
Current Status of Next-Generation Sequencing in Bone Genetic Diseases

Abstract
:1. Introduction
2. Genome Database
3. Single Nucleotide Polymorphism and Genome-Wide Association Study
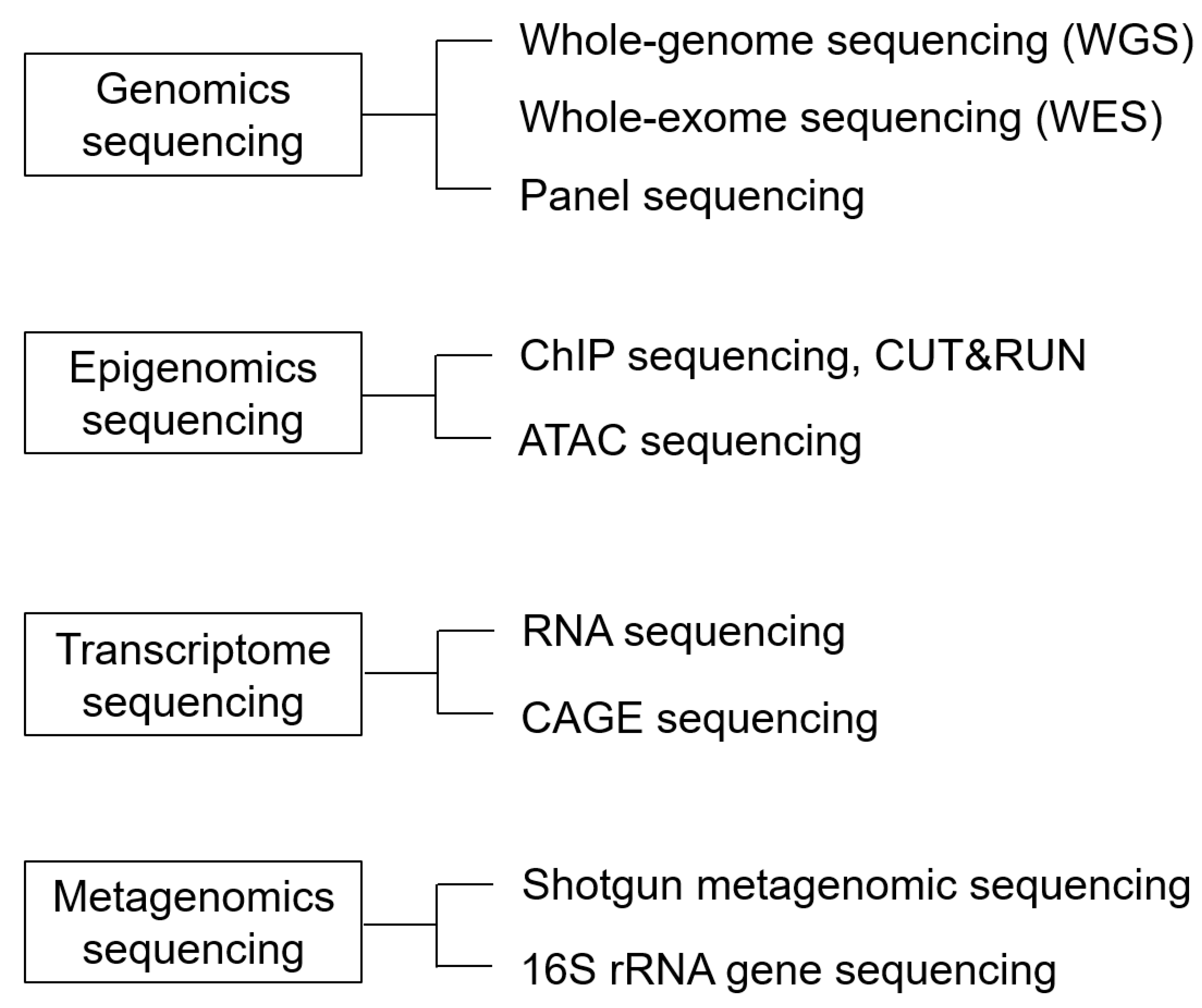
4. Next-Generation Sequencing
4.1. Genomics Analysis
4.1.1. Advances in Genetic Analysis: From Gene Chips and Panels to Whole-Exome Sequencing (WES), and Finally, WGS
4.1.2. Panel Sequencing
4.1.3. Whole-Exome Sequencing (WES)
4.1.4. Whole-Genome Sequencing (WGS)
4.2. Epigenomics Sequencing
4.2.1. ChIP-Sequencing
4.2.2. ATAC-Sequencing
4.3. Transcriptome Sequencing
4.3.1. RNA Sequencing
4.3.2. CAGE Sequencing
4.4. Metagenomic Sequencing
5. Single-Cell Analysis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-generation sequencing technology: Current trends and advancements. Biology 2023, 12, 997. [Google Scholar] [CrossRef]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Aartsma-Rus, A.; Cohen, J.L.; Cowsert, L.M.; Howell, R.R.; Yu, T.W.; Wasserstein, M.P.; Defay, T. Whole-genome sequencing holds the key to the success of gene-targeted therapies. Am. J. Med. Genet. C Semin. Med. Genet. 2023, 193, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Voelkerding, K.V.; Dames, S.A.; Durtschi, J.D. Next-generation sequencing: From basic research to diagnostics. Clin. Chem. 2009, 55, 641–658. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child. Educ. Pract. Ed. 2013, 98, 236–238. [Google Scholar] [CrossRef]
- Schloss, J.A.; Gibbs, R.A.; Makhijani, V.B.; Marziali, A. Cultivating DNA sequencing technology after the human genome project. Annu. Rev. Genomics Hum. Genet. 2020, 21, 117–138. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age; ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Unger, S.; Ferreira, C.R.; Mortier, G.R.; Ali, H.; Bertola, D.R.; Calder, A.; Cohn, D.H.; Cormier-Daire, V.; Girisha, K.M.; Hall, C.; et al. Nosology of genetic skeletal disorders: 2023 revision. Am. J. Med. Genet. A 2023, 191, 1164–1209. [Google Scholar] [CrossRef]
- Need, A.C.; Shashi, V.; Hitomi, Y.; Schoch, K.; Shianna, K.V.; McDonald, M.T.; Meisler, M.H.; Goldstein, D.B. Clinical application of exome sequencing in undiagnosed genetic conditions. J. Med. Genet. 2012, 49, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Marwaha, S.; Knowles, J.W.; Ashley, E.A. A guide for the diagnosis of rare and undiagnosed disease: Beyond the exome. Genome Med. 2022, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Mardis, E.R.; Ding, L.; Fulton, B.; McLellan, M.D.; Chen, K.; Dooling, D.; Dunford-Shore, B.H.; McGrath, S.; Hickenbotham, M.; et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008, 456, 66–72. [Google Scholar] [CrossRef]
- Meldrum, C.; Doyle, M.A.; Tothill, R.W. Next-generation sequencing for cancer diagnostics: A practical perspective. Clin. Biochem. Rev. 2011, 32, 177–195. [Google Scholar] [PubMed]
- Langerhorst, P.; Noori, S.; Zajec, M.; De Rijke, Y.B.; Gloerich, J.; van Gool, A.J.; Caillon, H.; Joosten, I.; Luider, T.M.; Corre, J.; et al. Multiple Myeloma Minimal Residual Disease Detection: Targeted Mass Spectrometry in Blood vs. Next-Generation Sequencing in Bone Marrow. Clin. Chem. 2021, 67, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Hutter, C.; Zenklusen, J.C. The cancer genome Atlas: Creating lasting value beyond its data. Cell 2018, 173, 283–285. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- ICGC/TCGA Pan-cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Stewart, E.; McEvoy, J.; Wang, H.; Chen, X.; Honnell, V.; Ocarz, M.; Gordon, B.; Dapper, J.; Blankenship, K.; Yang, Y.; et al. Identification of therapeutic targets in rhabdomyosarcoma through integrated genomic, epigenomic, and proteomic analyses. Cancer Cell 2018, 34, 411–426.e19. [Google Scholar] [CrossRef]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- Syvänen, A.C. Toward genome-wide SNP genotyping. Nat. Genet. 2005, 37, S5–S10. [Google Scholar] [CrossRef]
- Li, M.; Li, C.; Guan, W. Evaluation of coverage variation of SNP chips for genome-wide association studies. Eur. J. Hum. Genet. 2008, 16, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Cardon, L.R. Evaluating coverage of genome-wide association studies. Nat. Genet. 2006, 38, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Bello, J.; Jiménez-Morales, M. Functional implications of single nucleotide polymorphisms (SNPs) in protein-coding and non-coding RNA genes in multifactorial diseases. Gac. Med. Mex. 2017, 153, 238–250. [Google Scholar]
- Halushka, M.K.; Fan, J.B.; Bentley, K.; Hsie, L.; Shen, N.; Weder, A.; Cooper, R.; Lipshutz, R.; Chakravarti, A. Patterns of single-nucleotide polymorphisms in candidate genes for blood-pressure homeostasis. Nat. Genet. 1999, 22, 239–247. [Google Scholar] [CrossRef]
- Brookes, A.J. The essence of SNPs. Gene 1999, 234, 177–186. [Google Scholar] [CrossRef]
- De Gobbi, M.; Viprakasit, V.; Hughes, J.R.; Fisher, C.; Buckle, V.J.; Ayyub, H.; Gibbons, R.J.; Vernimmen, D.; Yoshinaga, Y.; de Jong, P.; et al. A regulatory SNP causes a human genetic disease by creating a new transcriptional promoter. Science 2006, 312, 1215–1217. [Google Scholar] [CrossRef]
- Hirschhorn, J.N.; Daly, M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 2005, 6, 95–108. [Google Scholar] [CrossRef]
- Ozaki, K.; Ohnishi, Y.; Iida, A.; Sekine, A.; Yamada, R.; Tsunoda, T.; Sato, H.; Sato, H.; Hori, M.; Nakamura, Y.; et al. Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction. Nat. Genet. 2002, 32, 650–654. [Google Scholar] [CrossRef]
- International HapMap Consortium; Frazer, K.A.; Ballinger, D.G.; Cox, D.R.; Hinds, D.A.; Stuve, L.L.; Gibbs, R.A.; Belmont, J.W.; Boudreau, A.; Hardenbol, P.; et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007, 449, 851–861. [Google Scholar]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; 1000 Genomes Project Consortium; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [PubMed]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef] [PubMed]
- Urano, T.; Inoue, S. Genetics of osteoporosis. Biochem. Biophys. Res. Commun. 2014, 452, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of next-generation sequencing technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Davis, E.E.; Katsanis, N. The ciliopathies: A transitional model into systems biology of human genetic disease. Curr. Opin. Genet. 2012, 22, 290–303. [Google Scholar] [CrossRef]
- Semler, O.; Garbes, L.; Keupp, K.; Swan, D.; Zimmermann, K.; Becker, J.; Iden, S.; Wirth, B.; Eysel, P.; Koerber, F.; et al. A mutation in the 5′-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am. J. Hum. Genet. 2012, 91, 349–357. [Google Scholar] [CrossRef]
- Becker, J.; Semler, O.; Gilissen, C.; Li, Y.; Bolz, H.J.; Giunta, C.; Bergmann, C.; Rohrbach, M.; Koerber, F.; Zimmermann, K.; et al. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2011, 88, 362–371. [Google Scholar] [CrossRef]
- Morello, R.; Bertin, T.K.; Chen, Y.; Hicks, J.; Tonachini, L.; Monticone, M.; Castagnola, P.; Rauch, F.; Glorieux, F.H.; Vranka, J.; et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006, 127, 291–304. [Google Scholar] [CrossRef]
- Caparrós-Martin, J.A.; Valencia, M.; Pulido, V.; Martínez-Glez, V.; Rueda-Arenas, I.; Amr, K.; Farra, C.; Lapunzina, P.; Ruiz-Perez, V.L.; Temtamy, S.; et al. Clinical and molecular analysis in families with autosomal recessive osteogenesis imperfecta identifies mutations in five genes and suggests genotype-phenotype correlations. Am. J. Med. Genet. A 2013, 161A, 1354–1369. [Google Scholar] [CrossRef]
- Cabral, W.A.; Chang, W.; Barnes, A.M.; Weis, M.; Scott, M.A.; Leikin, S.; Makareeva, E.; Kuznetsova, N.V.; Rosenbaum, K.N.; Tifft, C.J.; et al. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 2007, 39, 359–365. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, F.S.; Nesbitt, I.M.; Zwikstra, E.H.; Nikkels, P.G.; Piersma, S.R.; Fratantoni, S.A.; Jimenez, C.R.; Huizer, M.; Morsman, A.C.; Cobben, J.M.; et al. PPIB mutations cause severe osteogenesis imperfecta. Am. J. Hum. Genet. 2009, 85, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, H.E.; Schwarze, U.; Pyott, S.M.; AlSwaid, A.; Al Balwi, M.; Alrasheed, S.; Pepin, M.G.; Weis, M.A.; Eyre, D.R.; Byers, P.H. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010, 86, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Alanay, Y.; Avaygan, H.; Camacho, N.; Utine, G.E.; Boduroglu, K.; Aktas, D.; Alikasifoglu, M.; Tuncbilek, E.; Orhan, D.; Bakar, F.T.; et al. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010, 86, 551–559. [Google Scholar] [CrossRef]
- Lapunzina, P.; Aglan, M.; Temtamy, S.; Caparrós-Martín, J.A.; Valencia, M.; Letón, R.; Martínez-Glez, V.; Elhossini, R.; Amr, K.; Vilaboa, N.; et al. Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010, 87, 110–114. [Google Scholar] [CrossRef]
- Pihlajaniemi, T.; Dickson, L.A.; Pope, F.M.; Korhonen, V.R.; Nicholls, A.; Prockop, D.J.; Myers, J.C. Osteogenesis imperfecta: Cloning of a pro-alpha 2(I) collagen gene with a frameshift mutation. J. Biol. Chem. 1984, 259, 12941–12944. [Google Scholar] [CrossRef]
- Shaheen, R.; Alazami, A.M.; Alshammari, M.J.; Faqeih, E.; Alhashmi, N.; Mousa, N.; Alsinani, A.; Ansari, S.; Alzahrani, F.; Al-Owain, M.; et al. Study of autosomal recessive osteogenesis imperfecta in Arabia reveals a novel locus defined by TMEM38B mutation. J. Med. Genet. 2012, 49, 630–635. [Google Scholar] [CrossRef]
- Keupp, K.; Beleggia, F.; Kayserili, H.; Barnes, A.M.; Steiner, M.; Semler, O.; Fischer, B.; Yigit, G.; Janda, C.Y.; Becker, J.; et al. Mutations in WNT1 cause different forms of bone fragility. Am. J. Hum. Genet. 2013, 92, 565–574. [Google Scholar] [CrossRef]
- Symoens, S.; Malfait, F.; D’hondt, S.; Callewaert, B.; Dheedene, A.; Steyaert, W.; Bächinger, H.P.; De Paepe, A.; Kayserili, H.; Coucke, P.J. Deficiency for the ER-stress transducer OASIS causes severe recessive osteogenesis imperfecta in humans. Orphanet J. Rare Dis. 2013, 8, 154. [Google Scholar] [CrossRef]
- Mendoza-Londono, R.; Fahiminiya, S.; Majewski, J.; Care4Rare Canada Consortium; Tétreault, M.; Nadaf, J.; Kannu, P.; Sochett, E.; Howard, A.; Stimec, J.; et al. Recessive osteogenesis imperfecta caused by missense mutations in SPARC. Am. J. Hum. Genet. 2015, 96, 979–985. [Google Scholar] [CrossRef]
- Doyard, M.; Bacrot, S.; Huber, C.; Di Rocco, M.; Goldenberg, A.; Aglan, M.S.; Brunelle, P.; Temtamy, S.; Michot, C.; Otaify, G.A.; et al. FAM46A mutations are responsible for autosomal recessive osteogenesis imperfecta. J. Med. Genet. 2018, 55, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Lindert, U.; Cabral, W.A.; Ausavarat, S.; Tongkobpetch, S.; Ludin, K.; Barnes, A.M.; Yeetong, P.; Weis, M.; Krabichler, B.; Srichomthong, C.; et al. MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta. Nat. Commun. 2016, 7, 11920. [Google Scholar] [CrossRef] [PubMed]
- Moosa, S.; Yamamoto, G.L.; Garbes, L.; Keupp, K.; Beleza-Meireles, A.; Moreno, C.A.; Valadares, E.R.; de Sousa, S.B.; Maia, S.; Saraiva, J.; et al. Autosomal-Recessive Mutations in MESD Cause Osteogenesis Imperfecta. Am. J. Hum. Genet. 2019, 105, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Garbes, L.; Kim, K.; Rieß, A.; Hoyer-Kuhn, H.; Beleggia, F.; Bevot, A.; Kim, M.J.; Huh, Y.H.; Kweon, H.S.; Savarirayan, R.; et al. Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta. Am. J. Hum. Genet. 2015, 96, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Dubail, J.; Brunelle, P.; Baujat, G.; Huber, C.; Doyard, M.; Michot, C.; Chavassieux, P.; Khairouni, A.; Topouchian, V.; Monnot, S.; et al. Homozygous Loss-of-Function Mutations in CCDC134 Are Responsible for a Severe Form of Osteogenesis Imperfecta. J. Bone Miner. Res. 2020, 35, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, D.; Zheng, W.; Wang, O.; Jiang, Y.; Xia, W.; Xing, X.; Li, M. A novel missense mutation in P4HB causes mild osteogenesis imperfecta. Biosci. Rep. 2019, 39, BSR20182118. [Google Scholar] [CrossRef] [PubMed]
- Ha-Vinh, R.; Alanay, Y.; Bank, R.A.; Campos-Xavier, A.B.; Zankl, A.; Superti-Furga, A.; Bonafe, L. Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in PLOD2. Am. J. Med. Genet. A 2004, 131, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wu, H.; Zhang, C.; Feng, J.; Chen, L.; Xie, R.; Wang, F.; Chen, X.; Zhou, H.; Sun, H.; et al. Clinical, Genetics, and Bioinformatic Characterization of Mutations Affecting an Essential Region of PLS3 in Patients with BMND18. Int. J. Endocrinol. 2018, 2018, 8953217. [Google Scholar] [CrossRef]
- van Dijk, F.S.; Semler, O.; Etich, J.; Köhler, A.; Jimenez-Estrada, J.A.; Bravenboer, N.; Claeys, L.; Riesebos, E.; Gegic, S.; Piersma, S.R.; et al. Interaction between KDELR2 and HSP47 as a Key Determinant in Osteogenesis Imperfecta Caused by Bi-allelic Variants in KDELR2. Am. J. Hum. Genet. 2020, 107, 989–999. [Google Scholar] [CrossRef]
- Nagahashi, M.; Shimada, Y.; Ichikawa, H.; Kameyama, H.; Takabe, K.; Okuda, S.; Wakai, T. Next generation sequencing-based gene panel tests for the management of solid tumors. Cancer Sci. 2019, 110, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.T.; Campeau, P.M.; Lee, B.H. Genotype-phenotype correlation--promiscuity in the era of next-generation sequencing. N. Engl. J. Med. 2014, 371, 593–596. [Google Scholar] [CrossRef]
- Qin, D. Next-generation sequencing and its clinical application. Cancer Biol. Med. 2019, 16, 4–10. [Google Scholar] [PubMed]
- Nakamura, Y.; Onodera, S.; Takano, M.; Katakura, A.; Nomura, T.; Azuma, T. Development of a targeted gene panel for the diagnosis of Gorlin syndrome. Int. J. Oral Maxillofac. Surg. 2022, 51, 1431–1444. [Google Scholar] [CrossRef]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016, 18, 696–704. [Google Scholar] [CrossRef]
- Maddirevula, S.; Alsahli, S.; Alhabeeb, L.; Patel, N.; Alzahrani, F.; Shamseldin, H.E.; Anazi, S.; Ewida, N.; Alsaif, H.S.; Mohamed, J.Y.; et al. Expanding the phenome and variome of skeletal dysplasia. Genet. Med. 2018, 20, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.; Ding, L.; Shen, D.; Luo, J.; Suman, V.J.; Wallis, J.W.; Van Tine, B.A.; Hoog, J.; Goiffon, R.J.; Goldstein, T.C.; et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 2012, 486, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Andersson, K.; Malmgren, B.; Åström, E.; Nordgren, A.; Taylan, F.; Dahllöf, G. Mutations in COL1A1/A2 and CREB3L1 are associated with oligodontia in osteogenesis imperfecta. Orphanet J. Rare Dis. 2020, 15, 80. [Google Scholar] [CrossRef] [PubMed]
- Laird, P.W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef]
- Park, P.J. ChIP-seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Aguilar, R.; Henriquez, B.; Lian, J.B.; Stein, J.L.; Stein, G.S.; van Wijnen, A.J.; van Zundert, B.; Allende, M.L.; Montecino, M. Epigenetic control of the bone-master Runx2 gene during osteoblast-lineage commitment by the histone demethylase JARID1B/KDM5B. J. Biol. Chem. 2015, 290, 28329–28342. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chen, K.; Ye, G.; Wang, S.; Wang, P.; Li, J.; Zheng, G.; Liu, W.; Lin, J.; Su, Z.; et al. SNP-adjacent super enhancer network mediates enhanced osteogenic differentiation of MSCs in ankylosing spondylitis. Hum. Mol. Genet. 2021, 26, 277–293. [Google Scholar] [CrossRef]
- Sun, Y.; Miao, N.; Sun, T. Detect accessible chromatin using ATAC-sequencing, from principle to applications. Hereditas 2019, 156, 29. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Zhang, Z.; Grasfeder, L.L.; Boyle, A.P.; Giresi, P.G.; Lee, B.K.; Sheffield, N.C.; Gräf, S.; Huss, M.; Keefe, D.; et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011, 21, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Liu, Y.; Chang, J.C.; Hon, C.C.; Fukui, N.; Tanaka, N.; Zhang, Z.; Lee, M.T.M.; Minoda, A. Chromatin accessibility landscape of articular knee cartilage reveals aberrant enhancer regulation in osteoarthritis. Sci. Rep. 2018, 8, 15499. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef]
- Zhang, H.; He, L.; Cai, L. Transcriptome sequencing: RNA-seq. Methods Mol. Biol. 2018, 1754, 15–27. [Google Scholar]
- Kridel, R.; Meissner, B.; Rogic, S.; Boyle, M.; Telenius, A.; Woolcock, B.; Gunawardana, J.; Jenkins, C.E.; Cochrane, C.; Ben-Neriah, S.; et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood 2012, 119, 1963–1971. [Google Scholar] [CrossRef]
- Onodera, S.; Saito, A.; Hojo, H.; Nakamura, T.; Zujur, D.; Watanabe, K.; Morita, N.; Hasegawa, D.; Masaki, H.; Nakauchi, H.; et al. Hedgehog activation regulates human osteoblastogenesis. Stem Cell Rep. 2020, 15, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Hojo, H.; Saito, T.; He, X.; Guo, Q.; Onodera, S.; Azuma, T.; Koebis, M.; Nakao, K.; Aiba, A.; Seki, M.; et al. Runx2 regulates chromatin accessibility to direct the osteoblast program at neonatal stages. Cell Rep. 2022, 40, 111315. [Google Scholar] [CrossRef] [PubMed]
- de Hoon, M.; Hayashizaki, Y. Deep cap analysis gene expression (CAGE): Genome-wide identification of promoters, quantification of their expression, and network inference. BioTechniques 2008, 44, 627–628, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Yoshizaki, K.; Tian, T.; Miyazaki, K.; Martin, D.; Genomics and Computational Biology Core; Saito, K.; Yamada, A.; Fukumoto, S. Integration of Single-Cell RNA- and CAGE-seq Reveals Tooth-Enriched Genes. J. Dent. Res. 2021, 101, 220345211049785. [Google Scholar] [CrossRef]
- Kawaji, H.; Lizio, M.; Itoh, M.; Kanamori-Katayama, M.; Kaiho, A.; Nishiyori-Sueki, H.; Shin, J.W.; Kojima-Ishiyama, M.; Kawano, M.; Murata, M.; et al. Comparison of CAGE and RNA-seq transcriptome profiling using clonally amplified and single-molecule next-generation sequencing. Genome Res. 2014, 24, 708–717. [Google Scholar] [CrossRef]
- Ooki, A.; Onodera, S.; Saito, A.; Oguchi, A.; Murakawa, Y.; Sakamoto, T.; Sueishi, K.; Nishii, Y.; Azuma, T. CAGE-seq analysis of osteoblast derived from cleidocranial dysplasia human induced pluripotent stem cells. Bone 2020, 141, 115582. [Google Scholar] [CrossRef]
- Seth, S.; Välimäki, N.; Kaski, S.; Honkela, A. Exploration and retrieval of whole-metagenome sequencing samples. Bioinformatics 2014, 30, 2471–2479. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, G.; Lau, H.C.; Yu, J. Metagenomic sequencing for microbial DNA in human samples: Emerging technological advances. Int. J. Mol. Sci. 2022, 23, 2181. [Google Scholar] [CrossRef]
- Ye, P.; Xie, C.; Wu, C.; Yu, C.; Chen, Y.; Liang, Z.; Chen, Y.; Chen, Q.; Kong, Y. The application of metagenomic next-generation sequencing for detection of pathogens from dialysis effluent in peritoneal dialysis-associated peritonitis. Perit. Dial. Int. 2022, 42, 585–590. [Google Scholar] [CrossRef]
- Huang, Y.; Zhao, X.; Cui, L.; Huang, S. Metagenomic and metatranscriptomic insight into oral biofilms in periodontitis and related systemic diseases. Front. Microbiol. 2021, 12, 728585. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, T.; Wu, K.; Li, Z.; Chen, X.; Jiang, S.; Du, L.; Lu, S.; Lin, C.; Wu, J.; et al. Metagenomic analysis reveals A possible association between respiratory infection and periodontitis. Genom. Proteom. Bioinform. 2022, 20, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qi, J.; Zhao, H.; He, S.; Zhang, Y.; Wei, S.; Zhao, F. Metagenomic sequencing reveals microbiota and its functional potential associated with periodontal disease. Sci. Rep. 2013, 3, 1843. [Google Scholar] [CrossRef]
- Wen, L.; Tang, F. Single-cell sequencing in stem cell biology. Genome Biol. 2016, 15, 71. [Google Scholar] [CrossRef]
- Macaulay, I.C.; Voet, T. Single cell genomics: Advances and future perspectives. PLoS Genet. 2014, 30, e1004126. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.E.; Westermann, A.J.; Gorski, S.A.; Vogel, J. Single-cell RNA-seq: Advances and future challenges. Nucleic Acids Res. 2014, 42, 8845–8860. [Google Scholar] [CrossRef] [PubMed]
- Ayturk, U.M.; Scollan, J.P.; Goz Ayturk, D.; Suh, E.S.; Vesprey, A.; Jacobsen, C.M.; Divieti Pajevic, P.; Warman, M.L. Single-cell RNA sequencing of calvarial and long-bone endocortical cells. J. Bone Miner. Res. 2020, 35, 1981–1991. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
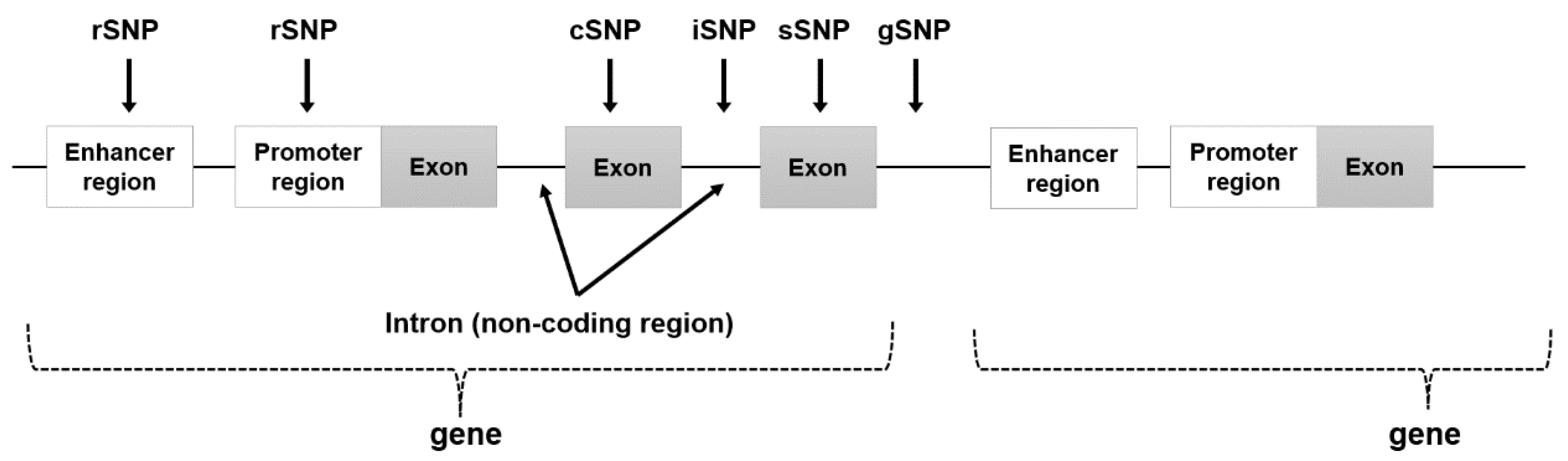
| Classification | Location | Features |
|---|---|---|
| rSNP (regulatory SNP) | enhancer and promoter region (regulatory region) |
|
| cSNP (cording SNP) | exon region (translation region) |
|
| sSNP (silent SNP) | exon region (translation region) |
|
| iSNP (intron SNP) | intron region (regulatory region) |
|
| gSNP (genomics SNP) | junk region |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aida, N.; Saito, A.; Azuma, T. Current Status of Next-Generation Sequencing in Bone Genetic Diseases. Int. J. Mol. Sci. 2023, 24, 13802. https://doi.org/10.3390/ijms241813802
Aida N, Saito A, Azuma T. Current Status of Next-Generation Sequencing in Bone Genetic Diseases. International Journal of Molecular Sciences. 2023; 24(18):13802. https://doi.org/10.3390/ijms241813802
Chicago/Turabian StyleAida, Natsuko, Akiko Saito, and Toshifumi Azuma. 2023. "Current Status of Next-Generation Sequencing in Bone Genetic Diseases" International Journal of Molecular Sciences 24, no. 18: 13802. https://doi.org/10.3390/ijms241813802