

Ascites-Derived Organoids to Depict Platinum Resistance in Gynaecological Serous Carcinomas
 ,
,
Abstract
:1. Introduction
2. Results
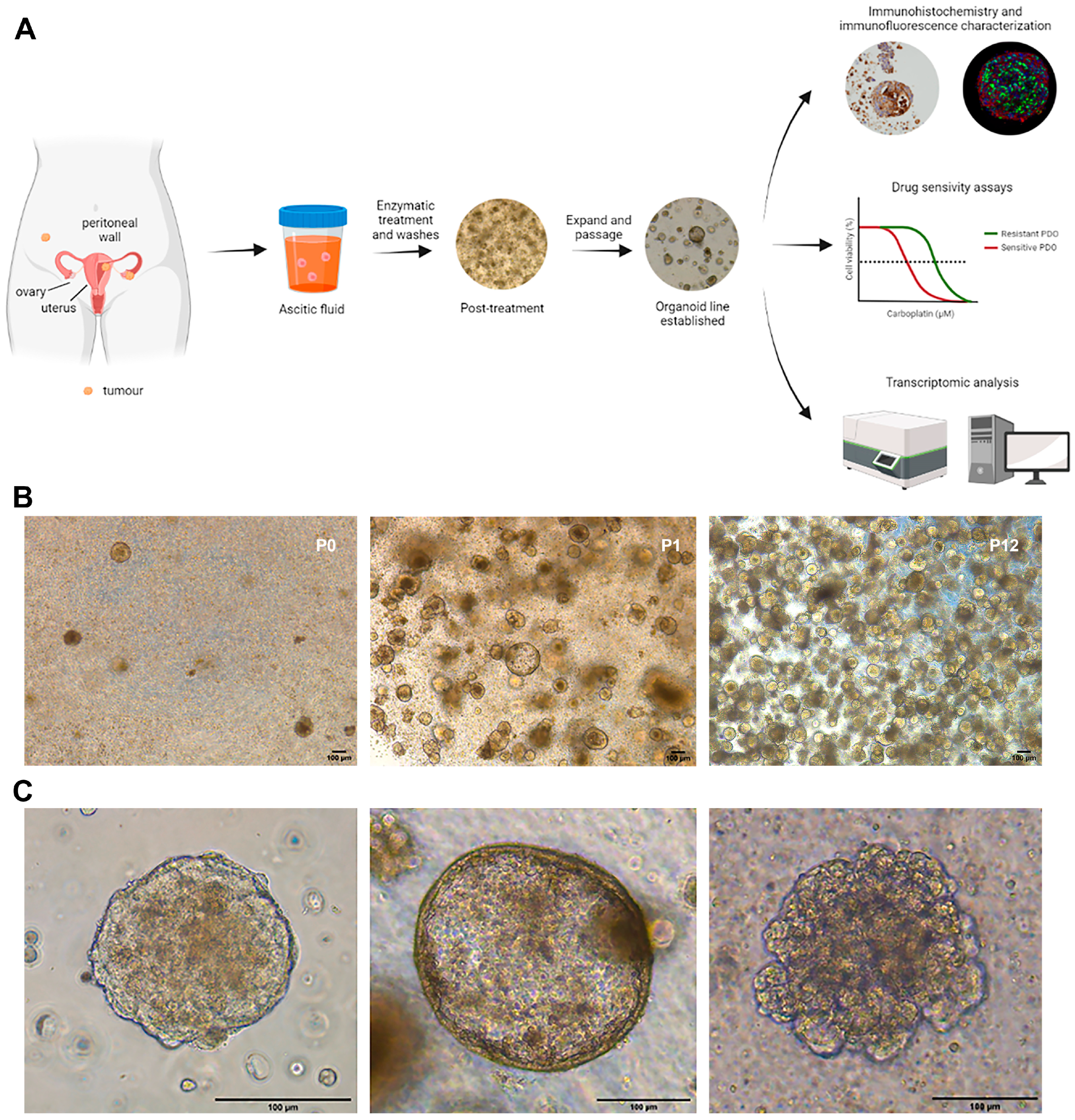
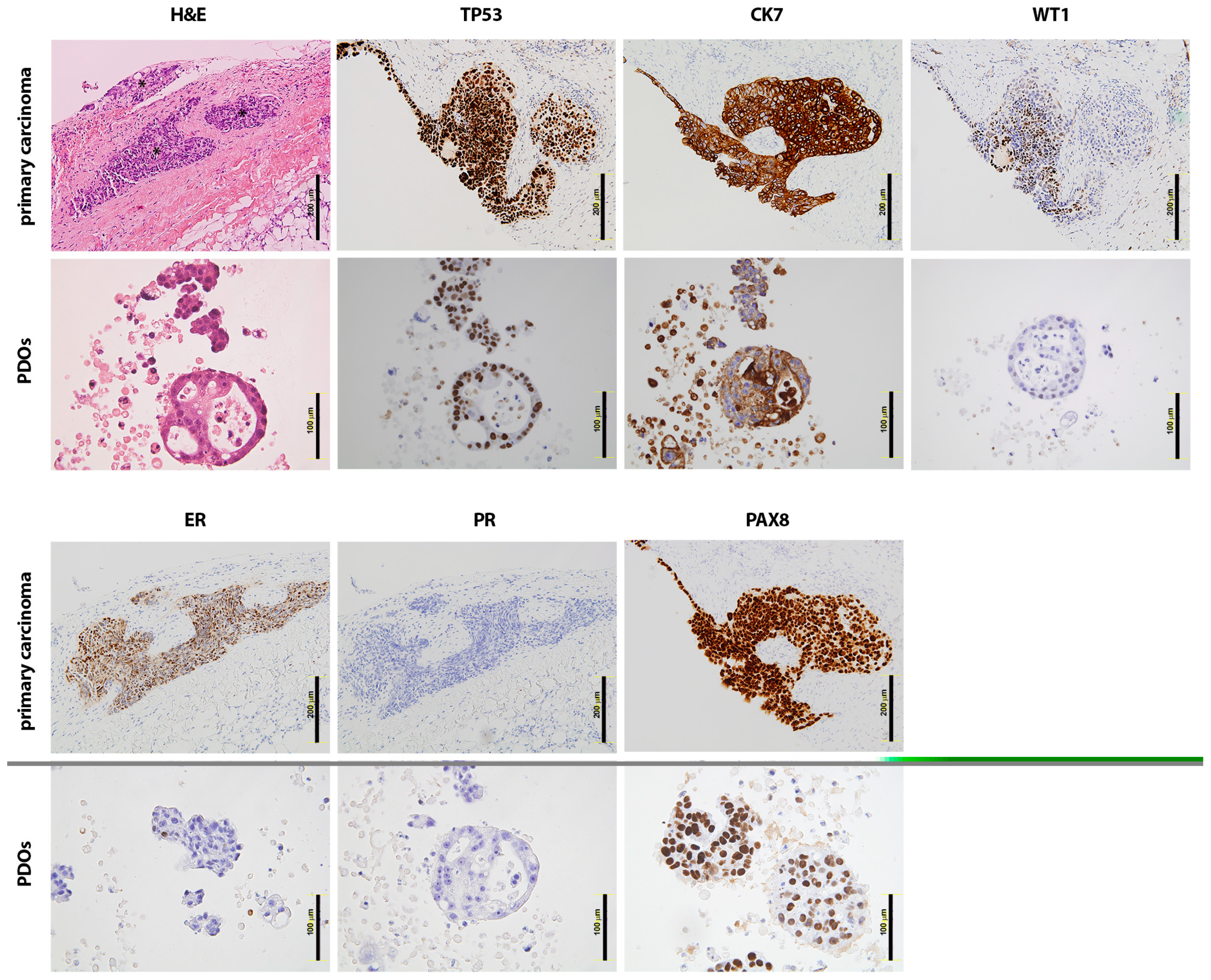
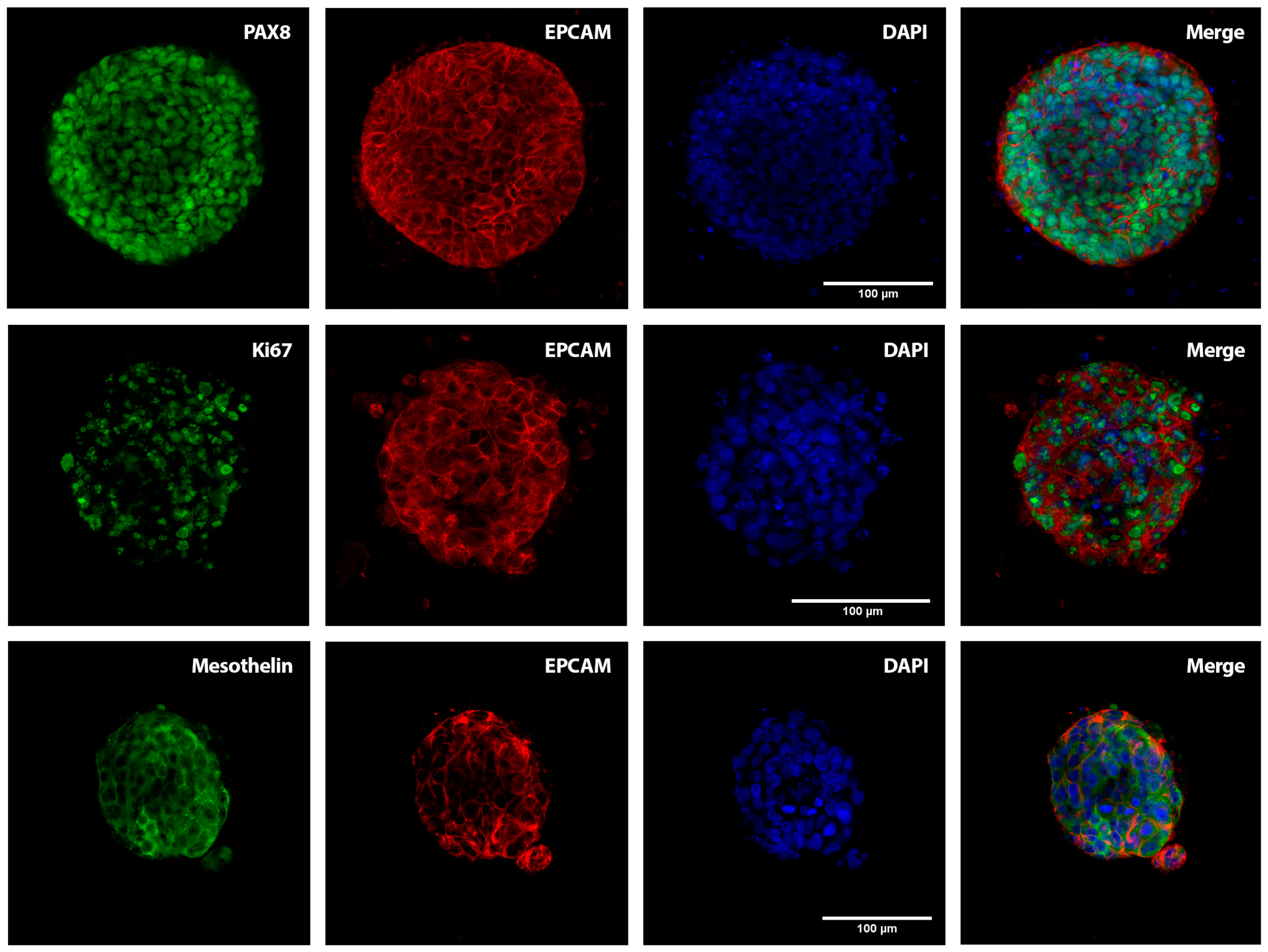
2.1. Patient-Derived Organoids Generated from the Ascites of GSC Patients Recapitulate the Histological Features of the Tumours of Origin
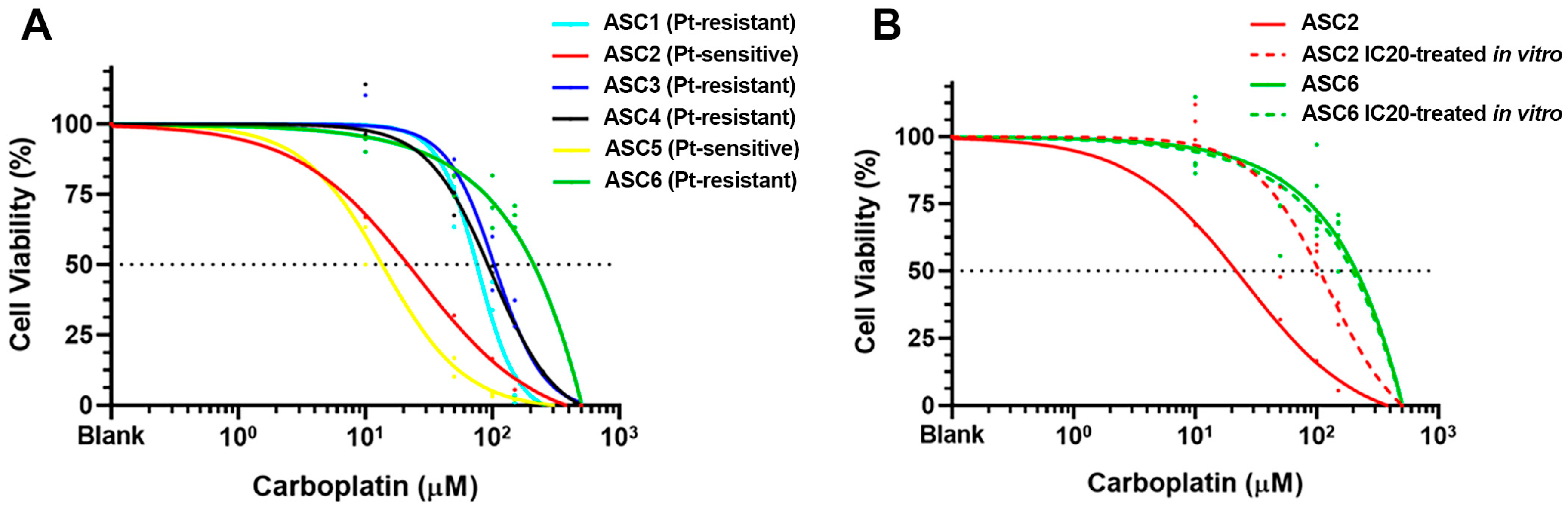
2.2. Sensitivity to Carboplatin in PDOs Generated from Platinum-Sensitive and Platinum-Resistant GSC Patients
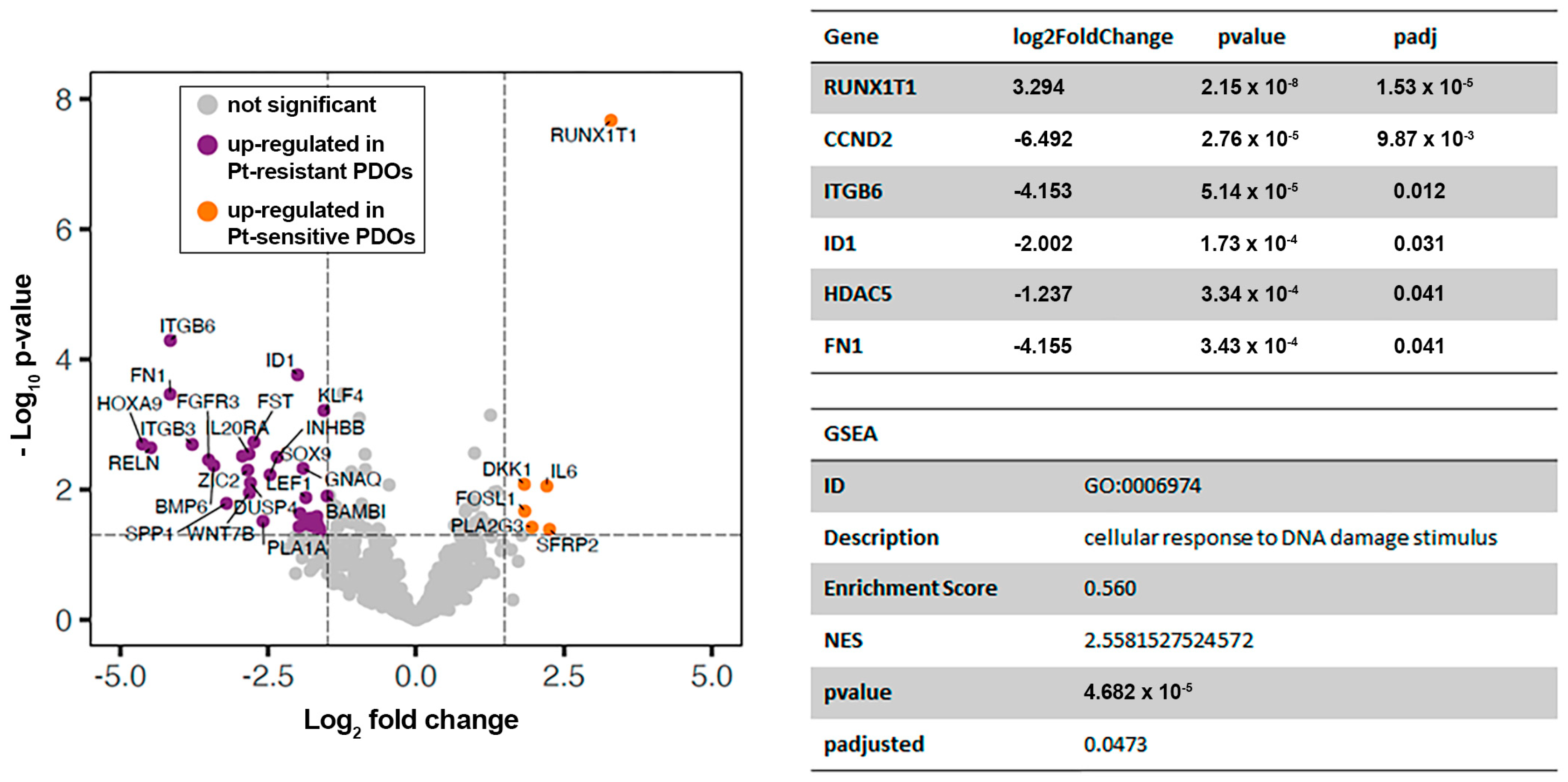
2.3. Comparative Transcriptomic Analysis of Platinum-Sensitive and Platinum-Resistant PDOs Generated from the Ascites of GSC Patients
3. Discussion
4. Materials and Methods
4.1. Establishing Organoid Cultures from Ascitic Fluids
4.2. Immunohistochemical Analysis
4.3. Immunofluorescence Analysis
4.4. Drug Sensitivity Assay
4.5. Nanostring nCounter Expression Assay
4.6. Differential Gene Expression Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Narod, S. Can Advanced-Stage Ovarian Cancer Be Cured? Nat. Rev. Clin. Oncol. 2016, 13, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Berek, J.S.; Renz, M.; Kehoe, S.; Kumar, L.; Friedlander, M. Cancer of the Ovary, Fallopian Tube, and Peritoneum: 2021 Update. Int. J. Gynaecol. Obstet. 2021, 155 (Suppl. S1), 61–85. [Google Scholar] [CrossRef] [PubMed]
- Harter, P.; Sehouli, J.; Vergote, I.; Ferron, G.; Reuss, A.; Meier, W.; Greggi, S.; Mosgaard, B.J.; Selle, F.; Guyon, F.; et al. Randomized Trial of Cytoreductive Surgery for Relapsed Ovarian Cancer. N. Engl. J. Med. 2021, 385, 2123–2131. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Savage, S.R.; Calinawan, A.P.; Lin, C.; Zhang, B.; Wang, P.; Starr, T.K.; Birrer, M.J.; Paulovich, A.G. A Highly Annotated Database of Genes Associated with Platinum Resistance in Cancer. Oncogene 2021, 40, 6395–6405. [Google Scholar] [CrossRef]
- Lengyel, E. Ovarian Cancer Development and Metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Kipps, E.; Tan, D.S.P.; Kaye, S.B. Meeting the Challenge of Ascites in Ovarian Cancer: New Avenues for Therapy and Research. Nat. Rev. Cancer 2013, 13, 273–282. [Google Scholar] [CrossRef]
- Nero, C.; Vizzielli, G.; Lorusso, D.; Cesari, E.; Daniele, G.; Loverro, M.; Scambia, G.; Sette, C. Patient-Derived Organoids and High Grade Serous Ovarian Cancer: From Disease Modeling to Personalized Medicine. J. Exp. Clin. Cancer Res. 2021, 40, 116. [Google Scholar] [CrossRef]
- Graham, O.; Rodriguez, J.; van Biljon, L.; Fashemi, B.; Graham, E.; Fuh, K.; Khabele, D.; Mullen, M. Generation and Culturing of High-Grade Serous Ovarian Cancer Patient-Derived Organoids. J. Vis. Exp. 2023, 191, e64878. [Google Scholar] [CrossRef]
- Maenhoudt, N.; Defraye, C.; Boretto, M.; Jan, Z.; Heremans, R.; Boeckx, B.; Hermans, F.; Arijs, I.; Cox, B.; Van Nieuwenhuysen, E.; et al. Developing Organoids from Ovarian Cancer as Experimental and Preclinical Models. Stem Cell Rep. 2020, 14, 717–729. [Google Scholar] [CrossRef]
- Boretto, M.; Maenhoudt, N.; Luo, X.; Hennes, A.; Boeckx, B.; Bui, B.; Heremans, R.; Perneel, L.; Kobayashi, H.; Van Zundert, I.; et al. Patient-Derived Organoids from Endometrial Disease Capture Clinical Heterogeneity and Are Amenable to Drug Screening. Nat. Cell Biol. 2019, 21, 1041–1051. [Google Scholar] [CrossRef]
- Nakamura, M.; Obata, T.; Daikoku, T.; Fujiwara, H. The Association and Significance of P53 in Gynecologic Cancers: The Potential of Targeted Therapy. Int. J. Mol. Sci. 2019, 20, 5482. [Google Scholar] [CrossRef]
- Fadare, O.; James, S.; Desouki, M.M.; Khabele, D. Coordinate Patterns of Estrogen Receptor, Progesterone Receptor, and Wilms Tumor 1 Expression in the Histopathologic Distinction of Ovarian from Endometrial Serous Adenocarcinomas. Ann. Diagn. Pathol. 2013, 17, 430–433. [Google Scholar] [CrossRef]
- Tong, G.-X.; Devaraj, K.; Hamele-Bena, D.; Yu, W.M.; Turk, A.; Chen, X.; Wright, J.D.; Greenebaum, E. Pax8: A Marker for Carcinoma of Müllerian Origin in Serous Effusions. Diagn. Cytopathol. 2011, 39, 567–574. [Google Scholar] [CrossRef]
- Giordano, G.; Ferioli, E.; Tafuni, A. The Role of Mesothelin Expression in Serous Ovarian Carcinoma: Impacts on Diagnosis, Prognosis, and Therapeutic Targets. Cancers 2022, 14, 2283. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Carballa, A.; Rivero-Calle, I.; Pardo-Seco, J.; Gómez-Rial, J.; Rivero-Velasco, C.; Rodríguez-Núñez, N.; Barbeito-Castiñeiras, G.; Pérez-Freixo, H.; Cebey-López, M.; Barral-Arca, R.; et al. A Multi-Tissue Study of Immune Gene Expression Profiling Highlights the Key Role of the Nasal Epithelium in COVID-19 Severity. Environ. Res. 2022, 210, 112890. [Google Scholar] [CrossRef] [PubMed]
- Baumann, K.H.; Wagner, U.; du Bois, A. The Changing Landscape of Therapeutic Strategies for Recurrent Ovarian Cancer. Future Oncol. 2012, 8, 1135–1147. [Google Scholar] [CrossRef]
- Choi, Y.P.; Shim, H.S.; Gao, M.-Q.; Kang, S.; Cho, N.H. Molecular Portraits of Intratumoral Heterogeneity in Human Ovarian Cancer. Cancer Lett. 2011, 307, 62–71. [Google Scholar] [CrossRef]
- Duesberg, P.; Stindl, R.; Hehlmann, R. Explaining the High Mutation Rates of Cancer Cells to Drug and Multidrug Resistance by Chromosome Reassortments That Are Catalyzed by Aneuploidy. Proc. Natl. Acad. Sci. USA 2000, 97, 14295–14300. [Google Scholar] [CrossRef]
- Cooke, S.L.; Ng, C.K.Y.; Melnyk, N.; Garcia, M.J.; Hardcastle, T.; Temple, J.; Langdon, S.; Huntsman, D.; Brenton, J.D. Genomic Analysis of Genetic Heterogeneity and Evolution in High-Grade Serous Ovarian Carcinoma. Oncogene 2010, 29, 4905–4913. [Google Scholar] [CrossRef]
- Damia, G.; Broggini, M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers 2019, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Ciucci, A.; Buttarelli, M.; Fagotti, A.; Scambia, G.; Gallo, D. Preclinical Models of Epithelial Ovarian Cancer: Practical Considerations and Challenges for a Meaningful Application. Cell Mol. Life Sci. 2022, 79, 364. [Google Scholar] [CrossRef] [PubMed]
- Psilopatis, I.; Sykaras, A.G.; Mandrakis, G.; Vrettou, K.; Theocharis, S. Patient-Derived Organoids: The Beginning of a New Era in Ovarian Cancer Disease Modeling and Drug Sensitivity Testing. Biomedicines 2022, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Kopper, O.; de Witte, C.J.; Lõhmussaar, K.; Valle-Inclan, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An Organoid Platform for Ovarian Cancer Captures Intra- and Interpatient Heterogeneity. Nat. Med. 2019, 25, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef]
- Spagnol, G.; Sensi, F.; De Tommasi, O.; Marchetti, M.; Bonaldo, G.; Xhindoli, L.; Noventa, M.; Agostini, M.; Tozzi, R.; Saccardi, C. Patient Derived Organoids (PDOs), Extracellular Matrix (ECM), Tumor Microenvironment (TME) and Drug Screening: State of the Art and Clinical Implications of Ovarian Cancer Organoids in the Era of Precision Medicine. Cancers 2023, 15, 2059. [Google Scholar] [CrossRef]
- Chen, H.; Gotimer, K.; De Souza, C.; Tepper, C.G.; Karnezis, A.N.; Leiserowitz, G.S.; Chien, J.; Smith, L.H. Short-Term Organoid Culture for Drug Sensitivity Testing of High-Grade Serous Carcinoma. Gynecol. Oncol. 2020, 157, 783–792. [Google Scholar] [CrossRef]
- Helleman, J.; Jansen, M.P.H.M.; Span, P.N.; van Staveren, I.L.; Massuger, L.F.A.G.; Meijer-van Gelder, M.E.; Sweep, F.C.G.J.; Ewing, P.C.; van der Burg, M.E.L.; Stoter, G.; et al. Molecular Profiling of Platinum Resistant Ovarian Cancer. Int. J. Cancer 2006, 118, 1963–1971. [Google Scholar] [CrossRef]
- Hazlehurst, L.A.; Dalton, W.S. Mechanisms Associated with Cell Adhesion Mediated Drug Resistance (CAM-DR) in Hematopoietic Malignancies. Cancer Metastasis Rev. 2001, 20, 43–50. [Google Scholar] [CrossRef]
- Yoshihara, M.; Kajiyama, H.; Yokoi, A.; Sugiyama, M.; Koya, Y.; Yamakita, Y.; Liu, W.; Nakamura, K.; Moriyama, Y.; Yasui, H.; et al. Ovarian Cancer-Associated Mesothelial Cells Induce Acquired Platinum-Resistance in Peritoneal Metastasis via the FN1/Akt Signaling Pathway. Int. J. Cancer 2020, 146, 2268–2280. [Google Scholar] [CrossRef]
- Sonego, M.; Pellizzari, I.; Dall’Acqua, A.; Pivetta, E.; Lorenzon, I.; Benevol, S.; Bomben, R.; Spessotto, P.; Sorio, R.; Gattei, V.; et al. Common Biological Phenotypes Characterize the Acquisition of Platinum-Resistance in Epithelial Ovarian Cancer Cells. Sci. Rep. 2017, 7, 7104. [Google Scholar] [CrossRef]
- Meng, J.; Liu, K.; Shao, Y.; Feng, X.; Ji, Z.; Chang, B.; Wang, Y.; Xu, L.; Yang, G. ID1 Confers Cancer Cell Chemoresistance through STAT3/ATF6-Mediated Induction of Autophagy. Cell Death Dis. 2020, 11, 137. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xing, H.; Lu, D.; Wang, J.; Li, B.; Tang, J.; Gu, F.; Hong, L. Role of Jagged1/STAT3 Signalling in Platinum-Resistant Ovarian Cancer. J. Cell Mol. Med. 2019, 23, 4005–4018. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Verma, V.; Mohania, D.; Gupta, S.; Babbar, A.K.; Rathi, B.; Dhanda, R.S.; Yadav, M. Leukemia Associated RUNX1T1 Gene Reduced Proliferation and Invasiveness of Glioblastoma Cells. J. Cell Biochem. 2021, 122, 1737–1748. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qiu, J.-G.; Jia, X.-Y.; Ke, Y.; Zhang, M.-K.; Stieg, D.; Liu, W.-J.; Liu, L.-Z.; Wang, L.; Jiang, B.-H. METTL3-Mediated N6-Methyladenosine Modification and HDAC5/YY1 Promote IFFO1 Downregulation in Tumor Development and Chemo-Resistance. Cancer Lett. 2023, 553, 215971. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, H.-J.; Li, Y.-Y.; Wang, X.; Liu, X.-X.; Mai, J. Molecular Mechanisms of Platinum-based Chemotherapy Resistance in Ovarian Cancer (Review). Oncol. Rep. 2022, 47, 82. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Rajal, A.; Marzec, K.A.; McCloy, R.A.; Nobis, M.; Chin, V.; Hastings, J.F.; Lai, K.; Kennerson, M.; Hughes, W.E.; Vaghjiani, V.; et al. A Non-Genetic, Cell Cycle-Dependent Mechanism of Platinum Resistance in Lung Adenocarcinoma. eLife 2021, 10, e65234. [Google Scholar] [CrossRef]
- Casas-Arozamena, C.; Cortegoso, A.; Piñeiro-Perez, R.; Abalo, A.; Arias, E.; Sampayo, V.; Vilar, A.; Bouso, M.; Diaz, E.; Moreno-Bueno, G.; et al. Improving the Management of Endometrial Cancer Patients through the Use of Liquid Biopsy Analyses: A Case Report. Int. J. Mol. Sci. 2022, 23, 8539. [Google Scholar] [CrossRef]
- Liston, D.R.; Davis, M. Clinically Relevant Concentrations of Anticancer Drugs: A Guide for Nonclinical Studies. Clin. Cancer Res. 2017, 23, 3489–3498. [Google Scholar] [CrossRef]
- Berg, H.F.; Hjelmeland, M.E.; Lien, H.; Espedal, H.; Fonnes, T.; Srivastava, A.; Stokowy, T.; Strand, E.; Bozickovic, O.; Stefansson, I.M.; et al. Patient-derived organoids reflect the genetic profile of endometrial tumors and predict patient prognosis. Commun. Med. 2021, 1, 20. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | Diagnosis a | Lines of Therapy | End of Treatment | Date of Progression | Date of Collection of the Sample | Organoid Morphology |
|---|---|---|---|---|---|---|
| ASC1 | HGSOC IV | 1st line: CarboTaxol 2nd line: Niraparib | 07/2022 02/2023 | 08/2022 | 25/02/2022 | Dense/low-cohesive |
| ASC2 | HGPPC | 1st line: CarboTaxol 2nd line: Bevacizumab | 09/2022 10/2022 | - | 29/03/2022 | Dense/cystic |
| ASC3 | HGSOC IIIB-IV | 1st line: CarboTaxol 2nd line: Caelyx 3rd line: Paclitaxel | 09/2022 01/2023 04/2023 | 11/2022 | 15/12/2022 | Dense |
| ASC4 | (OA) IIIB | 1st line: CarboTaxol 2nd line: Carbo-Caelyx 3rd line: Niraparib 4th line: Carboplatin | 01/2021 12/2021 05/2022 06/2022 | 09/2021 | 12/08/2022 | Dense/low-cohesive |
| ASC5 | USC IV | 1st line: CarboTaxol | 07/2022 | Dissociated response | 13/12/2022 | Dense |
| ASC6 | USC III | 1st line: CarboTaxol 2nd line: CarboTaxol | 02/2022 12/2021 | 06/2022 | b (05/08/2021) | Dense |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arias-Diaz, A.E.; Ferreiro-Pantin, M.; Barbazan, J.; Perez-Beliz, E.; Ruiz-Bañobre, J.; Casas-Arozamena, C.; Muinelo-Romay, L.; Lopez-Lopez, R.; Vilar, A.; Curiel, T.; et al. Ascites-Derived Organoids to Depict Platinum Resistance in Gynaecological Serous Carcinomas. Int. J. Mol. Sci. 2023, 24, 13208. https://doi.org/10.3390/ijms241713208
Arias-Diaz AE, Ferreiro-Pantin M, Barbazan J, Perez-Beliz E, Ruiz-Bañobre J, Casas-Arozamena C, Muinelo-Romay L, Lopez-Lopez R, Vilar A, Curiel T, et al. Ascites-Derived Organoids to Depict Platinum Resistance in Gynaecological Serous Carcinomas. International Journal of Molecular Sciences. 2023; 24(17):13208. https://doi.org/10.3390/ijms241713208
Chicago/Turabian StyleArias-Diaz, Andrea Estrella, Miriam Ferreiro-Pantin, Jorge Barbazan, Edurne Perez-Beliz, Juan Ruiz-Bañobre, Carlos Casas-Arozamena, Laura Muinelo-Romay, Rafael Lopez-Lopez, Ana Vilar, Teresa Curiel, and et al. 2023. "Ascites-Derived Organoids to Depict Platinum Resistance in Gynaecological Serous Carcinomas" International Journal of Molecular Sciences 24, no. 17: 13208. https://doi.org/10.3390/ijms241713208