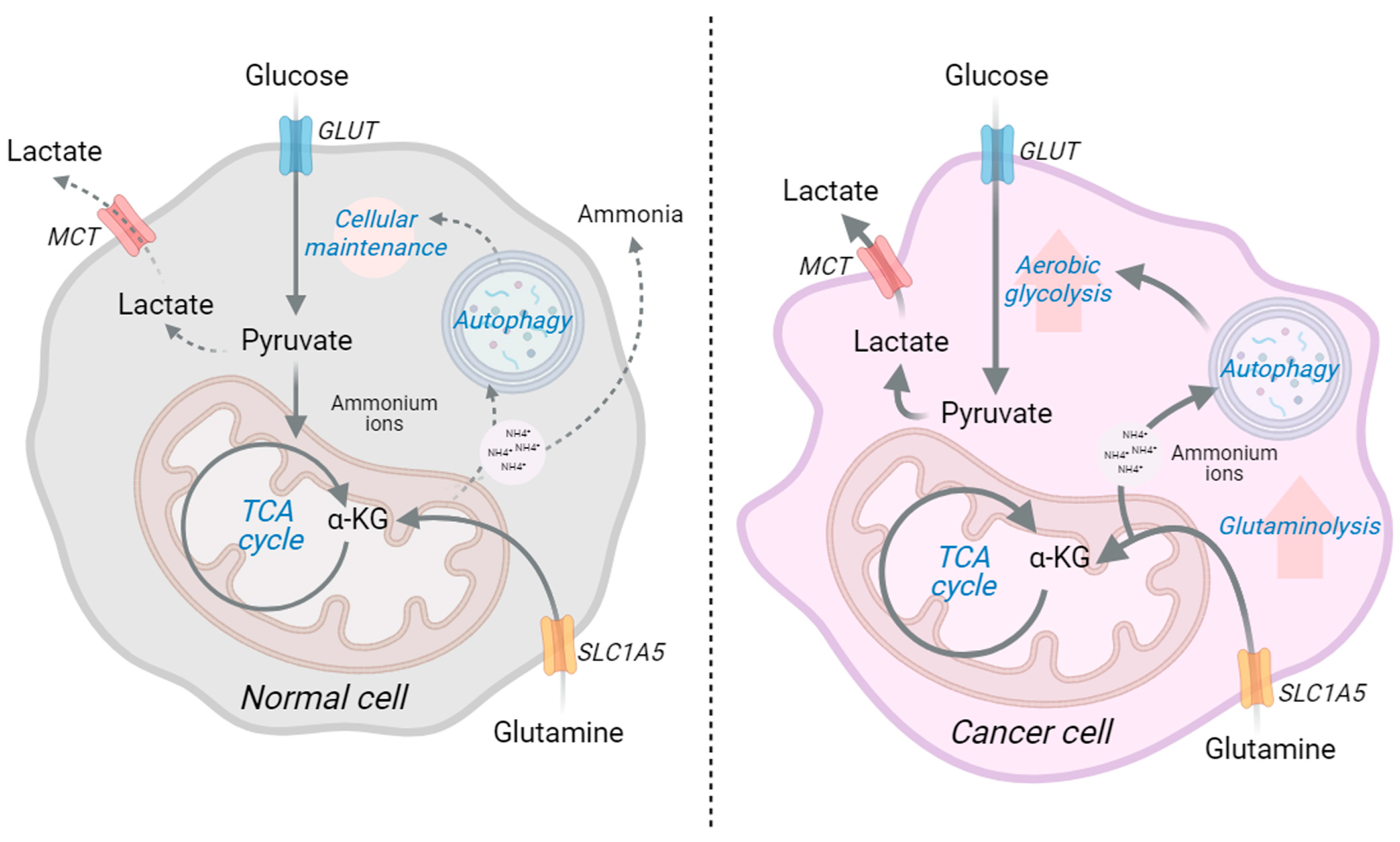
:1. Introduction
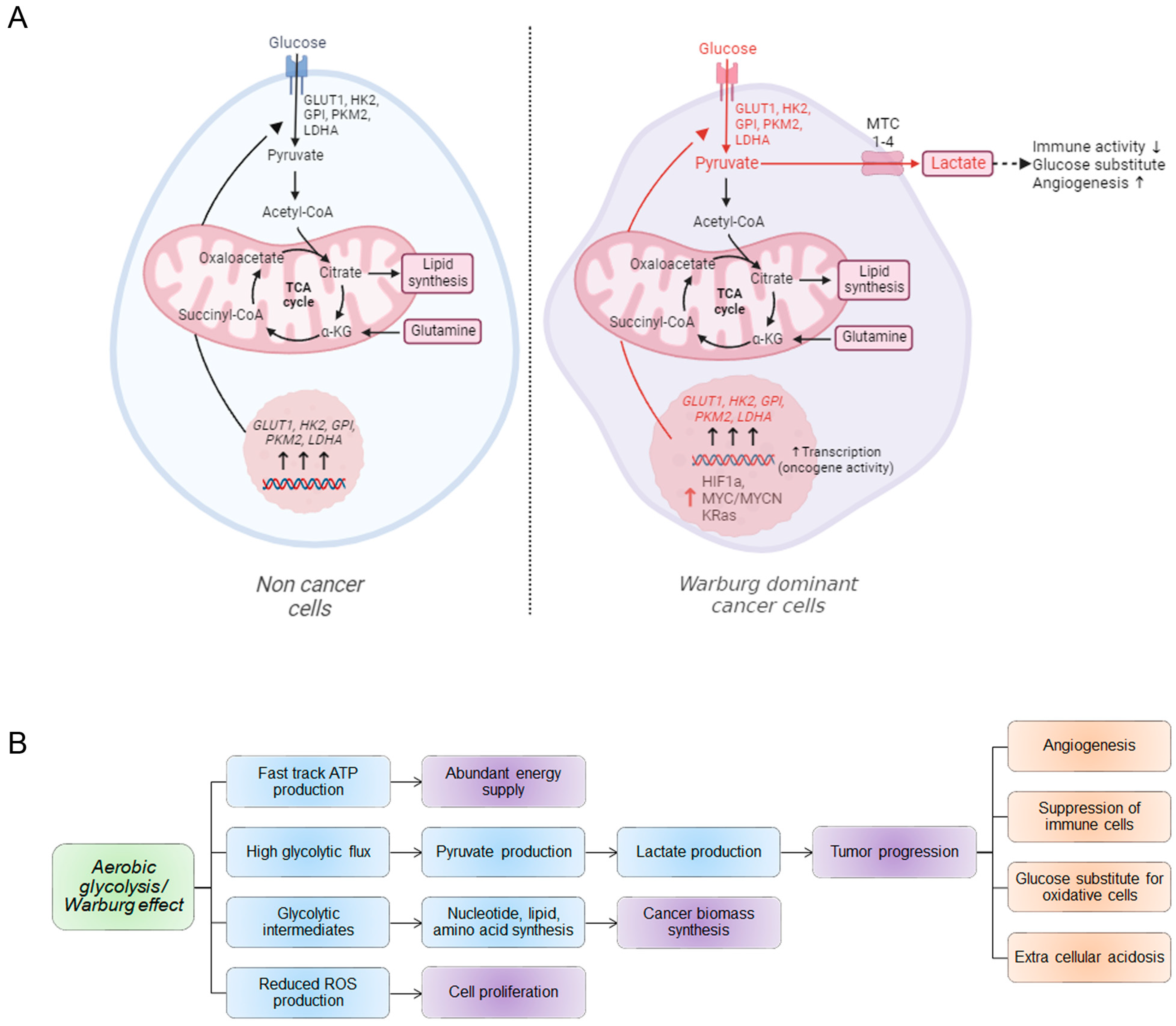
2. Warburg Effect on Cancer Cell Proliferation
3. Transcriptional Regulation of Warburg Genes
4. Activation of Pentose Phosphate Pathway in Warburg Dominant Cancer Cells
5. Flux Controlling between Warburg and Pentose Phosphate Pathway
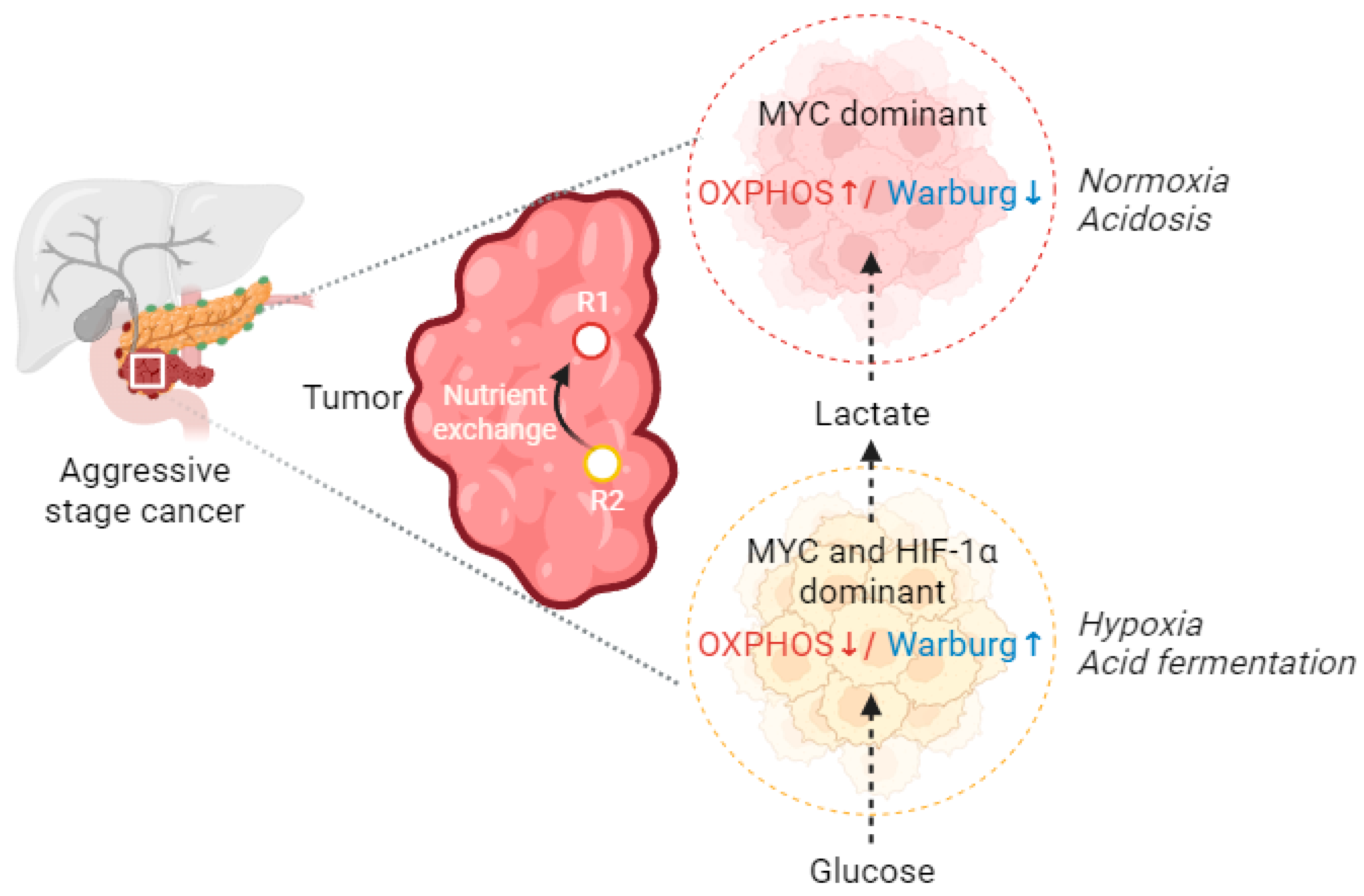
6. Metabolic Profiling of the Warburg Dominant Cells
7. Reversing the Warburg Pathway
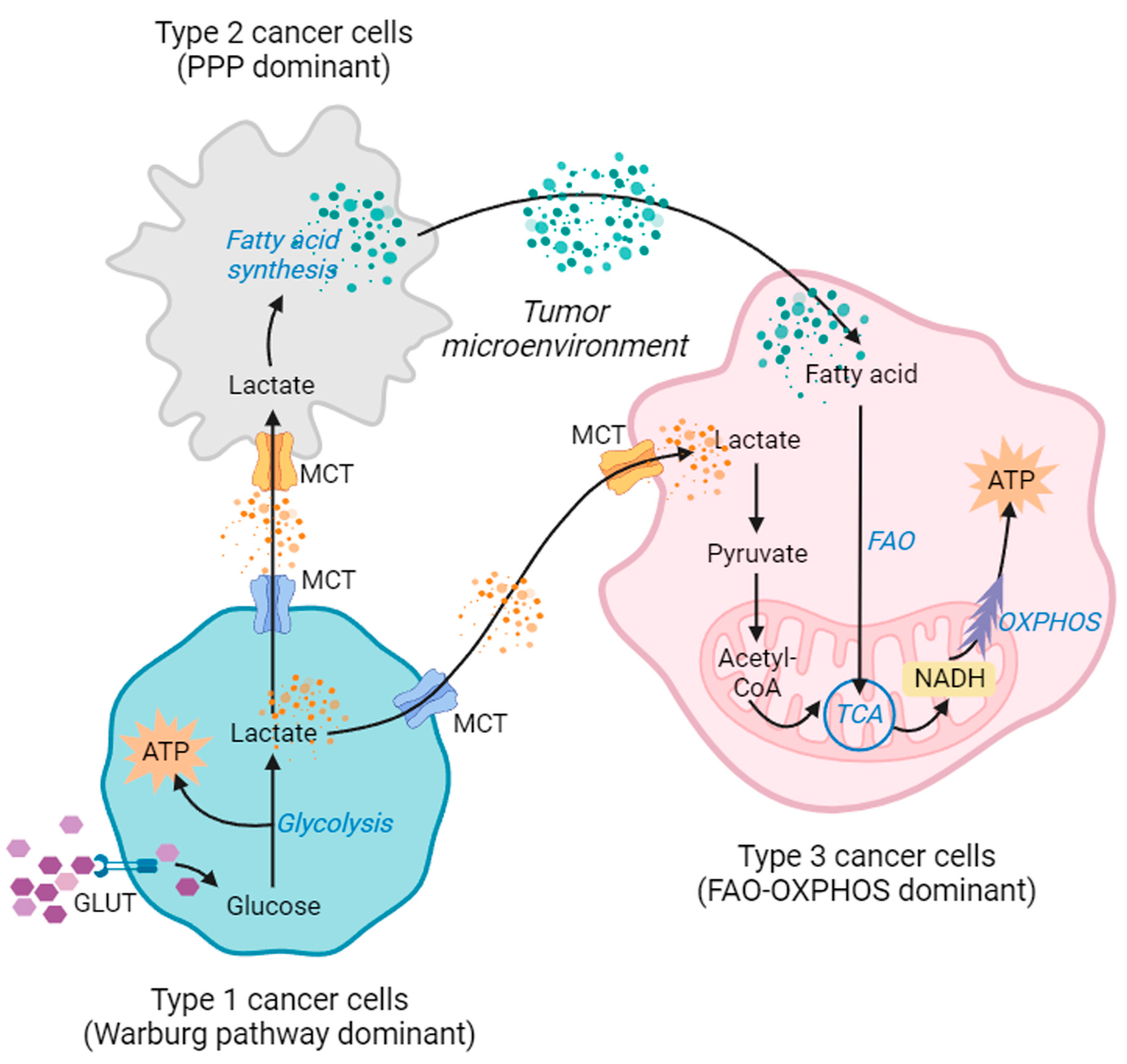
8. Metabolic Plasticity, a Switch between Warburg and Other Metabolic Pathways
8.1. Warburg–Oxphos Switch
8.2. Fatty Acid Synthesis and Oxidation
8.3. Glutaminolysis
9. Glucose Metabolism-Based Drug Development
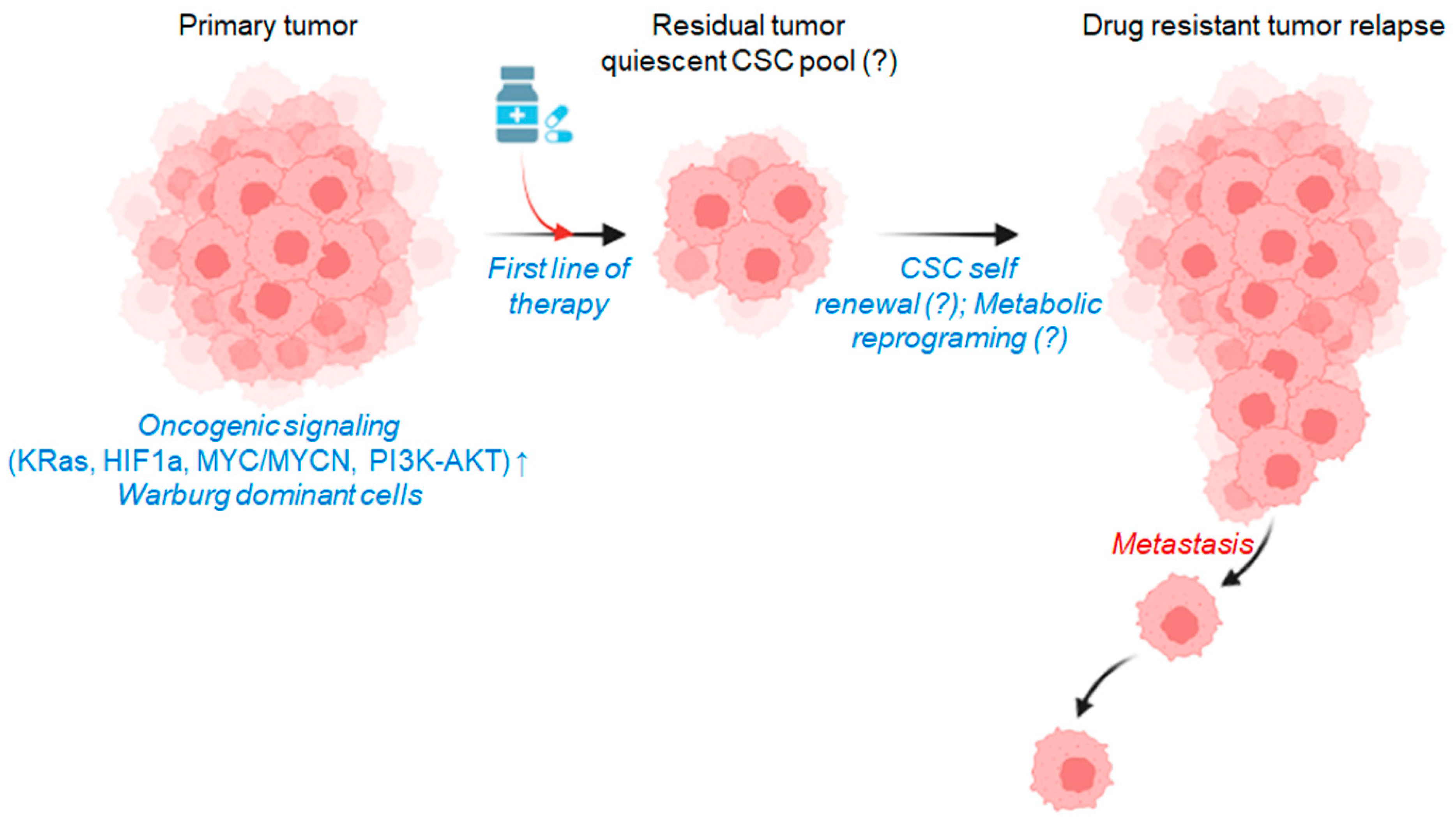
10. Glucose Metabolic Reprogramming and Therapy Resistance
11. Metabolic Alterations in Cancer Cells at Low Glycolytic State
12. Therapeutic Strategies
13. Future Directions
14. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Lee, P.Y.; Yeoh, Y.; Low, T.Y. A Recent Update on Small-Molecule Kinase Inhibitors for Targeted Cancer Therapy and Their Therapeutic Insights from Mass Spectrometry-Based Proteomic Analysis. FEBS J. 2023, 290, 2845–2864. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-Targeted Cancer Therapies: Progress, Challenges and Future Directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer Chemotherapy and beyond: Current Status, Drug Candidates, Associated Risks and Progress in Targeted Therapeutics. Genes Dis. 2023, 10, 1367–1401. [Google Scholar] [CrossRef]
- Baba, A.I.; Câtoi, C. Comparative Oncology; The Publishing House of the Romanian Academy: Bucharest, Romania, 2007; ISBN 973-27-1457-3. [Google Scholar]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug Resistance in Cancer: An Overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Phan, L.M.; Yeung, S.-C.J.; Lee, M.-H. Cancer Metabolic Reprogramming: Importance, Main Features, and Potentials for Precise Targeted Anti-Cancer Therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Cazzaniga, M.; Bonanni, B. Relationship Between Metabolic Reprogramming and Mitochondrial Activity in Cancer Cells. Understanding the Anticancer Effect of Metformin and Its Clinical Implications. Anticancer Res. 2015, 35, 5789–5796. [Google Scholar]
- Xu, Y.; He, L.; Fu, Q.; Hu, J. Metabolic Reprogramming in the Tumor Microenvironment with Immunocytes and Immune Checkpoints. Front. Oncol. 2021, 11, 759015. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J. Metabolic Reprogramming: The Emerging Concept and Associated Therapeutic Strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef]
- Bishayee, K.; Nazim, U.M.; Kumar, V.; Kang, J.; Kim, J.; Huh, S.-O.; Sadra, A. Reversing the HDAC-Inhibitor Mediated Metabolic Escape in MYCN-Amplified Neuroblastoma. Biomed. Pharmacother. 2022, 150, 113032. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Torrini, C.; Zhao, J.; Bianchetti, E.; Mela, A.; Humala, N.; Mahajan, A.; et al. HDAC Inhibitors Elicit Metabolic Reprogramming by Targeting Super-Enhancers in Glioblastoma Models. J. Clin. Investig. 2020, 130, 3699–3716. [Google Scholar] [CrossRef]
- Yadav, U.P.; Singh, T.; Kumar, P.; Sharma, P.; Kaur, H.; Sharma, S.; Singh, S.; Kumar, S.; Mehta, K. Metabolic Adaptations in Cancer Stem Cells. Front. Oncol. 2020, 10, 1010. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer Stem Cells (CSCs): Metabolic Strategies for Their Identification and Eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef]
- Deshmukh, A.; Deshpande, K.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Cancer Stem Cell Metabolism: A Potential Target for Cancer Therapy. Mol. Cancer 2016, 15, 69. [Google Scholar] [CrossRef]
- Coller, H.A. The Paradox of Metabolism in Quiescent Stem Cells. FEBS Lett. 2019, 593, 2817–2839. [Google Scholar] [CrossRef]
- Wallner, E.I.; Wada, J.; Tramonti, G.; Lin, S.; Kanwar, Y.S. Status of Glucose Transporters in the Mammalian Kidney and Renal Development. Ren. Fail. 2001, 23, 301–310. [Google Scholar] [CrossRef]
- Ancey, P.-B.; Contat, C.; Meylan, E. Glucose Transporters in Cancer—From Tumor Cells to the Tumor Microenvironment. FEBS J. 2018, 285, 2926–2943. [Google Scholar] [CrossRef] [PubMed]
- Adekola, K.; Rosen, S.T.; Shanmugam, M. Glucose Transporters in Cancer Metabolism. Curr. Opin. Oncol. 2012, 24, 650–654. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Verduzco, D.; Rauch, C.; Muddathir, A.K.; Adil, H.H.B.; Elhassan, G.O.; Ibrahim, M.E.; David Polo Orozco, J.; Cardone, R.A.; Reshkin, S.J.; et al. Glycolysis, Tumor Metabolism, Cancer Growth and Dissemination. A New PH-Based Etiopathogenic Perspective and Therapeutic Approach to an Old Cancer Question. Oncoscience 2014, 1, 777–802. [Google Scholar] [CrossRef]
- Liu, C.; Jin, Y.; Fan, Z. The Mechanism of Warburg Effect-Induced Chemoresistance in Cancer. Front. Oncol. 2021, 11, 698023. [Google Scholar] [CrossRef]
- Lin, X.; Xiao, Z.; Chen, T.; Liang, S.H.; Guo, H. Glucose Metabolism on Tumor Plasticity, Diagnosis, and Treatment. Front. Oncol. 2020, 10, 317. [Google Scholar] [CrossRef]
- de la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg Effect: Essential Part of Metabolic Reprogramming and Central Contributor to Cancer Progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. The Warburg Effect: Historical Dogma Versus Current Rationale. Adv. Exp. Med. Biol. 2021, 1269, 169–177. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg Effect: Historical Dogma versus Current Understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef]
- Danhier, P.; Bański, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer Metabolism in Space and Time: Beyond the Warburg Effect. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 556–572. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Cantley, L.C. Cancer’s Fuel Choice: New Flavors for a Picky Eater. Mol. Cell 2015, 60, 514–523. [Google Scholar] [CrossRef]
- Raskov, H.; Gaggar, S.; Tajik, A.; Orhan, A.; Gögenur, I. Metabolic Switch in Cancer—Survival of the Fittest. Eur. J. Cancer 2023, 180, 30–51. [Google Scholar] [CrossRef]
- Dey, P.; Kimmelman, A.C.; DePinho, R.A. Metabolic Codependencies in the Tumor Microenvironment. Cancer Discov. 2021, 11, 1067–1081. [Google Scholar] [CrossRef]
- Rohani, N.; Hao, L.; Alexis, M.S.; Joughin, B.A.; Krismer, K.; Moufarrej, M.N.; Soltis, A.R.; Lauffenburger, D.A.; Yaffe, M.B.; Burge, C.B.; et al. Acidification of Tumor at Stromal Boundaries Drives Transcriptome Alterations Associated with Aggressive Phenotypes. Cancer Res. 2019, 79, 1952–1966. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-H.; Peng, W.-B.; Zhang, P.; Yang, X.-P.; Zhou, Q. Lactate in the Tumour Microenvironment: From Immune Modulation to Therapy. eBioMedicine 2021, 73, 103627. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Jiang, Y.; Cao, H.; Yang, J.; Shu, Y.; Feng, H.; Yang, X.; Sun, X.; Shao, M. Lactate: A Regulator of Immune Microenvironment and a Clinical Prognosis Indicator in Colorectal Cancer. Front. Immunol. 2022, 13, 876195. [Google Scholar] [CrossRef]
- Yin, T.-T.; Huang, M.-X.; Wang, F.; Jiang, Y.-H.; Long, J.; Li, L.; Cao, J. Lactate Score Predicts Survival, Immune Cell Infiltration and Response to Immunotherapy in Breast Cancer. Front. Genet. 2022, 13, 943849. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Xie, Z.; Zhang, N.; Zhang, Y.; Xiao, D.; Liu, S.; Zhuang, W.; Li, L.; Tao, Y. Signaling Pathways in Cancer Metabolism: Mechanisms and Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 196. [Google Scholar] [CrossRef]
- Lennicke, C.; Cochemé, H.M. Redox Metabolism: ROS as Specific Molecular Regulators of Cell Signaling and Function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine Reliance in Cell Metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.-Q.; Lin, J.-F.; Tian, T.; Xie, D.; Xu, R.-H. NADPH Homeostasis in Cancer: Functions, Mechanisms and Therapeutic Implications. Signal Transduct. Target. Ther. 2020, 5, 231. [Google Scholar] [CrossRef]
- Mullarky, E.; Cantley, L.C. Diverting Glycolysis to Combat Oxidative Stress. In Innovative Medicine: Basic Research and Development; Nakao, K., Minato, N., Uemoto, S., Eds.; Springer: Tokyo, Japan, 2015; pp. 3–23. ISBN 978-4-431-55650-3. [Google Scholar]
- Ge, T.; Gu, X.; Jia, R.; Ge, S.; Chai, P.; Zhuang, A.; Fan, X. Crosstalk between Metabolic Reprogramming and Epigenetics in Cancer: Updates on Mechanisms and Therapeutic Opportunities. Cancer Commun. 2022, 42, 1049–1082. [Google Scholar] [CrossRef]
- Hsieh, W.-C.; Sutter, B.M.; Ruess, H.; Barnes, S.D.; Malladi, V.S.; Tu, B.P. Glucose Starvation Induces a Switch in the Histone Acetylome for Activation of Gluconeogenic and Fat Metabolism Genes. Mol. Cell 2022, 82, 60–74.e5. [Google Scholar] [CrossRef] [PubMed]
- Jo, C.; Park, S.; Oh, S.; Choi, J.; Kim, E.-K.; Youn, H.-D.; Cho, E.-J. Histone Acylation Marks Respond to Metabolic Perturbations and Enable Cellular Adaptation. Exp. Mol. Med. 2020, 52, 2005–2019. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liang, Y.; Kang, L.; Liu, Y.; Gao, S.; Chen, S.; Li, Y.; You, W.; Dong, Q.; Hong, T.; et al. Transcriptional Regulation of the Warburg Effect in Cancer by SIX1. Cancer Cell 2018, 33, 368–385.e7. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef] [PubMed]
- Cargill, K.R.; Stewart, C.A.; Park, E.M.; Ramkumar, K.; Gay, C.M.; Cardnell, R.J.; Wang, Q.; Diao, L.; Shen, L.; Fan, Y.-H.; et al. Targeting MYC-Enhanced Glycolysis for the Treatment of Small Cell Lung Cancer. Cancer Metab. 2021, 9, 33. [Google Scholar] [CrossRef]
- Dalton, K.M.; Lochmann, T.L.; Floros, K.V.; Calbert, M.L.; Kurupi, R.; Stein, G.T.; McClanaghan, J.; Murchie, E.; Egan, R.K.; Greninger, P.; et al. Catastrophic ATP Loss Underlies a Metabolic Combination Therapy Tailored for MYCN-Amplified Neuroblastoma. Proc. Natl. Acad. Sci. USA 2021, 118, e2009620118. [Google Scholar] [CrossRef]
- Pupo, E.; Avanzato, D.; Middonti, E.; Bussolino, F.; Lanzetti, L. KRAS-Driven Metabolic Rewiring Reveals Novel Actionable Targets in Cancer. Front. Oncol. 2019, 9, 848. [Google Scholar] [CrossRef]
- Jiang, B. Aerobic Glycolysis and High Level of Lactate in Cancer Metabolism and Microenvironment. Genes Dis. 2017, 4, 25–27. [Google Scholar] [CrossRef]
- Wagner, W.; Ciszewski, W.M.; Kania, K.D. L- and D-Lactate Enhance DNA Repair and Modulate the Resistance of Cervical Carcinoma Cells to Anticancer Drugs via Histone Deacetylase Inhibition and Hydroxycarboxylic Acid Receptor 1 Activation. Cell Commun. Signal. 2015, 13, 36. [Google Scholar] [CrossRef]
- Rodríguez-Enríquez, S.; Marín-Hernández, Á.; Gallardo-Pérez, J.C.; Pacheco-Velázquez, S.C.; Belmont-Díaz, J.A.; Robledo-Cadena, D.X.; Vargas-Navarro, J.L.; Corona de la Peña, N.A.; Saavedra, E.; Moreno-Sánchez, R. Transcriptional Regulation of Energy Metabolism in Cancer Cells. Cells 2019, 8, 1225. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 Mediates Metabolic Responses to Intratumoral Hypoxia and Oncogenic Mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef]
- Pawlus, M.R.; Wang, L.; Hu, C.-J. STAT3 and HIF1α Cooperatively Activate HIF1 Target Genes in MDA-MB-231 and RCC4 Cells. Oncogene 2014, 33, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Felipe-Abrio, B.; Verdugo-Sivianes, E.M.; Carnero, A. C-MYB- and PGC1a-Dependent Metabolic Switch Induced by MYBBP1A Loss in Renal Cancer. Mol. Oncol. 2019, 13, 1519–1533. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α Mediates Mitochondrial Biogenesis and Oxidative Phosphorylation in Cancer Cells to Promote Metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS Oncogenes: Weaving a Tumorigenic Web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liang, Z.; Zhou, C.; Zeng, Z.; Wang, F.; Hu, T.; He, X.; Wu, X.; Wu, X.; Lan, P. Mutant KRAS Triggers Functional Reprogramming of Tumor-Associated Macrophages in Colorectal Cancer. Signal Transduct. Target. Ther. 2021, 6, 144. [Google Scholar] [CrossRef]
- Song, J.; Sun, H.; Zhang, S.; Shan, C. The Multiple Roles of Glucose-6-Phosphate Dehydrogenase in Tumorigenesis and Cancer Chemoresistance. Life 2022, 12, 271. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Ahmed, S.B.M.; Elliott, R.L.; Benoit, A.; Alqahtani, S.S.; Ibrahim, M.E.; Bashir, A.H.H.; Alhoufie, S.T.S.; Elhassan, G.O.; Wales, C.C.; et al. The Pentose Phosphate Pathway Dynamics in Cancer and Its Dependency on Intracellular PH. Metabolites 2020, 10, 285. [Google Scholar] [CrossRef]
- Fornalewicz, K.; Wieczorek, A.; Węgrzyn, G.; Łyżeń, R. Silencing of the Pentose Phosphate Pathway Genes Influences DNA Replication in Human Fibroblasts. Gene 2017, 635, 33–38. [Google Scholar] [CrossRef]
- Jin, L.; Zhou, Y. Crucial Role of the Pentose Phosphate Pathway in Malignant Tumors. Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [CrossRef]
- Wang, J.; Duan, Z.; Nugent, Z.; Zou, J.X.; Borowsky, A.D.; Zhang, Y.; Tepper, C.G.; Li, J.J.; Fiehn, O.; Xu, J.; et al. Reprogramming Metabolism by Histone Methyltransferase NSD2 Drives Endocrine Resistance via Coordinated Activation of Pentose Phosphate Pathway Enzymes. Cancer Lett. 2016, 378, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.S.; Cha, Y.H.; Kim, H.S.; Kim, N.H.; Yook, J.I. The Pentose Phosphate Pathway as a Potential Target for Cancer Therapy. Biomol. Ther. 2018, 26, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Kowalik, M.A.; Columbano, A.; Perra, A. Emerging Role of the Pentose Phosphate Pathway in Hepatocellular Carcinoma. Front. Oncol. 2017, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.-H.; Li, S.; Du, Z.-X.; Liu, C.; Liu, B.-Q.; Li, C.; Zong, Z.-H.; Wang, H.-Q. BAG3 Elevation Inhibits Cell Proliferation via Direct Interaction with G6PD in Hepatocellular Carcinomas. Oncotarget 2016, 7, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Benito, A.; Polat, I.H.; Noé, V.; Ciudad, C.J.; Marin, S.; Cascante, M. Glucose-6-Phosphate Dehydrogenase and Transketolase Modulate Breast Cancer Cell Metabolic Reprogramming and Correlate with Poor Patient Outcome. Oncotarget 2017, 8, 106693–106706. [Google Scholar] [CrossRef]
- Yang, H.-C.; Wu, Y.-H.; Yen, W.-C.; Liu, H.-Y.; Hwang, T.-L.; Stern, A.; Chiu, D.T.-Y. The Redox Role of G6PD in Cell Growth, Cell Death, and Cancer. Cells 2019, 8, 1055. [Google Scholar] [CrossRef]
- Ma, H.; Zhang, J.; Zhou, L.; Wen, S.; Tang, H.-Y.; Jiang, B.; Zhang, F.; Suleman, M.; Sun, D.; Chen, A.; et al. C-Src Promotes Tumorigenesis and Tumor Progression by Activating PFKFB3. Cell Rep. 2020, 30, 4235–4249.e6. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.C.; Pohlmann, P.R.; Clarke, R.; Sengupta, S. Treatment against Glucose-Dependent Cancers through Metabolic PFKFB3 Targeting of Glycolytic Flux. Cancer Metastasis Rev. 2022, 41, 447–458. [Google Scholar] [CrossRef]
- Yamamoto, T.; Takano, N.; Ishiwata, K.; Ohmura, M.; Nagahata, Y.; Matsuura, T.; Kamata, A.; Sakamoto, K.; Nakanishi, T.; Kubo, A.; et al. Reduced Methylation of PFKFB3 in Cancer Cells Shunts Glucose towards the Pentose Phosphate Pathway. Nat. Commun. 2014, 5, 3480. [Google Scholar] [CrossRef]
- Yi, M.; Ban, Y.; Tan, Y.; Xiong, W.; Li, G.; Xiang, B. 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3 and 4: A Pair of Valves for Fine-Tuning of Glucose Metabolism in Human Cancer. Mol. Metab. 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.-M.; Krüger, A.; Tauqeer Alam, M.; et al. The Return of Metabolism: Biochemistry and Physiology of the Pentose Phosphate Pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef]
- Hurbain, J.; Thommen, Q.; Anquez, F.; Pfeuty, B. Quantitative Modeling of Pentose Phosphate Pathway Response to Oxidative Stress Reveals a Cooperative Regulatory Strategy. iScience 2022, 25, 104681. [Google Scholar] [CrossRef] [PubMed]
- Britt, E.C.; Lika, J.; Giese, M.A.; Schoen, T.J.; Seim, G.L.; Huang, Z.; Lee, P.Y.; Huttenlocher, A.; Fan, J. Switching to the Cyclic Pentose Phosphate Pathway Powers the Oxidative Burst in Activated Neutrophils. Nat. Metab. 2022, 4, 389–403. [Google Scholar] [CrossRef]
- Tran, Q.; Lee, H.; Park, J.; Kim, S.-H.; Park, J. Targeting Cancer Metabolism—Revisiting the Warburg Effects. Toxicol. Res. 2016, 32, 177–193. [Google Scholar] [CrossRef]
- Ying, M.; You, D.; Zhu, X.; Cai, L.; Zeng, S.; Hu, X. Lactate and Glutamine Support NADPH Generation in Cancer Cells under Glucose Deprived Conditions. Redox Biol. 2021, 46, 102065. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting Cancer Metabolism in the Era of Precision Oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, J.; Yadav, D.K.; Bai, X.; Liang, T. Glucose Metabolism: The Metabolic Signature of Tumor Associated Macrophage. Front. Immunol. 2021, 12, 702580. [Google Scholar] [CrossRef]
- Pal, S.; Sharma, A.; Mathew, S.P.; Jaganathan, B.G. Targeting Cancer-Specific Metabolic Pathways for Developing Novel Cancer Therapeutics. Front. Immunol. 2022, 13, 955476. [Google Scholar] [CrossRef]
- Lee, H.; Woo, S.M.; Jang, H.; Kang, M.; Kim, S.-Y. Cancer Depends on Fatty Acids for ATP Production: A Possible Link between Cancer and Obesity. Semin. Cancer Biol. 2022, 86, 347–357. [Google Scholar] [CrossRef]
- Nava, G.M.; Madrigal Perez, L.A. Metabolic Profile of the Warburg Effect as a Tool for Molecular Prognosis and Diagnosis of Cancer. Expert Rev. Mol. Diagn. 2022, 22, 439–447. [Google Scholar] [CrossRef]
- Whillier, S.; Garcia, B.; Chapman, B.E.; Kuchel, P.W.; Raftos, J.E. Glutamine and α-Ketoglutarate as Glutamate Sources for Glutathione Synthesis in Human Erythrocytes. FEBS J. 2011, 278, 3152–3163. [Google Scholar] [CrossRef]
- Li, F.; Simon, M.C. Cancer Cells Don’t Live Alone: Metabolic Communication within Tumor Microenvironments. Dev. Cell 2020, 54, 183–195. [Google Scholar] [CrossRef]
- Matula, Z.; Mikala, G.; Lukácsi, S.; Matkó, J.; Kovács, T.; Monostori, É.; Uher, F.; Vályi-Nagy, I. Stromal Cells Serve Drug Resistance for Multiple Myeloma via Mitochondrial Transfer: A Study on Primary Myeloma and Stromal Cells. Cancers 2021, 13, 3461. [Google Scholar] [CrossRef]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Goetzman, E.S.; Prochownik, E.V. The Role for Myc in Coordinating Glycolysis, Oxidative Phosphorylation, Glutaminolysis, and Fatty Acid Metabolism in Normal and Neoplastic Tissues. Front. Endocrinol. 2018, 9, 129. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Nong, S.; Han, X.; Xiang, Y.; Qian, Y.; Wei, Y.; Zhang, T.; Tian, K.; Shen, K.; Yang, J.; Ma, X. Metabolic Reprogramming in Cancer: Mechanisms and Therapeutics. MedComm 2023, 4, e218. [Google Scholar] [CrossRef]
- Lu, J.; Tan, M.; Cai, Q. The Warburg Effect in Tumor Progression: Mitochondrial Oxidative Metabolism as an Anti-Metastasis Mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef]
- Wang, X.; Shen, X.; Yan, Y.; Li, H. Pyruvate Dehydrogenase Kinases (PDKs): An Overview toward Clinical Applications. Biosci. Rep. 2021, 41, BSR20204402. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Gatter, K.C.; Harris, A.L. Pyruvate Dehydrogenase and Pyruvate Dehydrogenase Kinase Expression in Non Small Cell Lung Cancer and Tumor-Associated Stroma. Neoplasia 2005, 7, 1–6. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S.; Geschwind, J.-F.H. Tumor Glycolysis as a Target for Cancer Therapy: Progress and Prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W.; Nagaraja, N.V.; Hutson, A.D. Efficacy of Dichloroacetate as a Lactate-Lowering Drug. J. Clin. Pharmacol. 2003, 43, 683–691. [Google Scholar] [CrossRef]
- Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Salehi, B.; Berkay Yılmaz, Y.; Antika, G.; Boyunegmez Tumer, T.; Fawzi Mahomoodally, M.; Lobine, D.; Akram, M.; Riaz, M.; Capanoglu, E.; Sharopov, F.; et al. Insights on the Use of α-Lipoic Acid for Therapeutic Purposes. Biomolecules 2019, 9, 356. [Google Scholar] [CrossRef]
- Jin, P.; Jiang, J.; Zhou, L.; Huang, Z.; Nice, E.C.; Huang, C.; Fu, L. Mitochondrial Adaptation in Cancer Drug Resistance: Prevalence, Mechanisms, and Management. J. Hematol. Oncol. 2022, 15, 97. [Google Scholar] [CrossRef]
- Bahar, E.; Han, S.-Y.; Kim, J.-Y.; Yoon, H. Chemotherapy Resistance: Role of Mitochondrial and Autophagic Components. Cancers 2022, 14, 1462. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Gopal, Y.N.V.; Rizos, H.; Chen, G.; Deng, W.; Frederick, D.T.; Cooper, Z.A.; Scolyer, R.A.; Pupo, G.; Komurov, K.; Sehgal, V.; et al. Inhibition of MTORC1/2 Overcomes Resistance to MAPK Pathway Inhibitors Mediated by PGC1α and Oxidative Phosphorylation in Melanoma. Cancer Res. 2014, 74, 7037–7047. [Google Scholar] [CrossRef]
- Cassim, S.; Vučetić, M.; Ždralević, M.; Pouyssegur, J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers 2020, 12, 1119. [Google Scholar] [CrossRef]
- Antonio, M.J.; Zhang, C.; Le, A. Different Tumor Microenvironments Lead to Different Metabolic Phenotypes. Adv. Exp. Med. Biol. 2021, 1311, 137–147. [Google Scholar] [CrossRef]
- Amemiya, T.; Yamaguchi, T. Oscillations and Dynamic Symbiosis in Cellular Metabolism in Cancer. Front. Oncol. 2022, 12, 783908. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, X.; Wang, L.; Hong, X.; Yang, J. Metabolic Reprogramming and Crosstalk of Cancer-Related Fibroblasts and Immune Cells in the Tumor Microenvironment. Front. Endocrinol. 2022, 13, 988295. [Google Scholar] [CrossRef]
- Wanandi, S.I.; Ningsih, S.S.; Asikin, H.; Hosea, R.; Neolaka, G.M.G. Metabolic Interplay between Tumour Cells and Cancer-Associated Fibroblasts (CAFs) under Hypoxia versus Normoxia. Malays. J. Med. Sci. 2018, 25, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Daverio, Z.; Balcerczyk, A.; Rautureau, G.J.P.; Panthu, B. How Warburg-Associated Lactic Acidosis Rewires Cancer Cell Energy Metabolism to Resist Glucose Deprivation. Cancers 2023, 15, 1417. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating Cancer Metabolic Plasticity by Coupling Gene Regulation with Metabolic Pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef]
- Dupuy, F.; Tabariès, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E.; et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef]
- Yu, L.; Lu, M.; Jia, D.; Ma, J.; Ben-Jacob, E.; Levine, H.; Kaipparettu, B.A.; Onuchic, J.N. Modeling the Genetic Regulation of Cancer Metabolism: Interplay between Glycolysis and Oxidative Phosphorylation. Cancer Res. 2017, 77, 1564–1574. [Google Scholar] [CrossRef]
- Dang, C. V Rethinking the Warburg Effect with Myc Micromanaging Glutamine Metabolism. Cancer Res. 2010, 70, 859–862. [Google Scholar] [CrossRef]
- Caino, M.C.; Ghosh, J.C.; Chae, Y.C.; Vaira, V.; Rivadeneira, D.B.; Faversani, A.; Rampini, P.; Kossenkov, A.V.; Aird, K.M.; Zhang, R.; et al. PI3K Therapy Reprograms Mitochondrial Trafficking to Fuel Tumor Cell Invasion. Proc. Natl. Acad. Sci. USA 2015, 112, 8638–8643. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Allen, A.E.; Ramesh, V.; Dai, Z.; Singleton, K.R.; Guo, Z.; Liu, J.O.; Wood, K.C.; Locasale, J.W. Evolved Resistance to Partial GAPDH Inhibition Results in Loss of the Warburg Effect and in a Different State of Glycolysis. J. Biol. Chem. 2020, 295, 111–124. [Google Scholar] [CrossRef]
- Zaidi, N.; Lupien, L.; Kuemmerle, N.B.; Kinlaw, W.B.; Swinnen, J.V.; Smans, K. Lipogenesis and Lipolysis: The Pathways Exploited by the Cancer Cells to Acquire Fatty Acids. Prog. Lipid Res. 2013, 52, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Simeone, P.; Tacconi, S.; Longo, S.; Lanuti, P.; Bravaccini, S.; Pirini, F.; Ravaioli, S.; Dini, L.; Giudetti, A.M. Expanding Roles of de Novo Lipogenesis in Breast Cancer. Int. J. Environ. Res. Public Health 2021, 18, 3575. [Google Scholar] [CrossRef] [PubMed]
- Fhu, C.W.; Ali, A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules 2020, 25, 3935. [Google Scholar] [CrossRef]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic Pathways Promoting Cancer Cell Survival and Growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wu, M. Regulation of the Pentose Phosphate Pathway in Cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef]
- Fu, J.; Yu, S.; Zhao, X.; Zhang, C.; Shen, L.; Liu, Y.; Yu, H. Inhibition of TIGAR Increases Exogenous P53 and Cisplatin Combination Sensitivity in Lung Cancer Cells by Regulating Glycolytic Flux. Int. J. Mol. Sci. 2022, 23, 16034. [Google Scholar] [CrossRef]
- Zhang, Z.; Deng, X.; Liu, Y.; Liu, Y.; Sun, L.; Chen, F. PKM2, Function and Expression and Regulation. Cell Biosci. 2019, 9, 52. [Google Scholar] [CrossRef]
- Toller-Kawahisa, J.E.; Hiroki, C.H.; Silva, C.M.d.S.; Nascimento, D.C.; Públio, G.A.; Martins, T.V.; Damasceno, L.E.A.; Veras, F.P.; Viacava, P.R.; Sukesada, F.Y.; et al. The Metabolic Function of Pyruvate Kinase M2 Regulates Reactive Oxygen Species Production and Microbial Killing by Neutrophils. Nat. Commun. 2023, 14, 4280. [Google Scholar] [CrossRef]
- Luan, W.; Wang, Y.; Chen, X.; Shi, Y.; Wang, J.; Zhang, J.; Qian, J.; Li, R.; Tao, T.; Wei, W.; et al. PKM2 Promotes Glucose Metabolism and Cell Growth in Gliomas through a Mechanism Involving a Let-7a/c-Myc/HnRNPA1 Feedback Loop. Oncotarget 2015, 6, 13006–13018. [Google Scholar] [CrossRef]
- Sugden, M.C.; Holness, M.J. The Pyruvate Carboxylase-Pyruvate Dehydrogenase Axis in Islet Pyruvate Metabolism: Going Round in Circles? Islets 2011, 3, 302–319. [Google Scholar] [CrossRef]
- Icard, P.; Coquerel, A.; Wu, Z.; Gligorov, J.; Fuks, D.; Fournel, L.; Lincet, H.; Simula, L. Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update. Int. J. Mol. Sci. 2021, 22, 6587. [Google Scholar] [CrossRef] [PubMed]
- Kiesel, V.A.; Sheeley, M.P.; Coleman, M.F.; Cotul, E.K.; Donkin, S.S.; Hursting, S.D.; Wendt, M.K.; Teegarden, D. Pyruvate Carboxylase and Cancer Progression. Cancer Metab. 2021, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Oh, H.E.; Kim, H.; Lee, J.-H.; Lee, E.S.; Kim, Y.-S.; Choi, J.-W. Upregulation of Fatty Acid Transporters Is Associated with Tumor Progression in Non-Muscle-Invasive Bladder Cancer. Pathol. Oncol. Res. 2021, 27, 594705. [Google Scholar] [CrossRef] [PubMed]
- Gyamfi, J.; Kim, J.; Choi, J. Cancer as a Metabolic Disorder. Int. J. Mol. Sci. 2022, 23, 1155. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of Fatty Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a Target for Cancer Therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef]
- Wise, D.R.; Ward, P.S.; Shay, J.E.S.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia Promotes Isocitrate Dehydrogenase-Dependent Carboxylation of α-Ketoglutarate to Citrate to Support Cell Growth and Viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, H.; Gao, M.; Zhang, Y.; Zhang, L.; Huang, D.; Tu, K.; Xu, Q. The Regulation of Amino Acid Metabolism in Tumor Cell Death: From the Perspective of Physiological Functions. Apoptosis 2023, 28, 1304–1314. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive Glutamine Metabolism by IDH1 Mediates Lipogenesis under Hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef]
- Fan, J.; Kamphorst, J.J.; Rabinowitz, J.D.; Shlomi, T. Fatty Acid Labeling from Glutamine in Hypoxia Can Be Explained by Isotope Exchange without Net Reductive Isocitrate Dehydrogenase (IDH) Flux. J. Biol. Chem. 2013, 288, 31363–31369. [Google Scholar] [CrossRef]
- Dang, C.V. Links between Metabolism and Cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef]
- Liu, D.; Wang, Y.; Li, X.; Wang, Y.; Zhang, Z.; Wang, Z.; Zhang, X. Participation of Protein Metabolism in Cancer Progression and Its Potential Targeting for the Management of Cancer. Amino Acids 2023. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc Regulates a Transcriptional Program That Stimulates Mitochondrial Glutaminolysis and Leads to Glutamine Addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Samuels, Y.; Li, Q.; Krokowski, D.; Guan, B.-J.; Wang, C.; Jin, Z.; Dong, B.; Cao, B.; Feng, X.; et al. Oncogenic PIK3CA Mutations Reprogram Glutamine Metabolism in Colorectal Cancer. Nat. Commun. 2016, 7, 11971. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J.; Ito, D.; Rees, M.G.; Seashore-Ludlow, B.; Puyang, X.; Ramos, A.H.; Cheah, J.H.; Clemons, P.A.; Warmuth, M.; Zhu, P.; et al. NAMPT Is the Cellular Target of STF-31-like Small-Molecule Probes. ACS Chem. Biol. 2014, 9, 2247–2254. [Google Scholar] [CrossRef]
- Reckzeh, E.S.; Karageorgis, G.; Schwalfenberg, M.; Ceballos, J.; Nowacki, J.; Stroet, M.C.M.; Binici, A.; Knauer, L.; Brand, S.; Choidas, A.; et al. Inhibition of Glucose Transporters and Glutaminase Synergistically Impairs Tumor Cell Growth. Cell Chem. Biol. 2019, 26, 1214–1228.e25. [Google Scholar] [CrossRef]
- Wu, Q.; Ba-Alawi, W.; Deblois, G.; Cruickshank, J.; Duan, S.; Lima-Fernandes, E.; Haight, J.; Tonekaboni, S.A.M.; Fortier, A.-M.; Kuasne, H.; et al. GLUT1 Inhibition Blocks Growth of RB1-Positive Triple Negative Breast Cancer. Nat. Commun. 2020, 11, 4205. [Google Scholar] [CrossRef]
- Ciscato, F.; Ferrone, L.; Masgras, I.; Laquatra, C.; Rasola, A. Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. Int. J. Mol. Sci. 2021, 22, 4716. [Google Scholar] [CrossRef]
- Li, R.; Mei, S.; Ding, Q.; Wang, Q.; Yu, L.; Zi, F. A Pan-Cancer Analysis of the Role of Hexokinase II (HK2) in Human Tumors. Sci. Rep. 2022, 12, 18807. [Google Scholar] [CrossRef]
- Liu, Y.; Li, M.; Zhang, Y.; Wu, C.; Yang, K.; Gao, S.; Zheng, M.; Li, X.; Li, H.; Chen, L. Structure Based Discovery of Novel Hexokinase 2 Inhibitors. Bioorg. Chem. 2020, 96, 103609. [Google Scholar] [CrossRef]
- Xu, D.; Wang, Y.; Liu, X.; Zhou, K.; Wu, J.; Chen, J.; Chen, C.; Chen, L.; Zheng, J. Development and Clinical Validation of a Novel 9-Gene Prognostic Model Based on Multi-Omics in Pancreatic Adenocarcinoma. Pharmacol. Res. 2021, 164, 105370. [Google Scholar] [CrossRef]
- Suzuki, A.; Puri, S.; Leland, P.; Puri, A.; Moudgil, T.; Fox, B.A.; Puri, R.K.; Joshi, B.H. Subcellular Compartmentalization of PKM2 Identifies Anti-PKM2 Therapy Response in Vitro and in Vivo Mouse Model of Human Non-Small-Cell Lung Cancer. PLoS ONE 2019, 14, e0217131. [Google Scholar] [CrossRef]
- Hillis, A.L.; Lau, A.N.; Devoe, C.X.; Dayton, T.L.; Danai, L.V.; Di Vizio, D.; Vander Heiden, M.G. PKM2 Is Not Required for Pancreatic Ductal Adenocarcinoma. Cancer Metab. 2018, 6, 17. [Google Scholar] [CrossRef]
- Lau, A.N.; Israelsen, W.J.; Roper, J.; Sinnamon, M.J.; Georgeon, L.; Dayton, T.L.; Hillis, A.L.; Yilmaz, O.H.; Di Vizio, D.; Hung, K.E.; et al. PKM2 Is Not Required for Colon Cancer Initiated by APC Loss. Cancer Metab. 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, Q.; Zheng, S.; Liu, T.; Yang, L.; Han, X.; Lu, X. Shikonin Inhibits Tumor Growth of ESCC by Suppressing PKM2 Mediated Aerobic Glycolysis and STAT3 Phosphorylation. J. Cancer 2021, 12, 4830–4840. [Google Scholar] [CrossRef]
- Zang, F.; Rao, Y.; Zhu, X.; Wu, Z.; Jiang, H. Shikonin Suppresses NEAT1 and Akt Signaling in Treating Paclitaxel-Resistant Non-Small Cell of Lung Cancer. Mol. Med. 2020, 26, 28. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari Movahed, Z.; Rastegari-Pouyani, M.; Mohammadi, M.H.; Mansouri, K. Cancer Cells Change Their Glucose Metabolism to Overcome Increased ROS: One Step from Cancer Cell to Cancer Stem Cell? Biomed. Pharmacother. 2019, 112, 108690. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Li, J.; Zhang, Y.; Liao, Y.; Zhu, Y.; Wang, C.; Hou, J. LDHA Promotes Oral Squamous Cell Carcinoma Progression through Facilitating Glycolysis and Epithelial-Mesenchymal Transition. Front. Oncol. 2019, 9, 1446. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Q.; Guan, Y.; Liao, D.; Jiang, D.; Xiong, H.; Zhan, H.; Pang, J. Circular RNA CircVAMP3 Promotes Aerobic Glycolysis and Proliferation by Regulating LDHA in Renal Cell Carcinoma. Cell Death Dis. 2022, 13, 443. [Google Scholar] [CrossRef] [PubMed]
- Billiard, J.; Dennison, J.B.; Briand, J.; Annan, R.S.; Chai, D.; Colón, M.; Dodson, C.S.; Gilbert, S.A.; Greshock, J.; Jing, J.; et al. Quinoline 3-Sulfonamides Inhibit Lactate Dehydrogenase A and Reverse Aerobic Glycolysis in Cancer Cells. Cancer Metab. 2013, 1, 19. [Google Scholar] [CrossRef]
- Boudreau, A.; Purkey, H.E.; Hitz, A.; Robarge, K.; Peterson, D.; Labadie, S.; Kwong, M.; Hong, R.; Gao, M.; Del Nagro, C.; et al. Metabolic Plasticity Underpins Innate and Acquired Resistance to LDHA Inhibition. Nat. Chem. Biol. 2016, 12, 779–786. [Google Scholar] [CrossRef]
- Ždralević, M.; Brand, A.; Di Ianni, L.; Dettmer, K.; Reinders, J.; Singer, K.; Peter, K.; Schnell, A.; Bruss, C.; Decking, S.-M.; et al. Double Genetic Disruption of Lactate Dehydrogenases A and B Is Required to Ablate the “Warburg Effect” Restricting Tumor Growth to Oxidative Metabolism. J. Biol. Chem. 2018, 293, 15947–15961. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Zhang, B.; Lin, X.; Fu, X.; An, Y.; Zou, Y.; Wang, J.-X.; Wang, Z.; Yu, T. Lactate Metabolism in Human Health and Disease. Signal Transduct. Target. Ther. 2022, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.; Banerjee, D. Lactate Dehydrogenases as Metabolic Links between Tumor and Stroma in the Tumor Microenvironment. Cancers 2019, 11, 750. [Google Scholar] [CrossRef]
- Tiersma, J.F.; Evers, B.; Bakker, B.M.; Jalving, M.; de Jong, S. Pyruvate Dehydrogenase Kinase Inhibition by Dichloroacetate in Melanoma Cells Unveils Metabolic Vulnerabilities. Int. J. Mol. Sci. 2022, 23, 3745. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Morris, M.E. In Vitro and In Vivo Efficacy of AZD3965 and Alpha-Cyano-4-Hydroxycinnamic Acid in the Murine 4T1 Breast Tumor Model. AAPS J. 2020, 22, 84. [Google Scholar] [CrossRef]
- Curtis, N.J.; Mooney, L.; Hopcroft, L.; Michopoulos, F.; Whalley, N.; Zhong, H.; Murray, C.; Logie, A.; Revill, M.; Byth, K.F.; et al. Pre-Clinical Pharmacology of AZD3965, a Selective Inhibitor of MCT1: DLBCL, NHL and Burkitt’s Lymphoma Anti-Tumor Activity. Oncotarget 2017, 8, 69219–69236. [Google Scholar] [CrossRef]
- Hua, Y.; Zheng, Y.; Yao, Y.; Jia, R.; Ge, S.; Zhuang, A. Metformin and Cancer Hallmarks: Shedding New Lights on Therapeutic Repurposing. J. Transl. Med. 2023, 21, 403. [Google Scholar] [CrossRef] [PubMed]
- Saraei, P.; Asadi, I.; Kakar, M.A.; Moradi-Kor, N. The Beneficial Effects of Metformin on Cancer Prevention and Therapy: A Comprehensive Review of Recent Advances. Cancer Manag. Res. 2019, 11, 3295–3313. [Google Scholar] [CrossRef]
- Kasznicki, J.; Sliwinska, A.; Drzewoski, J. Metformin in Cancer Prevention and Therapy. Ann. Transl. Med. 2014, 2, 57. [Google Scholar] [CrossRef]
- Aurora, A.B.; Khivansara, V.; Leach, A.; Gill, J.G.; Martin-Sandoval, M.; Yang, C.; Kasitinon, S.Y.; Bezwada, D.; Tasdogan, A.; Gu, W.; et al. Loss of Glucose 6-Phosphate Dehydrogenase Function Increases Oxidative Stress and Glutaminolysis in Metastasizing Melanoma Cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2120617119. [Google Scholar] [CrossRef]
- Pes, G.M.; Errigo, A.; Soro, S.; Longo, N.P.; Dore, M.P. Glucose-6-Phosphate Dehydrogenase Deficiency Reduces Susceptibility to Cancer of Endodermal Origin. Acta Oncol. 2019, 58, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Mele, L.; la Noce, M.; Paino, F.; Regad, T.; Wagner, S.; Liccardo, D.; Papaccio, G.; Lombardi, A.; Caraglia, M.; Tirino, V.; et al. Glucose-6-Phosphate Dehydrogenase Blockade Potentiates Tyrosine Kinase Inhibitor Effect on Breast Cancer Cells through Autophagy Perturbation. J. Exp. Clin. Cancer Res. 2019, 38, 160. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, R.; Dey, T.; Kumar, L.; Kar, S.; Sarkar, R.; Ghorai, M.; Malik, S.; Jha, N.K.; Vellingiri, B.; Kesari, K.K.; et al. Cellular Landscaping of Cisplatin Resistance in Cervical Cancer. Biomed. Pharmacother. 2022, 153, 113345. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, P.; Chen, Q.; Deng, S.; Liu, X.; Situ, H.; Zhong, S.; Hann, S.; Lin, Y. Targeting AMPK Signaling Pathway to Overcome Drug Resistance for Cancer Therapy. Curr. Drug Targets 2016, 17, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.-Y.; Lucavs, J.; Ballard, D.; Das, J.K.; Kumar, A.; Wang, L.; Ren, Y.; Xiong, X.; Song, J. Metabolic Reprogramming and Reactive Oxygen Species in T Cell Immunity. Front. Immunol. 2021, 12, 652687. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkova, N.N.; Starkov, A.A. Metabolic ROS Signaling: To Immunity and Beyond. Biochemistry 2020, 85, 1650–1667. [Google Scholar] [CrossRef]
- Gwangwa, M.V.; Joubert, A.M.; Visagie, M.H. Crosstalk between the Warburg Effect, Redox Regulation and Autophagy Induction in Tumourigenesis. Cell. Mol. Biol. Lett. 2018, 23, 20. [Google Scholar] [CrossRef]
- Pandey, A.; Yadav, P.; Shukla, S. Unfolding the Role of Autophagy in the Cancer Metabolism. Biochem. Biophys. Rep. 2021, 28, 101158. [Google Scholar] [CrossRef]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg Meets Autophagy: Cancer-Associated Fibroblasts Accelerate Tumor Growth and Metastasis via Oxidative Stress, Mitophagy, and Aerobic Glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Chang, Y.; Lu, W.; Sheng, X.; Wang, S.; Xu, H.; Ma, J. Regulation of Autophagy by Glycolysis in Cancer. Cancer Manag. Res. 2020, 12, 13259–13271. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.A.; So, E.-Y.; Simons, A.L.; Spitz, D.R.; Ouchi, T. DNA Damage Induces Reactive Oxygen Species Generation through the H2AX-Nox1/Rac1 Pathway. Cell Death Dis. 2012, 3, e249. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA Damage Response in Cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Tsochantaridis, I.; Kontopoulos, A.; Voulgaridou, G.-P.; Tsifintaris, M.; Triantafyllou, C.; Pappa, A. Aldehyde Dehydrogenase 1B1 Is Implicated in DNA Damage Response in Human Colorectal Adenocarcinoma. Cells 2022, 11, 2017. [Google Scholar] [CrossRef]
- Matsuura, K.; Canfield, K.; Feng, W.; Kurokawa, M. Metabolic Regulation of Apoptosis in Cancer. Int. Rev. Cell Mol. Biol. 2016, 327, 43–87. [Google Scholar] [CrossRef]
- Bansal, M.; Gupta, A.; Ding, H.-F. MYCN and Metabolic Reprogramming in Neuroblastoma. Cancers 2022, 14, 4113. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Tan-Sah, V.P.; Ding, E.Y.; Smith, J.M.; Miyamoto, S. Hexokinase-II Positively Regulates Glucose Starvation-Induced Autophagy through TORC1 Inhibition. Mol. Cell 2014, 53, 521–533. [Google Scholar] [CrossRef]
- Nazim, U.M.; Bishayee, K.; Kang, J.; Yoo, D.; Huh, S.-O.; Sadra, A. MTORC1-Inhibition Potentiating Metabolic Block by Tyrosine Kinase Inhibitor Ponatinib in Multiple Myeloma. Cancers 2022, 14, 2766. [Google Scholar] [CrossRef]
- Ding, X.-C.; Wang, L.-L.; Zhang, X.-D.; Xu, J.-L.; Li, P.-F.; Liang, H.; Zhang, X.-B.; Xie, L.; Zhou, Z.-H.; Yang, J.; et al. The Relationship between Expression of PD-L1 and HIF-1α in Glioma Cells under Hypoxia. J. Hematol. Oncol. 2021, 14, 92. [Google Scholar] [CrossRef]
- Shurin, M.R.; Umansky, V. Cross-Talk between HIF and PD-1/PD-L1 Pathways in Carcinogenesis and Therapy. J. Clin. Investig. 2022, 132, e159473. [Google Scholar] [CrossRef]
- Blank, C.; Gajewski, T.F.; Mackensen, A. Interaction of PD-L1 on Tumor Cells with PD-1 on Tumor-Specific T Cells as a Mechanism of Immune Evasion: Implications for Tumor Immunotherapy. Cancer Immunol. Immunother. 2005, 54, 307–314. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Westhoff, M.-A.; Karpel-Massler, G.; Siegelin, M.D. Targeting Super-Enhancers Reprograms Glioblastoma Central Carbon Metabolism. Oncotarget 2021, 12, 1309–1313. [Google Scholar] [CrossRef]
- Di Gregorio, J.; Petricca, S.; Iorio, R.; Toniato, E.; Flati, V. Mitochondrial and Metabolic Alterations in Cancer Cells. Eur. J. Cell Biol. 2022, 101, 151225. [Google Scholar] [CrossRef]
- Hollinshead, K.E.R.; Parker, S.J.; Eapen, V.V.; Encarnacion-Rosado, J.; Sohn, A.; Oncu, T.; Cammer, M.; Mancias, J.D.; Kimmelman, A.C. Respiratory Supercomplexes Promote Mitochondrial Efficiency and Growth in Severely Hypoxic Pancreatic Cancer. Cell Rep. 2020, 33, 108231. [Google Scholar] [CrossRef] [PubMed]
- Zhdanov, A.V.; Waters, A.H.C.; Golubeva, A.V.; Dmitriev, R.I.; Papkovsky, D.B. Availability of the Key Metabolic Substrates Dictates the Respiratory Response of Cancer Cells to the Mitochondrial Uncoupling. Biochim. Biophys. Acta 2014, 1837, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lim, S.K.; Liang, Q.; Iyer, S.V.; Wang, H.-Y.; Wang, Z.; Xie, X.; Sun, D.; Chen, Y.-J.; Tabar, V.; et al. Gboxin Is an Oxidative Phosphorylation Inhibitor That Targets Glioblastoma. Nature 2019, 567, 341–346. [Google Scholar] [CrossRef]
- Li, Y.-J.; Fahrmann, J.F.; Aftabizadeh, M.; Zhao, Q.; Tripathi, S.C.; Zhang, C.; Yuan, Y.; Ann, D.; Hanash, S.; Yu, H. Fatty Acid Oxidation Protects Cancer Cells from Apoptosis by Increasing Mitochondrial Membrane Lipids. Cell Rep. 2022, 39, 110870. [Google Scholar] [CrossRef]
- Prew, M.S.; Adhikary, U.; Choi, D.W.; Portero, E.P.; Paulo, J.A.; Gowda, P.; Budhraja, A.; Opferman, J.T.; Gygi, S.P.; Danial, N.N.; et al. MCL-1 Is a Master Regulator of Cancer Dependency on Fatty Acid Oxidation. Cell Rep. 2022, 41, 111445. [Google Scholar] [CrossRef]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.-Y.; Fang, X. Fatty Acid Oxidation: An Emerging Facet of Metabolic Transformation in Cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Halama, A.; Kulinski, M.; Dib, S.S.; Zaghlool, S.B.; Siveen, K.S.; Iskandarani, A.; Zierer, J.; Prabhu, K.S.; Satheesh, N.J.; Bhagwat, A.M.; et al. Accelerated Lipid Catabolism and Autophagy Are Cancer Survival Mechanisms under Inhibited Glutaminolysis. Cancer Lett. 2018, 430, 133–147. [Google Scholar] [CrossRef]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.-L.; et al. Fatty Acid Uptake and Lipid Storage Induced by HIF-1α Contribute to Cell Growth and Survival after Hypoxia-Reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Cardaci, S.; Desideri, E.; Ciriolo, M.R. Targeting Aerobic Glycolysis: 3-Bromopyruvate as a Promising Anticancer Drug. J. Bioenerg. Biomembr. 2012, 44, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Pisarsky, L.; Bill, R.; Fagiani, E.; Dimeloe, S.; Goosen, R.W.; Hagmann, J.; Hess, C.; Christofori, G. Targeting Metabolic Symbiosis to Overcome Resistance to Anti-Angiogenic Therapy. Cell Rep. 2016, 15, 1161–1174. [Google Scholar] [CrossRef]
- Hirpara, J.; Eu, J.Q.; Tan, J.K.M.; Wong, A.L.; Clement, M.-V.; Kong, L.R.; Ohi, N.; Tsunoda, T.; Qu, J.; Goh, B.C.; et al. Metabolic Reprogramming of Oncogene-Addicted Cancer Cells to OXPHOS as a Mechanism of Drug Resistance. Redox Biol. 2019, 25, 101076. [Google Scholar] [CrossRef]
- Zhang, Z.; Zeng, D.; Zhang, W.; Chen, A.; Lei, J.; Liu, F.; Deng, B.; Zhuo, J.; He, B.; Yan, M.; et al. Modulation of Oxidative Phosphorylation Augments Antineoplastic Activity of Mitotic Aurora Kinase Inhibition. Cell Death Dis. 2021, 12, 893. [Google Scholar] [CrossRef] [PubMed]
- Elkind-Hirsch, K.E.; Paterson, M.S.; Seidemann, E.L.; Gutowski, H.C. Short-Term Therapy with Combination Dipeptidyl Peptidase-4 Inhibitor Saxagliptin/Metformin Extended Release (XR) Is Superior to Saxagliptin or Metformin XR Monotherapy in Prediabetic Women with Polycystic Ovary Syndrome: A Single-Blind, Randomized, Pilot. Fertil. Steril. 2017, 107, 253–260.e1. [Google Scholar] [CrossRef] [PubMed]
- Nagabushan, S.; Lau, L.M.S.; Barahona, P.; Wong, M.; Sherstyuk, A.; Marshall, G.M.; Tyrrell, V.; Wegner, E.A.; Ekert, P.G.; Cowley, M.J.; et al. Efficacy of MEK Inhibition in a Recurrent Malignant Peripheral Nerve Sheath Tumor. npj Precis. Oncol. 2021, 5, 9. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCT Number | Study | Drug | Phase |
|---|---|---|---|
| NCT02022007 | PCOS Women with Impaired Glucose Homeostasis | Metformin XR, Saxagliptin | 3 |
| NCT03433183 | Malignant Peripheral Nerve Sheath Tumors | Selumetinib, Sirolimus | 2 |
| NCT03057600 | Advanced Triple Negative Breast Cancer (TNBC) | Paclitaxel, Telaglenestat (CB-839) | 2 |
| NCT02858921 | BRAF Mutant Stage III Melanoma | Dabrafenib, Trametinib, Pembrolizumab | 2 |
| NCT04776889 | Castrated Egyptian Prostate Cancer Patients | Rosuvastatin and surgical castration | 4 |
| NCT03428217 | Metastatic Renal Cell Carcinoma | CB-839, Cabozantinib | 2 |
| NCT05254171 | Pancreatic Cancer | SBP-101, Nab-paclitaxel, Gemcitabine | 2/3 |
| NCT03965845 | Solid Tumors | CB-839, Palbociclib | 1/2 |
| NCT02771626 | Melanoma, Clear Cell Renal Cell Carcinoma (ccRCC), and Non-Small Cell Lung Cancer (NSCLC) | CB-839, Nivolumab | 1/2 |
| NCT00859495 | Pleural Mesothelioma | Doxorubicin, Cisplatin, Pemetrexed and Radiotherapy | 2 |
| NCT03163667 | Renal Cell Carcinoma (RCC) | CB-839, Everolimus | 2 |
| NCT04207086 | Stage III Melanoma | Pembrolizumab, Lenvatinib | 2 |
| NCT02903914 | Advanced/Metastatic Solid Tumors | INCB001158, Pembrolizumab | 1/2 |
| NCT00360828 | Recurrent Anaplastic Astrocytomas, Mixed Malignant Gliomas, and Oligodendrogliomas | Irinotecan Hydrochloride | 2 |
| NCT03449901 | Soft Tissue Sarcoma, Osteosarcoma, Ewing’s Sarcoma, and Small Cell Lung Cancer | pegylated arginine deiminase, Gemcitabine, Docetaxel | 2 |
| NCT05796570 | AML, MDS, and Related Myeloid Malignancies | Decitabine, Filgrastim | 2 |
| NCT03875313 | Solid Tumors | CB-839, Talazoparib | 1/2 |
| NCT00634270 | Plexiform Neurofibromas | Sirolimus | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bishayee, K.; Lee, S.-H.; Park, Y.S. The Illustration of Altered Glucose Dependency in Drug-Resistant Cancer Cells. Int. J. Mol. Sci. 2023, 24, 13928. https://doi.org/10.3390/ijms241813928
Bishayee K, Lee S-H, Park YS. The Illustration of Altered Glucose Dependency in Drug-Resistant Cancer Cells. International Journal of Molecular Sciences. 2023; 24(18):13928. https://doi.org/10.3390/ijms241813928
Chicago/Turabian StyleBishayee, Kausik, Seung-Hee Lee, and Yong Soo Park. 2023. "The Illustration of Altered Glucose Dependency in Drug-Resistant Cancer Cells" International Journal of Molecular Sciences 24, no. 18: 13928. https://doi.org/10.3390/ijms241813928