
Hormonal Imbalances in Prader–Willi and Schaaf–Yang Syndromes Imply the Evolution of Specific Regulation of Hypothalamic Neuroendocrine Function in Mammals
 and
and
Abstract
:1. Introduction
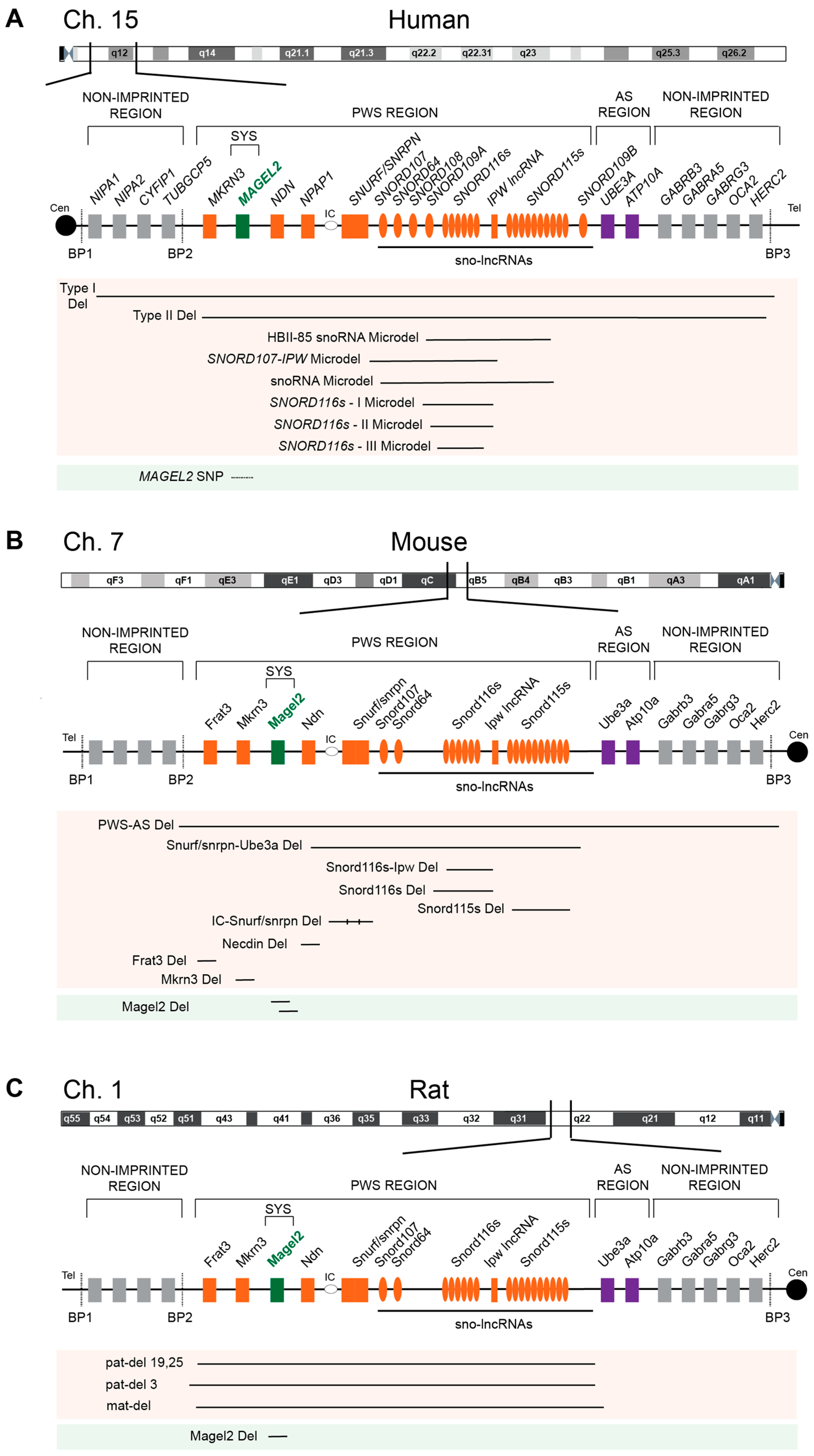
2. PWS-Associated Genes, Their Imprinting, and Expression Pattern
2.1. MKRN3
2.2. MAGEL2
2.3. NECDIN
2.4. NPAP1
2.5. SNURF/SNRPN
2.6. SNORD116
2.7. Genomic Imprinting
2.8. Expression Pattern
3. Hormonal Imbalance in PWS and SYS on an Organismal Level
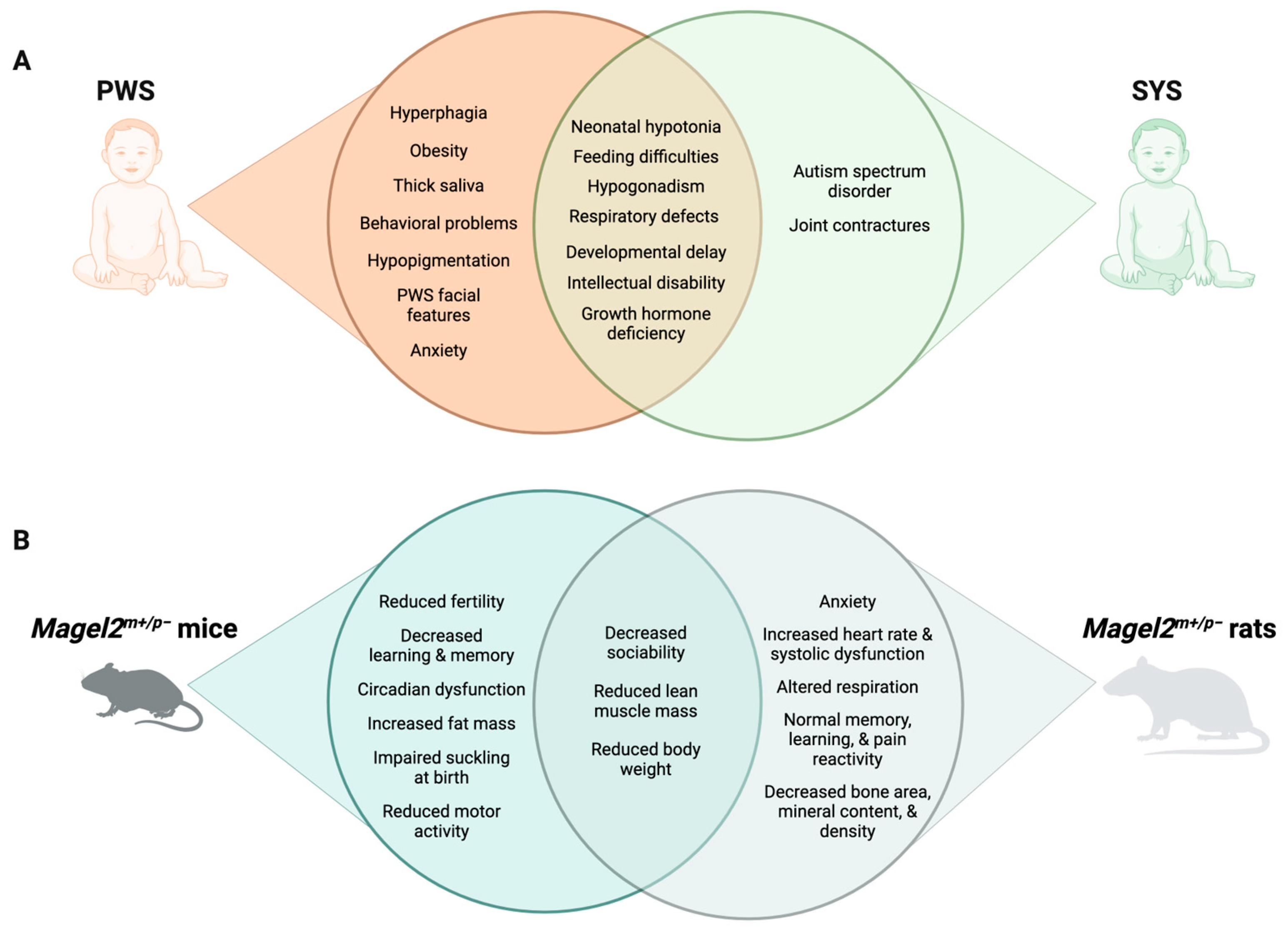
3.1. Symptoms and Treatment of PWS
3.2. Symptoms and Treatment of SYS
4. Hormonal Imbalance in PWS and SYS on a Molecular Level
4.1. Growth Hormone
4.2. Hypogonadism and GnRH
4.3. Hypothyroidism
4.4. Adrenal Insufficiency
4.5. Ghrelin
4.6. Oxytocin and Other Neuropeptides
5. MAGEL2 Regulates the Recycling of Core Components of Secretory Granules in the Hypothalamus and Enables Robust Endocrine Regulation
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef]
- Butler, M.G.; Hartin, S.N.; Hossain, W.A.; Manzardo, A.M.; Kimonis, V.; Dykens, E.; Gold, J.A.; Kim, S.J.; Weisensel, N.; Tamura, R.; et al. Molecular genetic classification in Prader-Willi syndrome: A multisite cohort study. J. Med. Genet. 2019, 56, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, R.D.; Knepper, J.L. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu. Rev. Genom. Hum. Genet. 2001, 2, 153–175. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G. Prader-Willi Syndrome and Chromosome 15q11.2 BP1-BP2 Region: A Review. Int. J. Mol. Sci. 2023, 24, 4271. [Google Scholar] [CrossRef] [PubMed]
- Marbach, F.; Elgizouli, M.; Rech, M.; Beygo, J.; Erger, F.; Velmans, C.; Stumpel, C.; Stegmann, A.P.A.; Beck-Wodl, S.; Gillessen-Kaesbach, G.; et al. The adult phenotype of Schaaf-Yang syndrome. Orphanet J. Rare Dis. 2020, 15, 294. [Google Scholar] [CrossRef]
- Driscoll, D.J.; Miller, J.L.; Cassidy, S.B. Prader-Willi Syndrome. In GeneReviews ((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Fon Tacer, K.; Potts, P.R. Cellular and disease functions of the Prader-Willi Syndrome gene MAGEL2. Biochem. J. 2017, 474, 2177–2190. [Google Scholar] [CrossRef]
- Schaaf, C.P.; Gonzalez-Garay, M.L.; Xia, F.; Potocki, L.; Gripp, K.W.; Zhang, B.; Peters, B.A.; McElwain, M.A.; Drmanac, R.; Beaudet, A.L.; et al. Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat. Genet. 2013, 45, 1405–1408. [Google Scholar] [CrossRef]
- Fountain, M.D.; Aten, E.; Cho, M.T.; Juusola, J.; Walkiewicz, M.A.; Ray, J.W.; Xia, F.; Yang, Y.; Graham, B.H.; Bacino, C.A.; et al. The phenotypic spectrum of Schaaf-Yang syndrome: 18 new affected individuals from 14 families. Genet. Med. 2017, 19, 45–52. [Google Scholar] [CrossRef]
- Koppes, E.A.; Johnson, M.A.; Moresco, J.J.; Luppi, P.; Lewis, D.W.; Stolz, D.B.; Diedrich, J.K.; Yates, J.R., 3rd; Wek, R.C.; Watkins, S.C.; et al. Insulin secretion deficits in a Prader-Willi syndrome beta-cell model are associated with a concerted downregulation of multiple endoplasmic reticulum chaperones. PLoS Genet. 2023, 19, e1010710. [Google Scholar] [CrossRef]
- Kummerfeld, D.M.; Raabe, C.A.; Brosius, J.; Mo, D.; Skryabin, B.V.; Rozhdestvensky, T.S. A Comprehensive Review of Genetically Engineered Mouse Models for Prader-Willi Syndrome Research. Int. J. Mol. Sci. 2021, 22, 3613. [Google Scholar] [CrossRef]
- Reznik, D.L.; Yang, M.V.; de la Haza, P.A.; Jain, A.; Spanjaard, M.; Theiss, S.; Schaaf, C.P.; Malovannaya, A.; Strong, T.V.; Veeraragavan, S.; et al. Magel2 truncation alters select behavioral and physiological outcomes in a rat model of Schaaf-Yang syndrome. Dis. Model. Mech. 2023, 16, dmm049829. [Google Scholar] [CrossRef]
- Meader, B.N.; Albano, A.; Sekizkardes, H.; Delaney, A. Heterozygous Deletions in MKRN3 Cause Central Precocious Puberty Without Prader-Willi Syndrome. J. Clin. Endocrinol. Metab. 2020, 105, 2732–2739. [Google Scholar] [CrossRef] [PubMed]
- Burnett, L.C.; LeDuc, C.A.; Sulsona, C.R.; Paull, D.; Rausch, R.; Eddiry, S.; Carli, J.F.; Morabito, M.V.; Skowronski, A.A.; Hubner, G.; et al. Deficiency in prohormone convertase PC1 impairs prohormone processing in Prader-Willi syndrome. J. Clin. Investig. 2017, 127, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Fon Tacer, K.; Montoya, M.C.; Oatley, M.J.; Lord, T.; Oatley, J.M.; Klein, J.; Ravichandran, R.; Tillman, H.; Kim, M.; Connelly, J.P.; et al. MAGE cancer-testis antigens protect the mammalian germline under environmental stress. Sci. Adv. 2019, 5, eaav4832. [Google Scholar] [CrossRef]
- Lee, A.K.; Klein, J.; Fon Tacer, K.; Lord, T.; Oatley, M.J.; Oatley, J.M.; Porter, S.N.; Pruett-Miller, S.M.; Tikhonova, E.B.; Karamyshev, A.L.; et al. Translational Repression of G3BP in Cancer and Germ Cells Suppresses Stress Granules and Enhances Stress Tolerance. Mol. Cell 2020, 79, 645–659.e9. [Google Scholar] [CrossRef]
- Florke Gee, R.R.; Chen, H.; Lee, A.K.; Daly, C.A.; Wilander, B.A.; Fon Tacer, K.; Potts, P.R. Emerging roles of the MAGE protein family in stress response pathways. J. Biol. Chem. 2020, 295, 16121–16155. [Google Scholar] [CrossRef]
- Su, A.I.; Cooke, M.P.; Ching, K.A.; Hakak, Y.; Walker, J.R.; Wiltshire, T.; Orth, A.P.; Vega, R.G.; Sapinoso, L.M.; Moqrich, A.; et al. Large-scale analysis of the human and mouse transcriptomes. Proc. Natl. Acad. Sci. USA 2002, 99, 4465–4470. [Google Scholar] [CrossRef]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G.; et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef] [PubMed]
- Cavaille, J.; Buiting, K.; Kiefmann, M.; Lalande, M.; Brannan, C.I.; Horsthemke, B.; Bachellerie, J.P.; Brosius, J.; Huttenhofer, A. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc. Natl. Acad. Sci. USA 2000, 97, 14311–14316. [Google Scholar] [CrossRef]
- Costa, R.A.; Ferreira, I.R.; Cintra, H.A.; Gomes, L.H.F.; Guida, L.D.C. Genotype-Phenotype Relationships and Endocrine Findings in Prader-Willi Syndrome. Front. Endocrinol. 2019, 10, 864. [Google Scholar] [CrossRef]
- Keshavarz, M.; Savriama, Y.; Refki, P.; Reeves, R.G.; Tautz, D. Natural copy number variation of tandemly repeated regulatory SNORD RNAs leads to individual phenotypic differences in mice. Mol. Ecol. 2021, 30, 4708–4722. [Google Scholar] [CrossRef] [PubMed]
- Kanber, D.; Giltay, J.; Wieczorek, D.; Zogel, C.; Hochstenbach, R.; Caliebe, A.; Kuechler, A.; Horsthemke, B.; Buiting, K. A paternal deletion of MKRN3, MAGEL2 and NDN does not result in Prader-Willi syndrome. Eur. J. Hum. Genet. 2009, 17, 582–590. [Google Scholar] [CrossRef]
- Bervini, S.; Herzog, H. Mouse models of Prader-Willi Syndrome: A systematic review. Front. Neuroendocr. 2013, 34, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Gray, T.A.; Saitoh, S.; Nicholls, R.D. An imprinted, mammalian bicistronic transcript encodes two independent proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 5616–5621. [Google Scholar] [CrossRef] [PubMed]
- Resnick, J.L.; Nicholls, R.D.; Wevrick, R.; Prader-Willi Syndrome Animal Models Working, G. Recommendations for the investigation of animal models of Prader-Willi syndrome. Mamm. Genome 2013, 24, 165–178. [Google Scholar] [CrossRef]
- Jong, M.T.; Gray, T.A.; Ji, Y.; Glenn, C.C.; Saitoh, S.; Driscoll, D.J.; Nicholls, R.D. A novel imprinted gene, encoding a RING zinc-finger protein, and overlapping antisense transcript in the Prader-Willi syndrome critical region. Hum. Mol. Genet. 1999, 8, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Jong, M.T.; Carey, A.H.; Caldwell, K.A.; Lau, M.H.; Handel, M.A.; Driscoll, D.J.; Stewart, C.L.; Rinchik, E.M.; Nicholls, R.D. Imprinting of a RING zinc-finger encoding gene in the mouse chromosome region homologous to the Prader-Willi syndrome genetic region. Hum. Mol. Genet. 1999, 8, 795–803. [Google Scholar] [CrossRef]
- Abreu, A.P.; Toro, C.A.; Song, Y.B.; Navarro, V.M.; Bosch, M.A.; Eren, A.; Liang, J.N.; Carroll, R.S.; Latronico, A.C.; Ronnekleiv, O.K.; et al. MKRN3 inhibits the reproductive axis through actions in kisspeptin-expressing neurons. J. Clin. Investig. 2020, 130, 4486–4500. [Google Scholar] [CrossRef] [PubMed]
- Gray, T.A.; Hernandez, L.; Carey, A.H.; Schaldach, M.A.; Smithwick, M.J.; Rus, K.; Marshall Graves, J.A.; Stewart, C.L.; Nicholls, R.D. The ancient source of a distinct gene family encoding proteins featuring RING and C(3)H zinc-finger motifs with abundant expression in developing brain and nervous system. Genomics 2000, 66, 76–86. [Google Scholar] [CrossRef]
- Macedo, D.B.; Abreu, A.P.; Reis, A.C.; Montenegro, L.R.; Dauber, A.; Beneduzzi, D.; Cukier, P.; Silveira, L.F.; Teles, M.G.; Carroll, R.S.; et al. Central precocious puberty that appears to be sporadic caused by paternally inherited mutations in the imprinted gene makorin ring finger 3. J. Clin. Endocrinol. Metab. 2014, 99, E1097–E1103. [Google Scholar] [CrossRef]
- Ludwig, N.G.; Radaeli, R.F.; Silva, M.M.; Romero, C.M.; Carrilho, A.J.; Bessa, D.; Macedo, D.B.; Oliveira, M.L.; Latronico, A.C.; Mazzuco, T.L. A boy with Prader-Willi syndrome: Unmasking precocious puberty during growth hormone replacement therapy. Arch. Endocrinol. Metab. 2016, 60, 596–600. [Google Scholar] [CrossRef]
- Lee, H.S.; Hwang, J.S. Central precocious puberty in a girl with Prader-Willi syndrome. J. Pediatr. Endocrinol. Metab. 2013, 26, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Han, T.; Li, Q.; Zhang, M.; Guo, R.; Yang, Y.; Lu, W.; Li, Z.; Peng, C.; Wu, P.; et al. MKRN3-mediated ubiquitination of Poly(A)-binding proteins modulates the stability and translation of GNRH1 mRNA in mammalian puberty. Nucleic Acids Res. 2021, 49, 3796–3813. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lu, W.; Yang, L.; Li, Z.; Zhou, X.; Guo, R.; Wang, J.; Wu, Z.; Dong, Z.; Ning, G.; et al. MKRN3 regulates the epigenetic switch of mammalian puberty via ubiquitination of MBD3. Natl. Sci. Rev. 2020, 7, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Naule, L.; Mancini, A.; Pereira, S.A.; Gassaway, B.M.; Lydeard, J.R.; Magnotto, J.C.; Kim, H.K.; Liang, J.; Matos, C.; Gygi, S.P.; et al. MKRN3 inhibits puberty onset via interaction with IGF2BP1 and regulation of hypothalamic plasticity. JCI Insight 2023, 8, e164178. [Google Scholar] [CrossRef]
- Liu, H.; Kong, X.; Chen, F. Mkrn3 functions as a novel ubiquitin E3 ligase to inhibit Nptx1 during puberty initiation. Oncotarget 2017, 8, 85102–85109. [Google Scholar] [CrossRef]
- Yellapragada, V.; Liu, X.; Lund, C.; Kansakoski, J.; Pulli, K.; Vuoristo, S.; Lundin, K.; Tuuri, T.; Varjosalo, M.; Raivio, T. MKRN3 Interacts With Several Proteins Implicated in Puberty Timing but Does Not Influence GNRH1 Expression. Front. Endocrinol. 2019, 10, 48. [Google Scholar] [CrossRef]
- Valadares, L.P.; Meireles, C.G.; De Toledo, I.P.; de Oliveira, R.S.; de Castro, L.C.G.; Abreu, A.P.; Carroll, R.S.; Latronico, A.C.; Kaiser, U.B.; Guerra, E.N.S.; et al. MKRN3 Mutations in Central Precocious Puberty: A Systematic Review and Meta-Analysis. J. Endocr. Soc. 2019, 3, 979–995. [Google Scholar] [CrossRef]
- Abreu, A.P.; Dauber, A.; Macedo, D.B.; Noel, S.D.; Brito, V.N.; Gill, J.C.; Cukier, P.; Thompson, I.R.; Navarro, V.M.; Gagliardi, P.C.; et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N. Engl. J. Med. 2013, 368, 2467–2475. [Google Scholar] [CrossRef]
- Doyle, J.M.; Gao, J.; Wang, J.; Yang, M.; Potts, P.R. MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol. Cell 2010, 39, 963–974. [Google Scholar] [CrossRef]
- Hao, Y.H.; Fountain, M.D., Jr.; Fon Tacer, K.; Xia, F.; Bi, W.; Kang, S.H.; Patel, A.; Rosenfeld, J.A.; Le Caignec, C.; Isidor, B.; et al. USP7 Acts as a Molecular Rheostat to Promote WASH-Dependent Endosomal Protein Recycling and Is Mutated in a Human Neurodevelopmental Disorder. Mol. Cell 2015, 59, 956–969. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.H.; Doyle, J.M.; Ramanathan, S.; Gomez, T.S.; Jia, D.; Xu, M.; Chen, Z.J.; Billadeau, D.D.; Rosen, M.K.; Potts, P.R. Regulation of WASH-dependent actin polymerization and protein trafficking by ubiquitination. Cell 2013, 152, 1051–1064. [Google Scholar] [CrossRef] [PubMed]
- Schaller, F.; Watrin, F.; Sturny, R.; Massacrier, A.; Szepetowski, P.; Muscatelli, F. A single postnatal injection of oxytocin rescues the lethal feeding behaviour in mouse newborns deficient for the imprinted Magel2 gene. Hum. Mol. Genet. 2010, 19, 4895–4905. [Google Scholar] [CrossRef]
- Wijesuriya, T.M.; De Ceuninck, L.; Masschaele, D.; Sanderson, M.R.; Carias, K.V.; Tavernier, J.; Wevrick, R. The Prader-Willi syndrome proteins MAGEL2 and necdin regulate leptin receptor cell surface abundance through ubiquitination pathways. Hum. Mol. Genet. 2017, 26, 4215–4230. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Dong, Y.; Li, J.D. Necdin regulates BMAL1 stability and circadian clock through SGT1-HSP90 chaperone machinery. Nucleic Acids Res. 2020, 48, 7944–7957. [Google Scholar] [CrossRef] [PubMed]
- Kuwajima, T.; Nishimura, I.; Yoshikawa, K. Necdin promotes GABAergic neuron differentiation in cooperation with Dlx homeodomain proteins. J. Neurosci. 2006, 26, 5383–5392. [Google Scholar] [CrossRef]
- Miller, N.L.; Wevrick, R.; Mellon, P.L. Necdin, a Prader-Willi syndrome candidate gene, regulates gonadotropin-releasing hormone neurons during development. Hum. Mol. Genet. 2009, 18, 248–260. [Google Scholar] [CrossRef]
- Napolitano, L.; Barone, B.; Morra, S.; Celentano, G.; La Rocca, R.; Capece, M.; Morgera, V.; Turco, C.; Caputo, V.F.; Spena, G.; et al. Hypogonadism in Patients with Prader Willi Syndrome: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 1993. [Google Scholar] [CrossRef]
- Wu, R.N.; Hung, W.C.; Chen, C.T.; Tsai, L.P.; Lai, W.S.; Min, M.Y.; Wong, S.B. Firing activity of locus coeruleus noradrenergic neurons decreases in necdin-deficient mice, an animal model of Prader-Willi syndrome. J. Neurodev. Disord. 2020, 12, 21. [Google Scholar] [CrossRef]
- Gerard, M.; Hernandez, L.; Wevrick, R.; Stewart, C.L. Disruption of the mouse necdin gene results in early post-natal lethality. Nat. Genet. 1999, 23, 199–202. [Google Scholar] [CrossRef]
- Rieusset, A.; Schaller, F.; Unmehopa, U.; Matarazzo, V.; Watrin, F.; Linke, M.; Georges, B.; Bischof, J.; Dijkstra, F.; Bloemsma, M.; et al. Stochastic loss of silencing of the imprinted Ndn/NDN allele, in a mouse model and humans with prader-willi syndrome, has functional consequences. PLoS Genet. 2013, 9, e1003752. [Google Scholar] [CrossRef] [PubMed]
- Watrin, F.; Roeckel, N.; Lacroix, L.; Mignon, C.; Mattei, M.G.; Disteche, C.; Muscatelli, F. The mouse Necdin gene is expressed from the paternal allele only and lies in the 7C region of the mouse chromosome 7, a region of conserved synteny to the human Prader-Willi syndrome region. Eur. J. Hum. Genet. 1997, 5, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Muscatelli, F.; Abrous, D.N.; Massacrier, A.; Boccaccio, I.; Le Moal, M.; Cau, P.; Cremer, H. Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum. Mol. Genet. 2000, 9, 3101–3110. [Google Scholar] [CrossRef] [PubMed]
- Matarazzo, V.; Caccialupi, L.; Schaller, F.; Shvarev, Y.; Kourdougli, N.; Bertoni, A.; Menuet, C.; Voituron, N.; Deneris, E.; Gaspar, P.; et al. Necdin shapes serotonergic development and SERT activity modulating breathing in a mouse model for Prader-Willi syndrome. Elife 2017, 6, e32640. [Google Scholar] [CrossRef]
- Farber, C.; Gross, S.; Neesen, J.; Buiting, K.; Horsthemke, B. Identification of a testis-specific gene (C15orf2) in the Prader-Willi syndrome region on chromosome 15. Genomics 2000, 65, 174–183. [Google Scholar] [CrossRef]
- Neumann, L.C.; Markaki, Y.; Mladenov, E.; Hoffmann, D.; Buiting, K.; Horsthemke, B. The imprinted NPAP1/C15orf2 gene in the Prader-Willi syndrome region encodes a nuclear pore complex associated protein. Hum. Mol. Genet. 2012, 21, 4038–4048. [Google Scholar] [CrossRef]
- Neumann, L.C.; Feiner, N.; Meyer, A.; Buiting, K.; Horsthemke, B. The imprinted NPAP1 gene in the Prader-Willi syndrome region belongs to a POM121-related family of retrogenes. Genome Biol. Evol. 2014, 6, 344–351. [Google Scholar] [CrossRef]
- Jonkers, J.; van Amerongen, R.; van der Valk, M.; Robanus-Maandag, E.; Molenaar, M.; Destree, O.; Berns, A. In vivo analysis of Frat1 deficiency suggests compensatory activity of Frat3. Mech. Dev. 1999, 88, 183–194. [Google Scholar] [CrossRef]
- Glenn, C.C.; Saitoh, S.; Jong, M.T.; Filbrandt, M.M.; Surti, U.; Driscoll, D.J.; Nicholls, R.D. Gene structure, DNA methylation, and imprinted expression of the human SNRPN gene. Am. J. Hum. Genet. 1996, 58, 335–346. [Google Scholar]
- Cheon, C.K. Genetics of Prader-Willi syndrome and Prader-Will-Like syndrome. Ann. Pediatr. Endocrinol. Metab. 2016, 21, 126–135. [Google Scholar] [CrossRef]
- Carias, K.V.; Wevrick, R. Preclinical Testing in Translational Animal Models of Prader-Willi Syndrome: Overview and Gap Analysis. Mol. Ther. Methods Clin. Dev. 2019, 13, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; AlHumaidi, S.S.; Faqeih, E.A.; Pitel, B.A.; Lundquist, P.; Aypar, U. A novel deletion of SNURF/SNRPN exon 1 in a patient with Prader-Willi-like phenotype. Eur. J. Med. Genet. 2017, 60, 416–420. [Google Scholar] [CrossRef]
- Huang, Y.; Grand, K.; Kimonis, V.; Butler, M.G.; Jain, S.; Huang, A.Y.; Martinez-Agosto, J.A.; Nelson, S.F.; Sanchez-Lara, P.A. Mosaic de novo SNRPN gene variant associated with Prader-Willi syndrome. J. Med. Genet. 2022, 59, 719–722. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Basak, A. Proteins and proteases of Prader-Willi syndrome: A comprehensive review and perspectives. Biosci. Rep. 2022, 42, BSR20220610. [Google Scholar] [CrossRef] [PubMed]
- Bieth, E.; Eddiry, S.; Gaston, V.; Lorenzini, F.; Buffet, A.; Conte Auriol, F.; Molinas, C.; Cailley, D.; Rooryck, C.; Arveiler, B.; et al. Highly restricted deletion of the SNORD116 region is implicated in Prader-Willi Syndrome. Eur. J. Hum. Genet. 2015, 23, 252–255. [Google Scholar] [CrossRef]
- Duker, A.L.; Ballif, B.C.; Bawle, E.V.; Person, R.E.; Mahadevan, S.; Alliman, S.; Thompson, R.; Traylor, R.; Bejjani, B.A.; Shaffer, L.G.; et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur. J. Hum. Genet. 2010, 18, 1196–1201. [Google Scholar] [CrossRef]
- Sahoo, T.; del Gaudio, D.; German, J.R.; Shinawi, M.; Peters, S.U.; Person, R.E.; Garnica, A.; Cheung, S.W.; Beaudet, A.L. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat. Genet. 2008, 40, 719–721. [Google Scholar] [CrossRef]
- de Smith, A.J.; Purmann, C.; Walters, R.G.; Ellis, R.J.; Holder, S.E.; Van Haelst, M.M.; Brady, A.F.; Fairbrother, U.L.; Dattani, M.; Keogh, J.M.; et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum. Mol. Genet. 2009, 18, 3257–3265. [Google Scholar] [CrossRef]
- Kim, S.J.; Miller, J.L.; Kuipers, P.J.; German, J.R.; Beaudet, A.L.; Sahoo, T.; Driscoll, D.J. Unique and atypical deletions in Prader-Willi syndrome reveal distinct phenotypes. Eur. J. Hum. Genet. 2012, 20, 283–290. [Google Scholar] [CrossRef]
- Runte, M.; Huttenhofer, A.; Gross, S.; Kiefmann, M.; Horsthemke, B.; Buiting, K. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum. Mol. Genet. 2001, 10, 2687–2700. [Google Scholar] [CrossRef]
- Qi, Y.; Purtell, L.; Fu, M.; Lee, N.J.; Aepler, J.; Zhang, L.; Loh, K.; Enriquez, R.F.; Baldock, P.A.; Zolotukhin, S.; et al. Snord116 is critical in the regulation of food intake and body weight. Sci. Rep. 2016, 6, 18614. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Victor, A.K.; Klein, J.; Tacer, K.F.; Tai, D.J.; de Esch, C.; Nuttle, A.; Temirov, J.; Burnett, L.C.; Rosenbaum, M.; et al. Loss of MAGEL2 in Prader-Willi syndrome leads to decreased secretory granule and neuropeptide production. JCI Insight 2020, 5, e138576. [Google Scholar] [CrossRef] [PubMed]
- Polex-Wolf, J.; Lam, B.Y.; Larder, R.; Tadross, J.; Rimmington, D.; Bosch, F.; Cenzano, V.J.; Ayuso, E.; Ma, M.K.; Rainbow, K.; et al. Hypothalamic loss of Snord116 recapitulates the hyperphagia of Prader-Willi syndrome. J. Clin. Investig. 2018, 128, 960–969. [Google Scholar] [CrossRef]
- Polvora-Brandao, D.; Joaquim, M.; Godinho, I.; Aprile, D.; Alvaro, A.R.; Onofre, I.; Raposo, A.C.; Pereira de Almeida, L.; Duarte, S.T.; da Rocha, S.T. Loss of hierarchical imprinting regulation at the Prader-Willi/Angelman syndrome locus in human iPSCs. Hum. Mol. Genet. 2018, 27, 3999–4011. [Google Scholar] [CrossRef] [PubMed]
- Salminen, I.I.; Crespi, B.J.; Mokkonen, M. Baby food and bedtime: Evidence for opposite phenotypes from different genetic and epigenetic alterations in Prader-Willi and Angelman syndromes. SAGE Open Med. 2019, 7, 2050312118823585. [Google Scholar] [CrossRef]
- Barlow, D.P. Genomic imprinting: A mammalian epigenetic discovery model. Annu. Rev. Genet. 2011, 45, 379–403. [Google Scholar] [CrossRef]
- Chung, M.S.; Langouet, M.; Chamberlain, S.J.; Carmichael, G.G. Prader-Willi syndrome: Reflections on seminal studies and future therapies. Open Biol. 2020, 10, 200195. [Google Scholar] [CrossRef]
- Horsthemke, B.; Wagstaff, J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am. J. Med. Genet. A 2008, 146, 2041–2052. [Google Scholar] [CrossRef]
- Buiting, K.; Lich, C.; Cottrell, S.; Barnicoat, A.; Horsthemke, B. A 5-kb imprinting center deletion in a family with Angelman syndrome reduces the shortest region of deletion overlap to 880 bp. Hum. Genet. 1999, 105, 665–666. [Google Scholar] [CrossRef]
- Shemer, R.; Hershko, A.Y.; Perk, J.; Mostoslavsky, R.; Tsuberi, B.; Cedar, H.; Buiting, K.; Razin, A. The imprinting box of the Prader-Willi/Angelman syndrome domain. Nat. Genet. 2000, 26, 440–443. [Google Scholar] [CrossRef]
- Saitoh, S.; Wada, T. Parent-of-Origin Specific Histone Acetylation and Reactivation of a Key Imprinted Gene Locus in Prader-Willi Syndrome. Am. J. Hum. Genet. 2000, 66, 1958–1962. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, J.; Zynger, D.; Francke, U. In vivo Nuclease Hypersensitivity Studies Reveal Multiple Sites of Parental Origin-Dependent Differential Chromatin Conformation in the 150 Kb SNRPN Transcription Unit. Hum. Mol. Genet. 1999, 8, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Perk, J.; Makedonski, K.; Lande, L.; Cedar, H.; Razin, A.; Shemer, R. The imprinting mechanism of the Prader-Willi/Angelman regional control center. EMBO J. 2002, 21, 5807–5814. [Google Scholar] [CrossRef] [PubMed]
- Brant, J.O.; Riva, A.; Resnick, J.L.; Yang, T.P. Influence of the Prader-Willi syndrome imprinting center on the DNA methylation landscape in the mouse brain. Epigenetics 2014, 9, 1540–1556. [Google Scholar] [CrossRef] [PubMed]
- El-Maarri, O.; Buiting, K.; Peery, E.G.; Kroisel, P.M.; Balaban, B.; Wagner, K.; Urman, B.; Heyd, J.; Lich, C.; Brannan, C.I.; et al. Maternal methylation imprints on human chromosome 15 are established during or after fertilization. Nat. Genet. 2001, 27, 341–344. [Google Scholar] [CrossRef]
- Huntriss, J.; Hinkins, M.; Oliver, B.; Harris, S.E.; Beazley, J.C.; Rutherford, A.J.; Gosden, R.G.; Lanzendorf, S.E.; Picton, H.M. Expression of mRNAs for DNA methyltransferases and methyl-CpG-binding proteins in the human female germ line, preimplantation embryos, and embryonic stem cells. Mol. Reprod. Dev. 2004, 67, 323–336. [Google Scholar] [CrossRef]
- Kantor, B.; Kaufman, Y.; Makedonski, K.; Razin, A.; Shemer, R. Establishing the epigenetic status of the Prader–Willi/Angelman imprinting center in the gametes and embryo. Hum. Mol. Genet. 2004, 13, 2767–2779. [Google Scholar] [CrossRef]
- DuBose, A.J.; Smith, E.Y.; Johnstone, K.A.; Resnick, J.L. Temporal and developmental requirements for the Prader-Willi imprinting center. Proc. Natl. Acad. Sci. USA 2012, 109, 3446–3450. [Google Scholar] [CrossRef]
- Jiang, C.; Yang, Y.; Huang, C.; Whitelaw, B. Promoter characterization and functional association with placenta of porcine MAGEL2. Gene 2014, 547, 63–69. [Google Scholar] [CrossRef]
- Renfree, M.B.; Hore, T.A.; Shaw, G.; Graves, J.A.; Pask, A.J. Evolution of genomic imprinting: Insights from marsupials and monotremes. Annu. Rev. Genom. Hum. Genet. 2009, 10, 241–262. [Google Scholar] [CrossRef]
- Plasschaert, R.N.; Bartolomei, M.S. Genomic imprinting in development, growth, behavior and stem cells. Development 2014, 141, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Peters, J. The role of genomic imprinting in biology and disease: An expanding view. Nat. Rev. Genet. 2014, 15, 517–530. [Google Scholar] [CrossRef]
- Moore, T.; Haig, D. Genomic imprinting in mammalian development: A parental tug-of-war. Trends Genet. 1991, 7, 45–49. [Google Scholar] [CrossRef]
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C.; Erice Imprinting, G. Genomic Imprinting and Physiological Processes in Mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Gregg, C.; Zhang, J.; Weissbourd, B.; Luo, S.; Schroth, G.P.; Haig, D.; Dulac, C. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science 2010, 329, 643–648. [Google Scholar] [CrossRef]
- Higgs, M.J.; Hill, M.J.; John, R.M.; Isles, A.R. Systematic investigation of imprinted gene expression and enrichment in the mouse brain explored at single-cell resolution. BMC Genom. 2022, 23, 754. [Google Scholar] [CrossRef] [PubMed]
- Broad, K.D.; Keverne, E.B. Placental protection of the fetal brain during short-term food deprivation. Proc. Natl. Acad. Sci. USA 2011, 108, 15237–15241. [Google Scholar] [CrossRef]
- Davies, W.; Lynn, P.M.; Relkovic, D.; Wilkinson, L.S. Imprinted genes and neuroendocrine function. Front. Neuroendocr. 2008, 29, 413–427. [Google Scholar] [CrossRef]
- Xie, Y.; Dorsky, R.I. Development of the hypothalamus: Conservation, modification and innovation. Development 2017, 144, 1588–1599. [Google Scholar] [CrossRef]
- Butler, M.G. Prader-Willi syndrome: Current understanding of cause and diagnosis. Am. J. Med. Genet. 1990, 35, 319–332. [Google Scholar] [CrossRef]
- Miller, J.L.; Lynn, C.H.; Driscoll, D.C.; Goldstone, A.P.; Gold, J.A.; Kimonis, V.; Dykens, E.; Butler, M.G.; Shuster, J.J.; Driscoll, D.J. Nutritional phases in Prader-Willi syndrome. Am. J. Med. Genet. A 2011, 155, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Miller, J.L.; Forster, J.L. Prader-Willi Syndrome-Clinical Genetics, Diagnosis and Treatment Approaches: An Update. Curr. Pediatr. Rev. 2019, 15, 207–244. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.V.; Whittington, J.E.; Holland, A.J.; Boer, H.; Clarke, D.; Webb, T. Prevalence of, and risk factors for, physical ill-health in people with Prader-Willi syndrome: A population-based study. Dev. Med. Child Neurol. 2002, 44, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Kim, J.; Cho, S.Y.; Jin, D.K. Prevalence and risk factors for type 2 diabetes mellitus with Prader-Willi syndrome: A single center experience. Orphanet J. Rare Dis. 2017, 12, 146. [Google Scholar] [CrossRef] [PubMed]
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocrinol. Investig. 2015, 38, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Teke Kısa, P.; Güzel, O.; Arslan, N.; Demir, K. Positive effects of ketogenic diet on weight control in children with obesity due to Prader–Willi syndrome. Clin. Endocrinol. 2023, 98, 332–341. [Google Scholar] [CrossRef]
- Louveau, C.; Turtuluci, M.-C.; Consoli, A.; Poitou, C.; Coupaye, M.; Krebs, M.-O.; Chaumette, B.; Iftimovici, A. Prader–Willi syndrome: Symptoms and topiramate response in light of genetics. Front. Neurosci. 2023, 17, 1126970. [Google Scholar] [CrossRef]
- Bhargava, S.A.; Putnam, P.E.; Kocoshis, S.A.; Rowe, M.; Hanchett, J.M. Rectal bleeding in Prader-Willi syndrome. Pediatrics 1996, 97, 265–267. [Google Scholar] [CrossRef]
- Boer, H.; Holland, A.; Whittington, J.; Butler, J.; Webb, T.; Clarke, D. Psychotic illness in people with Prader Willi syndrome due to chromosome 15 maternal uniparental disomy. Lancet 2002, 359, 135–136. [Google Scholar] [CrossRef]
- Soni, S.; Whittington, J.; Holland, A.J.; Webb, T.; Maina, E.; Boer, H.; Clarke, D. The course and outcome of psychiatric illness in people with Prader-Willi syndrome: Implications for management and treatment. J. Intellect. Disabil. Res. 2007, 51, 32–42. [Google Scholar] [CrossRef]
- Soni, S.; Whittington, J.; Holland, A.J.; Webb, T.; Maina, E.N.; Boer, H.; Clarke, D. The phenomenology and diagnosis of psychiatric illness in people with Prader-Willi syndrome. Psychol. Med. 2008, 38, 1505–1514. [Google Scholar] [CrossRef]
- Bennett, J.A.; Germani, T.; Haqq, A.M.; Zwaigenbaum, L. Autism spectrum disorder in Prader-Willi syndrome: A systematic review. Am. J. Med. Genet. A 2015, 167, 2936–2944. [Google Scholar] [CrossRef]
- Butler, M.G.; Manzardo, A.M.; Heinemann, J.; Loker, C.; Loker, J. Causes of death in Prader-Willi syndrome: Prader-Willi Syndrome Association (USA) 40-year mortality survey. Genet. Med. 2017, 19, 635–642. [Google Scholar] [CrossRef]
- McCandless, S.E.; Marissa, S.; Yin, D.; Yeh, M.; Czado, S.; Aghsaei, S.; Li, J.W.; Francis, K.; Hadker, N.; Stafford, D.E.J. SUN-604 U.S. Prevalence & Mortality of Prader-Willi Syndrome: A Population-Based Study of Medical Claims. J. Endocr. Soc. 2020, 4, SUN-604. [Google Scholar] [CrossRef]
- Manzardo, A.M.; Loker, J.; Heinemann, J.; Loker, C.; Butler, M.G. Survival trends from the Prader-Willi Syndrome Association (USA) 40-year mortality survey. Genet. Med. 2018, 20, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Erhardt, E.; Molnar, D. Prader-Willi Syndrome: Possibilities of Weight Gain Prevention and Treatment. Nutrients 2022, 14, 1950. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.B.; Driscoll, D.J. Prader-Willi syndrome. Eur. J. Hum. Genet. 2009, 17, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Grugni, G.; Sartorio, A.; Crino, A. Growth hormone therapy for Prader-willi syndrome: Challenges and solutions. Ther. Clin. Risk Manag. 2016, 12, 873–881. [Google Scholar] [CrossRef]
- Tauber, M.; Diene, G. Chapter 26-Prader–Willi syndrome: Hormone therapies. In Handbook of Clinical Neurology; Swaab, D.F., Buijs, R.M., Lucassen, P.J., Salehi, A., Kreier, F., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; Volume 181, pp. 351–367. [Google Scholar]
- Fountain, M.D.; Schaaf, C.P. Prader-Willi Syndrome and Schaaf-Yang Syndrome: Neurodevelopmental Diseases Intersecting at the MAGEL2 Gene. Diseases 2016, 4, 2. [Google Scholar] [CrossRef]
- Schaaf, C.P.; Marbach, F. Schaaf-Yang Syndrome. In GeneReviews ((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- McCarthy, J.; Lupo, P.J.; Kovar, E.; Rech, M.; Bostwick, B.; Scott, D.; Kraft, K.; Roscioli, T.; Charrow, J.; Schrier Vergano, S.A.; et al. Schaaf-Yang syndrome overview: Report of 78 individuals. Am. J. Med. Genet. A 2018, 176, 2564–2574. [Google Scholar] [CrossRef]
- Mejlachowicz, D.; Nolent, F.; Maluenda, J.; Ranjatoelina-Randrianaivo, H.; Giuliano, F.; Gut, I.; Sternberg, D.; Laquerriere, A.; Melki, J. Truncating Mutations of MAGEL2, a Gene within the Prader-Willi Locus, Are Responsible for Severe Arthrogryposis. Am. J. Hum. Genet. 2015, 97, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Negishi, Y.; Kurosawa, K.; Takano, K.; Matsubara, K.; Nishiyama, T.; Saitoh, S. A nationwide survey of Schaaf-Yang syndrome in Japan. J. Hum. Genet. 2022, 67, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Dykens, E.M.; Lee, E.; Roof, E. Prader-Willi syndrome and autism spectrum disorders: An evolving story. J. Neurodev. Disord. 2011, 3, 225–237. [Google Scholar] [CrossRef]
- Buiting, K.; Di Donato, N.; Beygo, J.; Bens, S.; von der Hagen, M.; Hackmann, K.; Horsthemke, B. Clinical phenotypes of MAGEL2 mutations and deletions. Orphanet J. Rare Dis. 2014, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Matarazzo, V.; Muscatelli, F. Natural breaking of the maternal silence at the mouse and human imprinted Prader-Willi locus: A whisper with functional consequences. Rare Dis. 2013, 1, e27228. [Google Scholar] [CrossRef] [PubMed]
- Juriaans, A.F.; Kerkhof, G.F.; Garrelfs, M.; Trueba-Timmermans, D.; Hokken-Koelega, A.C.S. Schaaf-Yang syndrome: Clinical phenotype and effects of 4 years of growth hormone treatment. Horm. Res. Paediatr. 2023. [Google Scholar] [CrossRef]
- Hebach, N.R.; Caro, P.; Martin-Giacalone, B.A.; Lupo, P.J.; Marbach, F.; Choukair, D.; Schaaf, C.P. A retrospective analysis of growth hormone therapy in children with Schaaf-Yang syndrome. Clin. Genet. 2021, 100, 298–307. [Google Scholar] [CrossRef]
- McCarthy, J.M.; McCann-Crosby, B.M.; Rech, M.E.; Yin, J.; Chen, C.A.; Ali, M.A.; Nguyen, H.N.; Miller, J.L.; Schaaf, C.P. Hormonal, metabolic and skeletal phenotype of Schaaf-Yang syndrome: A comparison to Prader-Willi syndrome. J. Med. Genet. 2018, 55, 307–315. [Google Scholar] [CrossRef]
- Tauber, M.; Hoybye, C. Endocrine disorders in Prader-Willi syndrome: A model to understand and treat hypothalamic dysfunction. Lancet Diabetes Endocrinol. 2021, 9, 235–246. [Google Scholar] [CrossRef]
- Alves, C.; Franco, R.R. Prader-Willi syndrome: Endocrine manifestations and management. Arch. Endocrinol. Metab. 2020, 64, 223–234. [Google Scholar] [CrossRef]
- Hirsch, H.J.; Eldar-Geva, T.; Bennaroch, F.; Pollak, Y.; Gross-Tsur, V. Sexual dichotomy of gonadal function in Prader-Willi syndrome from early infancy through the fourth decade. Hum. Reprod. 2015, 30, 2587–2596. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Kangawa, K. Ghrelin: Structure and function. Physiol. Rev. 2005, 85, 495–522. [Google Scholar] [CrossRef] [PubMed]
- Pellikaan, K.; Ben Brahim, Y.; Rosenberg, A.G.W.; Davidse, K.; Poitou, C.; Coupaye, M.; Goldstone, A.P.; Hoybye, C.; Markovic, T.P.; Grugni, G.; et al. Hypogonadism in Adult Males with Prader-Willi Syndrome-Clinical Recommendations Based on a Dutch Cohort Study, Review of the Literature and an International Expert Panel Discussion. J. Clin. Med. 2021, 10, 4361. [Google Scholar] [CrossRef] [PubMed]
- Turkkahraman, D.; Sirazi, E.C.; Aykal, G. Serum alpha-melanocyte-stimulating hormone (a-MSH), brain-derived neurotrophic factor (BDNF), and agouti-related protein (AGRP) levels in children with Prader-Willi or Bardet-Biedl syndromes. J. Endocrinol. Investig. 2022, 45, 1031–1037. [Google Scholar] [CrossRef]
- Correa-da-Silva, F.; Fliers, E.; Swaab, D.F.; Yi, C.X. Hypothalamic neuropeptides and neurocircuitries in Prader Willi syndrome. J. Neuroendocr. 2021, 33, e12994. [Google Scholar] [CrossRef]
- Bochukova, E.G.; Lawler, K.; Croizier, S.; Keogh, J.M.; Patel, N.; Strohbehn, G.; Lo, K.K.; Humphrey, J.; Hokken-Koelega, A.; Damen, L.; et al. A Transcriptomic Signature of the Hypothalamic Response to Fasting and BDNF Deficiency in Prader-Willi Syndrome. Cell Rep. 2018, 22, 3401–3408. [Google Scholar] [CrossRef]
- Bittel, D.C.; Kibiryeva, N.; McNulty, S.G.; Driscoll, D.J.; Butler, M.G.; White, R.A. Whole genome microarray analysis of gene expression in an imprinting center deletion mouse model of Prader-Willi syndrome. Am. J. Med. Genet. A 2007, 143, 422–429. [Google Scholar] [CrossRef]
- Stefan, M.; Ji, H.; Simmons, R.A.; Cummings, D.E.; Ahima, R.S.; Friedman, M.I.; Nicholls, R.D. Hormonal and metabolic defects in a prader-willi syndrome mouse model with neonatal failure to thrive. Endocrinology 2005, 146, 4377–4385. [Google Scholar] [CrossRef]
- Gajewska, J.; Szamotulska, K.; Klemarczyk, W.; Chelchowska, M.; Strucinska, M.; Ambroszkiewicz, J. Circulating Levels of Nesfatin-1 and Spexin in Children with Prader-Willi Syndrome during Growth Hormone Treatment and Dietary Intervention. Nutrients 2023, 15, 1240. [Google Scholar] [CrossRef]
- Bueno, M.; Esteba-Castillo, S.; Novell, R.; Gimenez-Palop, O.; Coronas, R.; Gabau, E.; Corripio, R.; Baena, N.; Vinas-Jornet, M.; Guitart, M.; et al. Lack of Postprandial Peak in Brain-Derived Neurotrophic Factor in Adults with Prader-Willi Syndrome. PLoS ONE 2016, 11, e0163468. [Google Scholar] [CrossRef]
- Eddiry, S.; Diene, G.; Molinas, C.; Salles, J.; Auriol, F.C.; Gennero, I.; Bieth, E.; Skryabin, B.V.; Rozhdestvensky, T.S.; Burnett, L.C.; et al. SNORD116 and growth hormone therapy impact IGFBP7 in Prader-Willi syndrome. Genet. Med. 2021, 23, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Bittel, D.C.; Talebizadeh, Z. Plasma peptide YY and ghrelin levels in infants and children with Prader-Willi syndrome. J. Pediatr. Endocrinol. Metab. 2004, 17, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Haqq, A.M.; Muehlbauer, M.; Svetkey, L.P.; Newgard, C.B.; Purnell, J.Q.; Grambow, S.C.; Freemark, M.S. Altered distribution of adiponectin isoforms in children with Prader-Willi syndrome (PWS): Association with insulin sensitivity and circulating satiety peptide hormones. Clin. Endocrinol. 2007, 67, 944–951. [Google Scholar] [CrossRef]
- Butler, M.G.; Nelson, T.A.; Driscoll, D.J.; Manzardo, A.M. Evaluation of Plasma Substance P and Beta-Endorphin Levels in Children with Prader-Willi Syndrome. J. Rare Disord. 2015, 3, 2. [Google Scholar]
- de Lind van Wijngaarden, R.F.; Otten, B.J.; Festen, D.A.; Joosten, K.F.; de Jong, F.H.; Sweep, F.C.; Hokken-Koelega, A.C. High prevalence of central adrenal insufficiency in patients with Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 1649–1654. [Google Scholar] [CrossRef]
- Nyunt, O.; Cotterill, A.M.; Archbold, S.M.; Wu, J.Y.; Leong, G.M.; Verge, C.F.; Crock, P.A.; Ambler, G.R.; Hofman, P.; Harris, M. Normal cortisol response on low-dose synacthen (1 microg) test in children with Prader Willi syndrome. J. Clin. Endocrinol. Metab. 2010, 95, E464–E467. [Google Scholar] [CrossRef]
- Grugni, G.; Beccaria, L.; Corrias, A.; Crino, A.; Cappa, M.; De Medici, C.; Di Candia, S.; Gargantini, L.; Ragusa, L.; Salvatoni, A.; et al. Central adrenal insufficiency in young adults with Prader-Willi syndrome. Clin. Endocrinol. 2013, 79, 371–378. [Google Scholar] [CrossRef]
- Cataldi, M.; Arnaldi, D.; Tucci, V.; De Carli, F.; Patti, G.; Napoli, F.; Pace, M.; Maghnie, M.; Nobili, L. Sleep disorders in Prader-Willi syndrome, evidence from animal models and humans. Sleep Med. Rev. 2021, 57, 101432. [Google Scholar] [CrossRef]
- Pace, M.; Falappa, M.; Freschi, A.; Balzani, E.; Berteotti, C.; Lo Martire, V.; Kaveh, F.; Hovig, E.; Zoccoli, G.; Amici, R.; et al. Loss of Snord116 impacts lateral hypothalamus, sleep, and food-related behaviors. JCI Insight 2020, 5, e137495. [Google Scholar] [CrossRef]
- Omokawa, M.; Ayabe, T.; Nagai, T.; Imanishi, A.; Omokawa, A.; Nishino, S.; Sagawa, Y.; Shimizu, T.; Kanbayashi, T. Decline of CSF orexin (hypocretin) levels in Prader-Willi syndrome. Am. J. Med. Genet. A 2016, 170, 1181–1186. [Google Scholar] [CrossRef]
- Manzardo, A.M.; Johnson, L.; Miller, J.L.; Driscoll, D.J.; Butler, M.G. Higher plasma orexin a levels in children with Prader-Willi syndrome compared with healthy unrelated sibling controls. Am. J. Med. Genet. A 2016, 170, 2328–2333. [Google Scholar] [CrossRef] [PubMed]
- Nevsimalova, S.; Vankova, J.; Stepanova, I.; Seemanova, E.; Mignot, E.; Nishino, S. Hypocretin deficiency in Prader-Willi syndrome. Eur. J. Neurol. 2005, 12, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.V.; Bogenpohl, J.W.; Howell, M.P.; Wevrick, R.; Panda, S.; Hogenesch, J.B.; Muglia, L.J.; Van Gelder, R.N.; Herzog, E.D.; Stewart, C.L. The imprinted gene Magel2 regulates normal circadian output. Nat. Genet. 2007, 39, 1266–1272. [Google Scholar] [CrossRef]
- Camerino, C. Oxytocin’s Regulation of Thermogenesis May Be the Link to Prader-Willi Syndrome. Curr. Issues Mol. Biol. 2023, 45, 4923–4935. [Google Scholar] [CrossRef]
- Miller, J.L.; Tamura, R.; Butler, M.G.; Kimonis, V.; Sulsona, C.; Gold, J.A.; Driscoll, D.J. Oxytocin treatment in children with Prader-Willi syndrome: A double-blind, placebo-controlled, crossover study. Am. J. Med. Genet. A 2017, 173, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Vaiani, E.; Herzovich, V.; Chaler, E.; Chertkoff, L.; Rivarola, M.A.; Torrado, M.; Belgorosky, A. Thyroid axis dysfunction in patients with Prader-Willi syndrome during the first 2 years of life. Clin. Endocrinol. 2010, 73, 546–550. [Google Scholar] [CrossRef]
- Sharkia, M.; Michaud, S.; Berthier, M.T.; Giguere, Y.; Stewart, L.; Deladoey, J.; Deal, C.; Van Vliet, G.; Chanoine, J.P. Thyroid function from birth to adolescence in Prader-Willi syndrome. J. Pediatr. 2013, 163, 800–805. [Google Scholar] [CrossRef]
- Lee, H.J.; Choe, Y.H.; Lee, J.H.; Sohn, Y.B.; Kim, S.J.; Park, S.W.; Son, J.S.; Kim, S.W.; Jin, D.K. Delayed response of amylin levels after an oral glucose challenge in children with Prader-Willi syndrome. Yonsei Med. J. 2011, 52, 257–262. [Google Scholar] [CrossRef]
- Butler, M.G.; Bittel, D.C. Plasma obestatin and ghrelin levels in subjects with Prader-Willi syndrome. Am. J. Med. Genet. A 2007, 143, 415–421. [Google Scholar] [CrossRef]
- Park, W.H.; Oh, Y.J.; Kim, G.Y.; Kim, S.E.; Paik, K.H.; Han, S.J.; Kim, A.H.; Chu, S.H.; Kwon, E.K.; Kim, S.W.; et al. Obestatin is not elevated or correlated with insulin in children with Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 229–234. [Google Scholar] [CrossRef]
- DelParigi, A.; Tschop, M.; Heiman, M.L.; Salbe, A.D.; Vozarova, B.; Sell, S.M.; Bunt, J.C.; Tataranni, P.A. High circulating ghrelin: A potential cause for hyperphagia and obesity in prader-willi syndrome. J. Clin. Endocrinol. Metab. 2002, 87, 5461–5464. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Li, H.H.; Zhang, S.; Solomon, N.M.; Camper, S.A.; Cohen, P.; Francke, U. SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PLoS ONE 2008, 3, e1709. [Google Scholar] [CrossRef] [PubMed]
- Eldar-Geva, T.; Hirsch, H.J.; Rabinowitz, R.; Benarroch, F.; Rubinstein, O.; Gross-Tsur, V. Primary ovarian dysfunction contributes to the hypogonadism in women with Prader-Willi Syndrome. Horm. Res. 2009, 72, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A.; Dahms, W.T.; Swerdloff, R.S.; Fiser, R.H.; Atkinson, R.L.; Carrel, R.E. The Prader-Willi syndrome: A study of 40 patients and a review of the literature. Medicine 1983, 62, 59–80. [Google Scholar] [CrossRef]
- Butler, M.G.; Theodoro, M.; Skouse, J.D. Thyroid function studies in Prader-Willi syndrome. Am. J. Med. Genet. A 2007, 143, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Festen, D.A.; Visser, T.J.; Otten, B.J.; Wit, J.M.; Duivenvoorden, H.J.; Hokken-Koelega, A.C. Thyroid hormone levels in children with Prader-Willi syndrome before and during growth hormone treatment. Clin. Endocrinol. 2007, 67, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Tauber, M.; Barbeau, C.; Jouret, B.; Pienkowski, C.; Malzac, P.; Moncla, A.; Rochiccioli, P. Auxological and endocrine evolution of 28 children with Prader-Willi syndrome: Effect of GH therapy in 14 children. Horm. Res. 2000, 53, 279–287. [Google Scholar] [CrossRef]
- McAlister, K.L.; Fisher, K.L.; Dumont-Driscoll, M.C.; Rubin, D.A. The relationship between metabolic syndrome, cytokines and physical activity in obese youth with and without Prader-Willi syndrome. J. Pediatr. Endocrinol. Metab. 2018, 31, 837–845. [Google Scholar] [CrossRef]
- Sohn, Y.B.; Kwak, M.J.; Kim, S.J.; Park, S.W.; Kim, C.H.; Kim, M.Y.; Kwon, E.K.; Paik, K.H.; Jin, D.K. Correlation of adiponectin receptor expression with cytokines and insulin sensitivity in growth hormone (GH)-treated children with Prader-Willi syndrome and in non-GH-treated obese children. J. Clin. Endocrinol. Metab. 2010, 95, 1371–1377. [Google Scholar] [CrossRef]
- Hoybye, C.; Bruun, J.M.; Richelsen, B.; Flyvbjerg, A.; Frystyk, J. Serum adiponectin levels in adults with Prader-Willi syndrome are independent of anthropometrical parameters and do not change with GH treatment. Eur. J. Endocrinol. 2004, 151, 457–461. [Google Scholar] [CrossRef]
- Pagano, C.; Marin, O.; Calcagno, A.; Schiappelli, P.; Pilon, C.; Milan, G.; Bertelli, M.; Fanin, E.; Andrighetto, G.; Federspil, G.; et al. Increased serum resistin in adults with prader-willi syndrome is related to obesity and not to insulin resistance. J. Clin. Endocrinol. Metab. 2005, 90, 4335–4340. [Google Scholar] [CrossRef] [PubMed]
- Haqq, A.M.; Muehlbauer, M.J.; Newgard, C.B.; Grambow, S.; Freemark, M. The metabolic phenotype of Prader-Willi syndrome (PWS) in childhood: Heightened insulin sensitivity relative to body mass index. J. Clin. Endocrinol. Metab. 2011, 96, E225–E232. [Google Scholar] [CrossRef]
- Lindgren, A.C.; Marcus, C.; Skwirut, C.; Elimam, A.; Hagenas, L.; Schalling, M.; Anvret, M.; Lonnqvist, F. Increased leptin messenger RNA and serum leptin levels in children with Prader-Willi syndrome and nonsyndromal obesity. Pediatr. Res. 1997, 42, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Moore, J.; Morawiecki, A.; Nicolson, M. Comparison of leptin protein levels in Prader-Willi syndrome and control individuals. Am. J. Med. Genet. 1998, 75, 7–12. [Google Scholar] [CrossRef]
- Goldstone, A.P.; Brynes, A.E.; Thomas, E.L.; Bell, J.D.; Frost, G.; Holland, A.; Ghatei, M.A.; Bloom, S.R. Resting metabolic rate, plasma leptin concentrations, leptin receptor expression, and adipose tissue measured by whole-body magnetic resonance imaging in women with Prader-Willi syndrome. Am. J. Clin. Nutr. 2002, 75, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Ma, Y.; Gu, M.; Zhang, Y.; Yan, S.; Li, N.; Wang, Y.; Ding, X.; Yin, J.; Fan, N.; et al. Spexin peptide is expressed in human endocrine and epithelial tissues and reduced after glucose load in type 2 diabetes. Peptides 2015, 71, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Tauber, M.; Cutfield, W. KIGS highlights: Growth hormone treatment in Prader-Willi Syndrome. Horm. Res. 2007, 68 (Suppl. S5), 48–50. [Google Scholar] [CrossRef]
- Noordam, C.; Hoybye, C.; Eiholzer, U. Prader-Willi Syndrome and Hypogonadism: A Review Article. Int. J. Mol. Sci. 2021, 22, 2705. [Google Scholar] [CrossRef]
- Mercer, R.E.; Wevrick, R. Loss of magel2, a candidate gene for features of Prader-Willi syndrome, impairs reproductive function in mice. PLoS ONE 2009, 4, e4291. [Google Scholar] [CrossRef]
- Li, K.; Zheng, X.; Tang, H.; Zang, Y.S.; Zeng, C.; Liu, X.; Shen, Y.; Pang, Y.; Wang, S.; Xie, F.; et al. E3 ligase MKRN3 is a tumor suppressor regulating PABPC1 ubiquitination in non-small cell lung cancer. J. Exp. Med. 2021, 218, e20210151. [Google Scholar] [CrossRef]
- Miller, J.L.; Goldstone, A.P.; Couch, J.A.; Shuster, J.; He, G.; Driscoll, D.J.; Liu, Y.; Schmalfuss, I.M. Pituitary abnormalities in Prader-Willi syndrome and early onset morbid obesity. Am. J. Med. Genet. A 2008, 146, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Nillni, E.A. Regulation of the hypothalamic thyrotropin releasing hormone (TRH) neuron by neuronal and peripheral inputs. Front. Neuroendocr. 2010, 31, 134–156. [Google Scholar] [CrossRef]
- Tennese, A.A.; Wevrick, R. Impaired hypothalamic regulation of endocrine function and delayed counterregulatory response to hypoglycemia in Magel2-null mice. Endocrinology 2011, 152, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Scaroni, C.; Ceccato, F.; Rizzati, S.; Mantero, F. Concomitant therapies (glucocorticoids and sex hormones) in adult patients with growth hormone deficiency. J. Endocrinol. Investig. 2008, 31, 61–65. [Google Scholar]
- Farholt, S.; Sode-Carlsen, R.; Christiansen, J.S.; Ostergaard, J.R.; Hoybye, C. Normal cortisol response to high-dose synacthen and insulin tolerance test in children and adults with Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2011, 96, E173–E180. [Google Scholar] [CrossRef] [PubMed]
- Cataletto, M.; Angulo, M.; Hertz, G.; Whitman, B. Prader-Willi syndrome: A primer for clinicians. Int. J. Pediatr. Endocrinol. 2011, 2011, 12. [Google Scholar] [CrossRef]
- Muller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef]
- Kweh, F.A.; Miller, J.L.; Sulsona, C.R.; Wasserfall, C.; Atkinson, M.; Shuster, J.J.; Goldstone, A.P.; Driscoll, D.J. Hyperghrelinemia in Prader-Willi syndrome begins in early infancy long before the onset of hyperphagia. Am. J. Med. Genet. A 2015, 167, 69–79. [Google Scholar] [CrossRef]
- Baribeau, D.A.; Anagnostou, E. Oxytocin and vasopressin: Linking pituitary neuropeptides and their receptors to social neurocircuits. Front. Neurosci. 2015, 9, 335. [Google Scholar] [CrossRef]
- Bachner-Melman, R.; Ebstein, R.P. The role of oxytocin and vasopressin in emotional and social behaviors. Handb. Clin. Neurol. 2014, 124, 53–68. [Google Scholar] [CrossRef]
- Swaab, D.F.; Purba, J.S.; Hofman, M.A. Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: A study of five cases. J. Clin. Endocrinol. Metab. 1995, 80, 573–579. [Google Scholar] [CrossRef]
- Hoybye, C.; Barkeling, B.; Espelund, U.; Petersson, M.; Thoren, M. Peptides associated with hyperphagia in adults with Prader-Willi syndrome before and during GH treatment. Growth Horm. IGF Res. 2003, 13, 322–327. [Google Scholar] [CrossRef]
- Johnson, L.; Manzardo, A.M.; Miller, J.L.; Driscoll, D.J.; Butler, M.G. Elevated plasma oxytocin levels in children with Prader-Willi syndrome compared with healthy unrelated siblings. Am. J. Med. Genet. A 2016, 170, 594–601. [Google Scholar] [CrossRef]
- Martin, A.; State, M.; Anderson, G.M.; Kaye, W.M.; Hanchett, J.M.; McConaha, C.W.; North, W.G.; Leckman, J.F. Cerebrospinal fluid levels of oxytocin in Prader-Willi syndrome: A preliminary report. Biol. Psychiatry 1998, 44, 1349–1352. [Google Scholar] [CrossRef] [PubMed]
- Dombret, C.; Nguyen, T.; Schakman, O.; Michaud, J.L.; Hardin-Pouzet, H.; Bertrand, M.J.; De Backer, O. Loss of Maged1 results in obesity, deficits of social interactions, impaired sexual behavior and severe alteration of mature oxytocin production in the hypothalamus. Hum. Mol. Genet. 2012, 21, 4703–4717. [Google Scholar] [CrossRef] [PubMed]
- Ates, T.; Oncul, M.; Dilsiz, P.; Topcu, I.C.; Civas, C.C.; Alp, M.I.; Aklan, I.; Ates Oz, E.; Yavuz, Y.; Yilmaz, B.; et al. Inactivation of Magel2 suppresses oxytocin neurons through synaptic excitation-inhibition imbalance. Neurobiol. Dis. 2019, 121, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Da Prato, L.C.; Zayan, U.; Abdallah, D.; Point, V.; Schaller, F.; Pallesi-Pocachard, E.; Montheil, A.; Canaan, S.; Gaiarsa, J.L.; Muscatelli, F.; et al. Early life oxytocin treatment improves thermo-sensory reactivity and maternal behavior in neonates lacking the autism-associated gene Magel2. Neuropsychopharmacology 2022, 47, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Bertoni, A.; Schaller, F.; Tyzio, R.; Gaillard, S.; Santini, F.; Xolin, M.; Diabira, D.; Vaidyanathan, R.; Matarazzo, V.; Medina, I.; et al. Oxytocin administration in neonates shapes hippocampal circuitry and restores social behavior in a mouse model of autism. Mol. Psychiatry 2021, 26, 7582–7595. [Google Scholar] [CrossRef]
- Rice, L.J.; Agu, J.; Carter, C.S.; Harris, J.C.; Nazarloo, H.P.; Naanai, H.; Einfeld, S.L. The relationship between endogenous oxytocin and vasopressin levels and the Prader-Willi syndrome behaviour phenotype. Front. Endocrinol. 2023, 14, 1183525. [Google Scholar] [CrossRef]
- Lopez, M.; Seoane, L.M.; Garcia Mdel, C.; Dieguez, C.; Senaris, R. Neuropeptide Y, but not agouti-related peptide or melanin-concentrating hormone, is a target peptide for orexin-A feeding actions in the rat hypothalamus. Neuroendocrinology 2002, 75, 34–44. [Google Scholar] [CrossRef]
- Hayashi, M.; Miyata, R.; Tanuma, N. Decrease in acetylcholinergic neurons in the pedunculopontine tegmental nucleus in a patient with Prader-Willi syndrome. Neuropathology 2011, 31, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Bischof, J.M.; Stewart, C.L.; Wevrick, R. Inactivation of the mouse Magel2 gene results in growth abnormalities similar to Prader-Willi syndrome. Hum. Mol. Genet. 2007, 16, 2713–2719. [Google Scholar] [CrossRef] [PubMed]
- Fountain, M.D.; Tao, H.; Chen, C.A.; Yin, J.; Schaaf, C.P. Magel2 knockout mice manifest altered social phenotypes and a deficit in preference for social novelty. Genes Brain Behav. 2017, 16, 592–600. [Google Scholar] [CrossRef]
- Yoon, H.; Enquist, L.W.; Dulac, C. Olfactory inputs to hypothalamic neurons controlling reproduction and fertility. Cell 2005, 123, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009, 10, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Seaman, M.N. The retromer complex-endosomal protein recycling and beyond. J. Cell Sci. 2012, 125, 4693–4702. [Google Scholar] [CrossRef] [PubMed]
- Borgonovo, B.; Ouwendijk, J.; Solimena, M. Biogenesis of secretory granules. Curr. Opin. Cell Biol. 2006, 18, 365–370. [Google Scholar] [CrossRef]
- Kogel, T.; Gerdes, H.H. Maturation of secretory granules. Results Probl. Cell Differ. 2010, 50, 1–20. [Google Scholar] [CrossRef]
- Wang, J.; Fedoseienko, A.; Chen, B.; Burstein, E.; Jia, D.; Billadeau, D.D. Endosomal receptor trafficking: Retromer and beyond. Traffic 2018, 19, 578–590. [Google Scholar] [CrossRef]
- Seaman, M.N.; Gautreau, A.; Billadeau, D.D. Retromer-mediated endosomal protein sorting: All WASHed up! Trends Cell Biol. 2013, 23, 522–528. [Google Scholar] [CrossRef]
- Myers, S.E.; Davis, A.; Whitman, B.Y.; Santiago, J.V.; Landt, M. Leptin concentrations in Prader-Willi syndrome before and after growth hormone replacement. Clin. Endocrinol. 2000, 52, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Teis, D. The ESCRT machinery. Curr. Biol. 2012, 22, R116–R120. [Google Scholar] [CrossRef]
- Mercer, R.E.; Michaelson, S.D.; Chee, M.J.; Atallah, T.A.; Wevrick, R.; Colmers, W.F. Magel2 is required for leptin-mediated depolarization of POMC neurons in the hypothalamic arcuate nucleus in mice. PLoS Genet. 2013, 9, e1003207. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.W.; Williams, K.W.; Ye, C.; Luo, J.; Balthasar, N.; Coppari, R.; Cowley, M.A.; Cantley, L.C.; Lowell, B.B.; Elmquist, J.K. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J. Clin. Investig. 2008, 118, 1796–1805. [Google Scholar] [CrossRef] [PubMed]
- Osborne, D.G.; Phillips-Krawczak, C.A.; Billadeau, D.D. Monitoring receptor trafficking following retromer and WASH deregulation. Methods Cell Biol. 2015, 130, 199–213. [Google Scholar] [CrossRef]
- Stern, C.; Schwarz, S.; Moser, G.; Cvitic, S.; Jantscher-Krenn, E.; Gauster, M.; Hiden, U. Placental Endocrine Activity: Adaptation and Disruption of Maternal Glucose Metabolism in Pregnancy and the Influence of Fetal Sex. Int. J. Mol. Sci. 2021, 22, 12722. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Tissue | Expression in PWS | References | |
|---|---|---|---|---|
| Patients | Mouse (Model) | |||
| Follicle-stimulating hormone (FSH) | Anterior pituitary | ↑ (variable in male and female) | -- | [49,134] |
| Growth hormone (GH) | Anterior pituitary | ↓ | ↓ (Magel2m+/pΔ) | [10,73,135] |
| Luteinizing hormone (LH) | Anterior pituitary | ↑ (variable in male and female) | ↓ (Magel2m+/pΔ) | [54,73,134,136] |
| Melanocyte-stimulating hormone (α-MSH, β-MSH) | Anterior pituitary | ↓ | ↓ (Magel2m+/pΔ) | [73,137,138] |
| Prolactin (PRL) | Anterior pituitary | -- | ↓ (Magel2m+/pΔ) | [73] |
| Proopiomelanocortin (POMC) * | Anterior pituitary | ↓ in CNS | ↑ (PWS IC deletion) | [138,139,140] |
| Cortisol/Corticosterone | Adrenal cortex | -- | ↑ (TgPWS) | [141] |
| Chromogranins (A, B) | Adrenal medulla and pancreas | ↓ | ↓ (Magel2m+/pΔ) | [73] |
| Proenkephalin (PENK) | CNS and adrenal medulla | -- | ↓ (Magel2m+/pΔ) | [65,73] |
| Nesfatin-1 | CNS, adipose, gonads, stomach, pancreas, and liver | ↑ | -- | [142] |
| Brain-derived neurotrophic factor (BDNF) | CNS, lungs, heart, spleen, GI tract, and liver | ↓ and ↑ are reported | -- | [137,138,139,143] |
| Insulin growth factor binding protein 7 (IGFBP7) | CNS, GI tract, liver, kidney, adrenal cortex, lung, testis, and ovary | ↑ in neuronal cells | ↑ (PWScrm+/p−) | [144] |
| Peptide YY (PYY) | GI tract | ↓ and ↑ are reported | -- | [145,146] |
| Substance P (SP) | GI tract | ↑ | -- | [147] |
| Adrenocorticotropic hormone (ACTH) | Anterior pituitary | ↓; no change | -- | [148,149,150] |
| Agouti-related protein (AgRP) | Hypothalamus (arcuate nucleus) | ↓ and no change are reported | ↓ and ↑ are reported (Snord116m+/p−) | [14,137,138] |
| β-endorphin (BE) | Hypothalamus and anterior pituitary | ↑ | -- | [147] |
| Galanin | Hypothalamus, pituitary, and GI tract | ↓ | ↓ (Magel2m+/pΔ) | [73] |
| Gonadotropin-releasing hormone (GnRH) | Hypothalamus | ↓ | ↓ (Magel2m+/pΔ) and no change (Mkrn3m+/p−) | [36,49,73] |
| Kisspeptin | Hypothalamus (arcuate nucleus) | -- | No change (Mkrn3m+/p−) | [36] |
| Melanin-concentrating hormone (MCH) | Hypothalamus | -- | No change (PWScrm+/p−) | [151,152] |
| Neurokinin B (NKB) | Hypothalamus (arcuate nucleus) | -- | ↑ (Mkrn3m+/p−) | [36] |
| Neuropeptide Y (NPY) | Hypothalamus (arcuate nucleus) | ↑; ↓ in CNS | ↑ (Snord116m+/p−) | [14,138] |
| Orexin/hypocretin | Hypothalamus | ↓ in CSF; ↑ in plasma | ↓ (Magel2m+/pΔ; PWScrm+/p−) | [65,152,153,154,155,156] |
| Oxytocin (OXT) | Hypothalamus (paraventricular and supraoptic nuclei) and posterior pituitary | ↓ and ↑ are reported | ↓ (Magel2m+/pΔ) | [73,138,157,158] |
| Somatostatin (SST) | Hypothalamus and GI tract | ↓ | ↓ (Magel2m+/pΔ) | [65,73,141] |
| Thyrotropin-releasing hormone (TRH) | Hypothalamus | -- | ↓ (Magel2m+/pΔ) | [73,159,160] |
| Vasopressin (AVP) | Hypothalamus | ↓ | ↓ (Magel2m+/pΔ) | [65,73] |
| Glucagon | Pancreas (α-cells) | -- | ↓ (TgPWS) | [141] |
| Amylin/Islet amyloid polypeptide (IAPP) | Pancreas (β-cells) | ↓ | -- | [10,161] |
| Insulin | Pancreas (β-cells) | ↓ and ↑ are reported | ↓ (TgPWS) | [10,65,138,146] |
| Obestatin | Stomach | ↑ and no change are reported | -- | [162,163] |
| Ghrelin | Stomach, hypothalamus (arcuate and paraventricular nuclei), pituitary, lung, adrenal cortex, and pancreas | ↑ | ↑ (TgPWS; Snord116m+/p−) | [14,138,141,163,164,165] |
| Testosterone | Testis | ↓ (male) | -- | [49,134,136] |
| Anti-Mullerian hormone (AMH)/Mullerian inhibiting hormone (MIH) | Testis and ovary | ↓ (female) | -- | [49,166] |
| Triiodothyronine (T3) | Thyroid gland | No change | -- | [159,167,168,169] |
| Thyroxine (T4) | Thyroid gland | ↓ (infants) | -- | [159,169,170] |
| Adiponectin | White adipose | ↑ and no change are reported | -- | [142,146,171,172,173] |
| Resistin | White adipose | ↑ and no change are reported | -- | [174,175] |
| Leptin | White adipose | ↑ and no change are reported | No change (Snord116m+/p−) | [74,142,143,176,177,178] |
| Spexin (Spx) | White adipose, hypothalamus, adrenal gland, pancreas, thyroid, and GI tract | ↓ | -- | [142,179] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoyos Sanchez, M.C.; Bayat, T.; Gee, R.R.F.; Fon Tacer, K. Hormonal Imbalances in Prader–Willi and Schaaf–Yang Syndromes Imply the Evolution of Specific Regulation of Hypothalamic Neuroendocrine Function in Mammals. Int. J. Mol. Sci. 2023, 24, 13109. https://doi.org/10.3390/ijms241713109
Hoyos Sanchez MC, Bayat T, Gee RRF, Fon Tacer K. Hormonal Imbalances in Prader–Willi and Schaaf–Yang Syndromes Imply the Evolution of Specific Regulation of Hypothalamic Neuroendocrine Function in Mammals. International Journal of Molecular Sciences. 2023; 24(17):13109. https://doi.org/10.3390/ijms241713109
Chicago/Turabian StyleHoyos Sanchez, Maria Camila, Tara Bayat, Rebecca R. Florke Gee, and Klementina Fon Tacer. 2023. "Hormonal Imbalances in Prader–Willi and Schaaf–Yang Syndromes Imply the Evolution of Specific Regulation of Hypothalamic Neuroendocrine Function in Mammals" International Journal of Molecular Sciences 24, no. 17: 13109. https://doi.org/10.3390/ijms241713109