
Partial Reversible Inhibition of Enzymes and Its Metabolic and Pharmaco-Toxicological Implications

Abstract
:1. Introduction
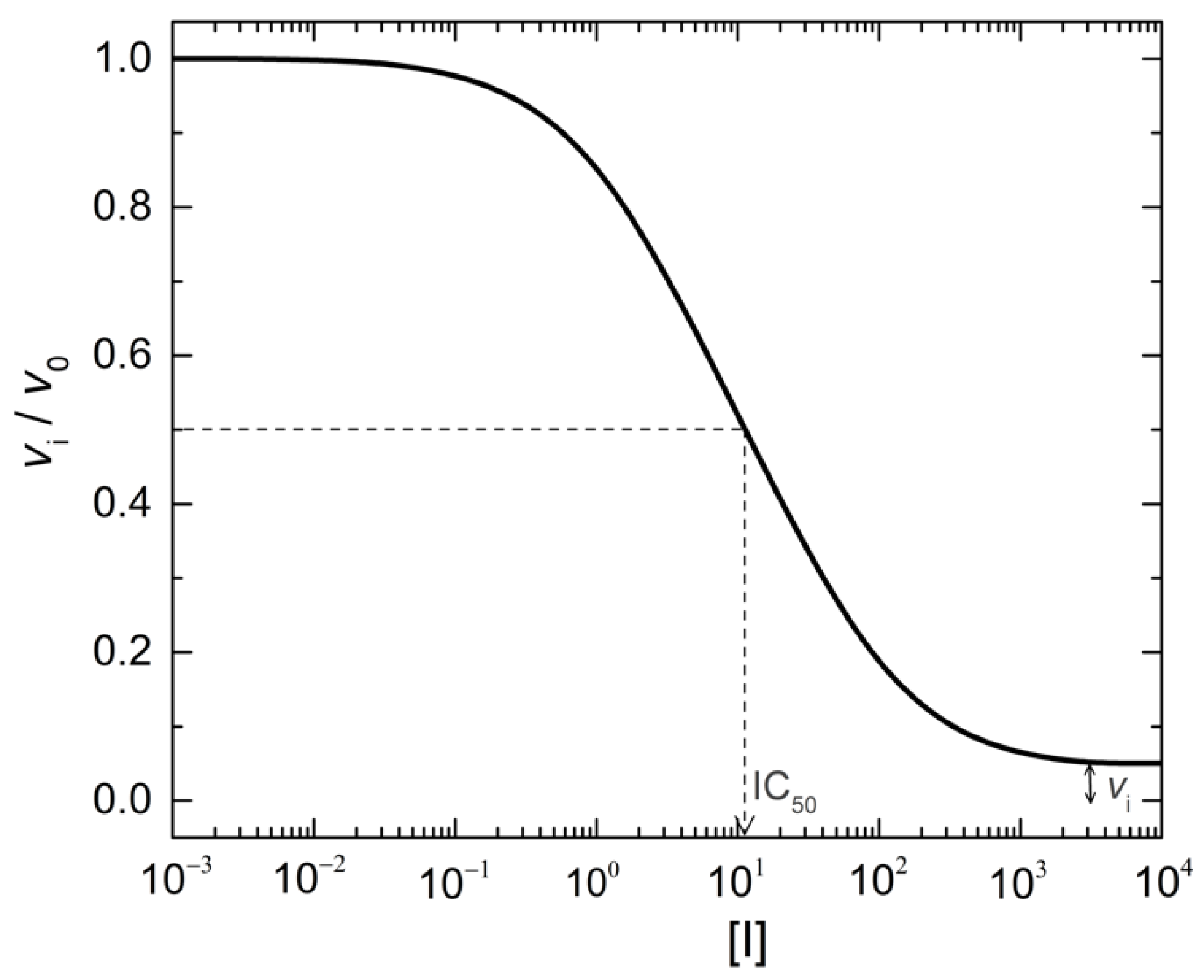
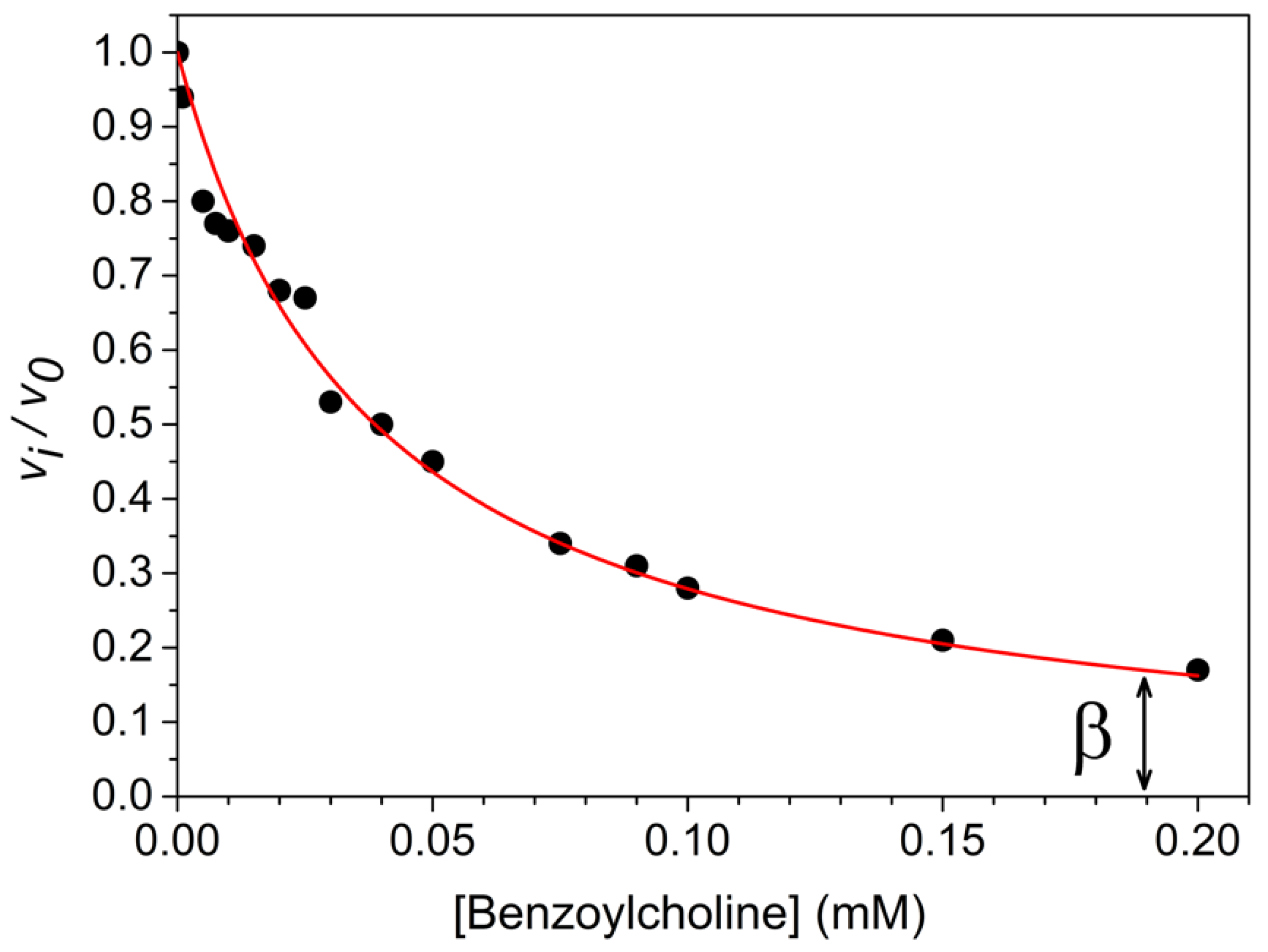
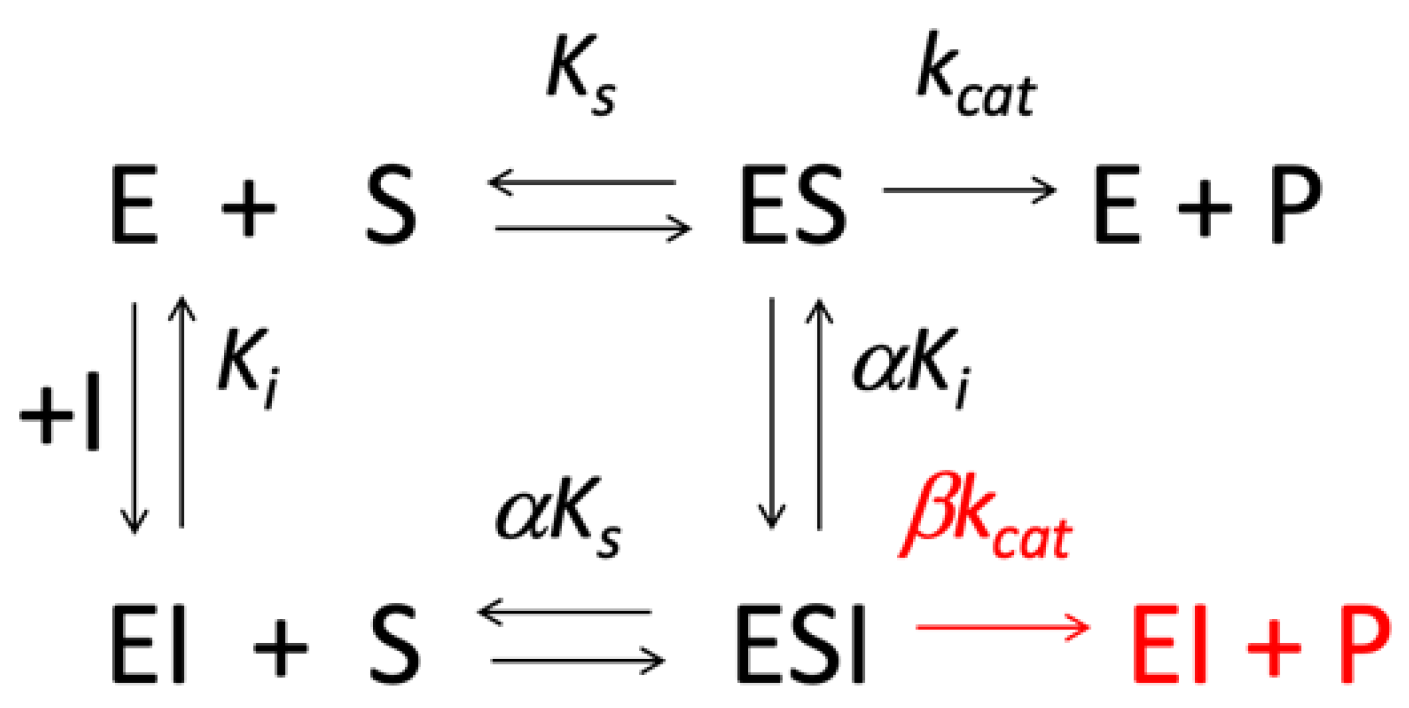
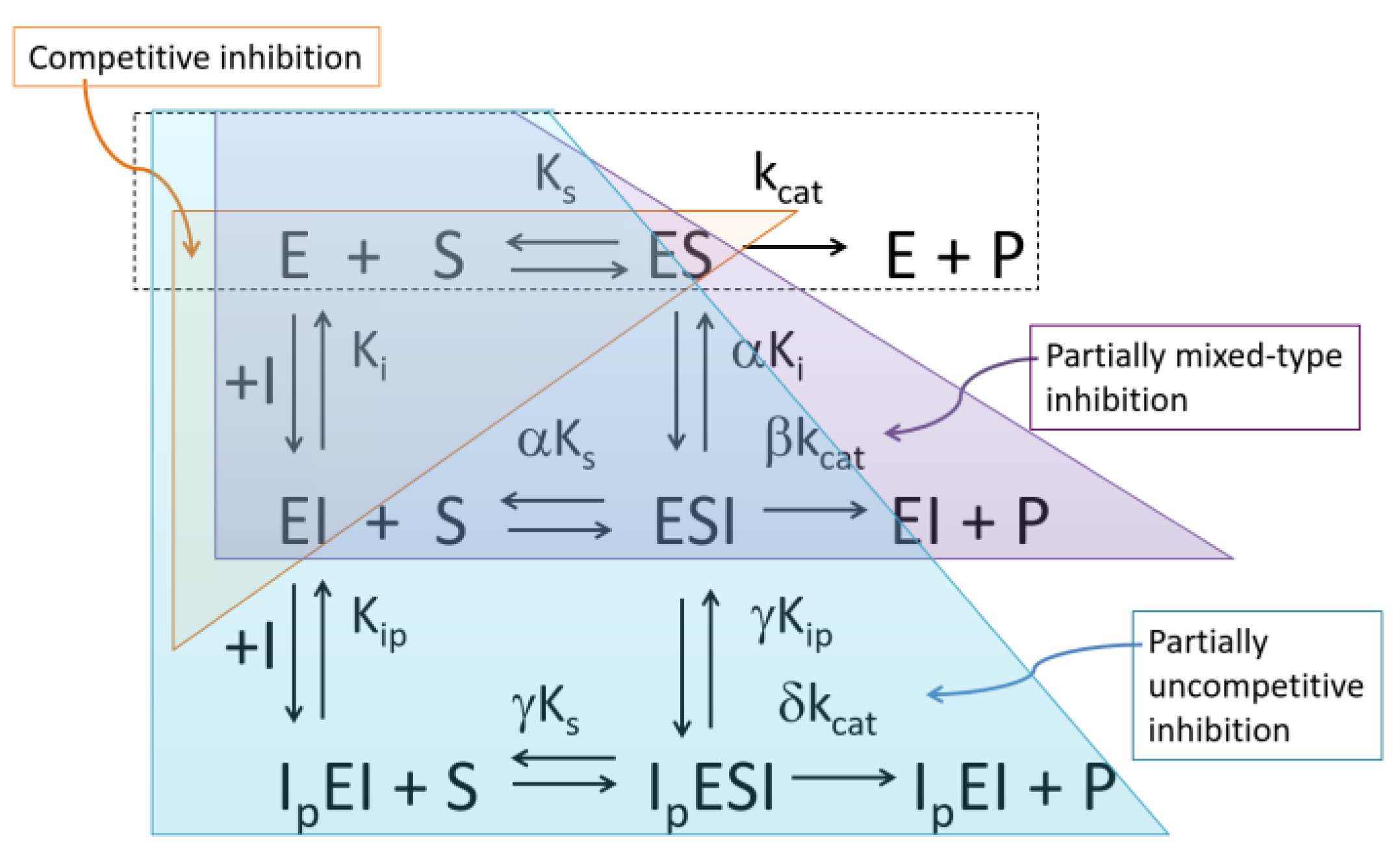
2. Formal Mechanisms, Analysis and Diagnosis of PRI
3. Possible Experimental Artefacts
4. Relevance of PRI
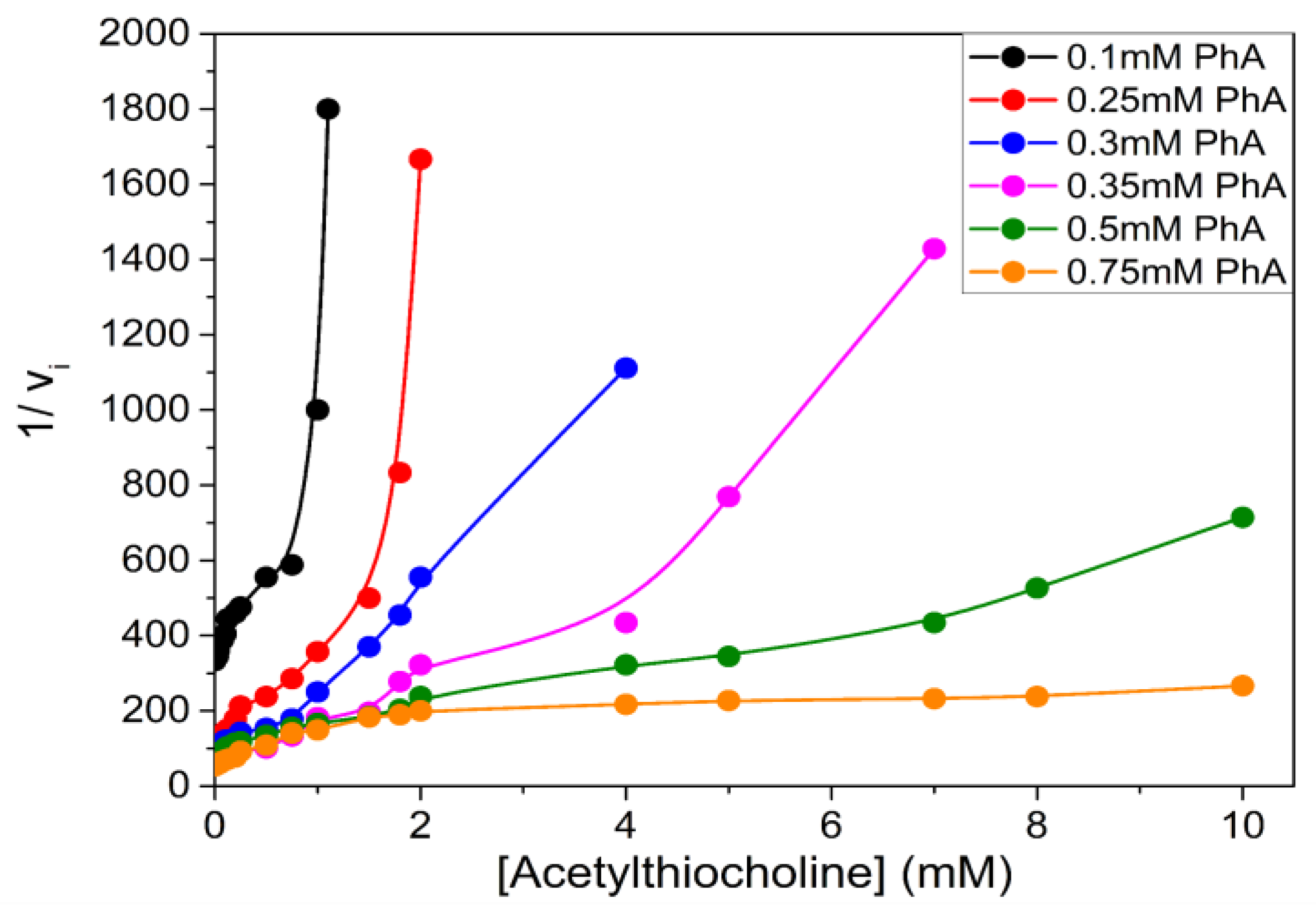
4.1. Metabolic Processes
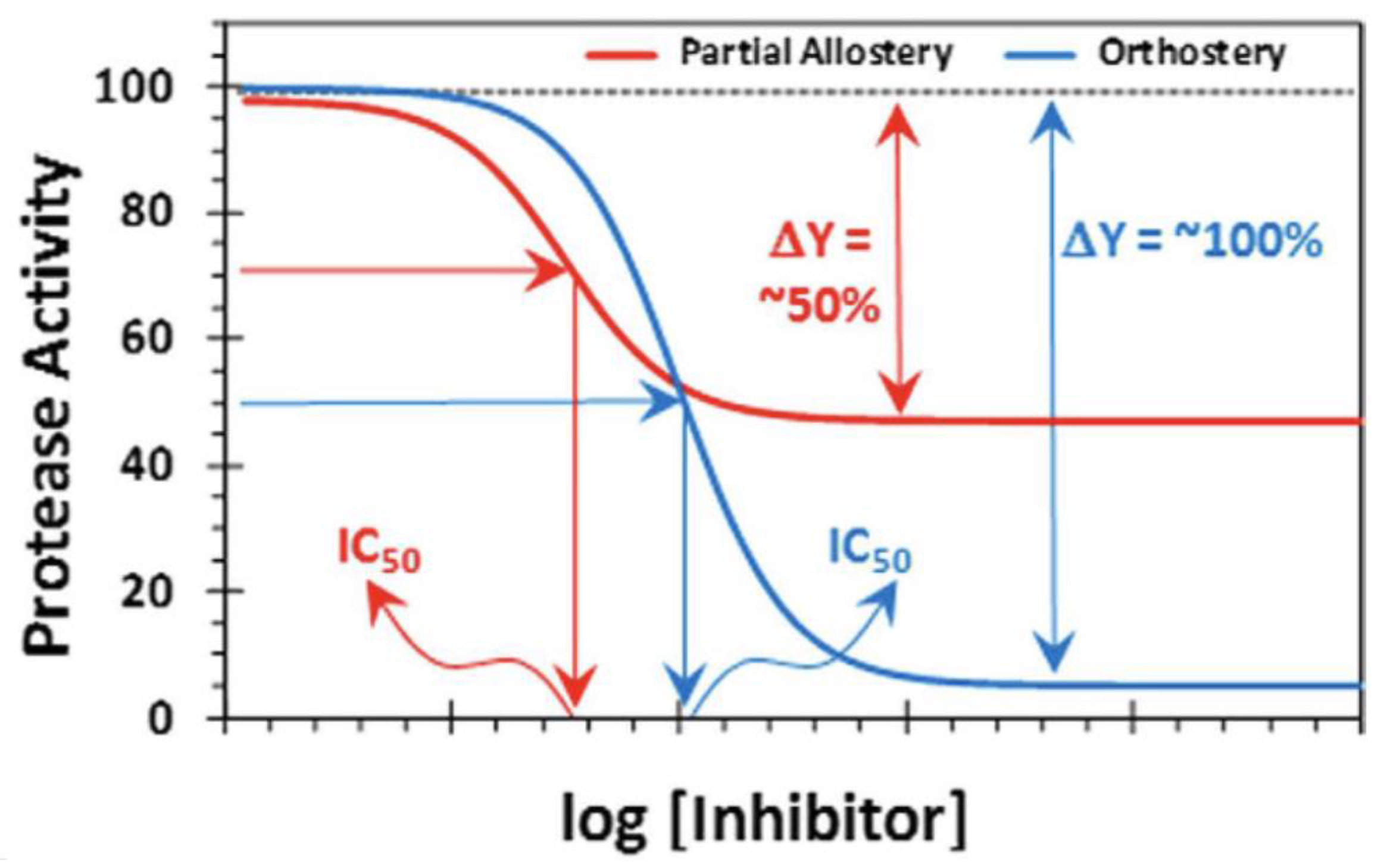
4.2. Pharmacology and Toxicology
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Cleland, W.W. The Kinetics of Enzyme-Catalyzed Reactions with Two or More Substrates or Products. Biochim. Biophys. Acta BBA—Spec. Sect. Enzymol. Subj. 1963, 67, 173–187. [Google Scholar] [CrossRef]
- Worcel, A.; Goldman, D.S.; Cleland, W.W. An Allosteric Reduced Nicotinamide Adenine Dinucleotide Oxidase from Mycobacterium Tuberculosis. J. Biol. Chem. 1965, 240, 3399–3407. [Google Scholar] [CrossRef]
- Wang, J.H.; Tu, J.-I.; Lo, F.M. Effect of Glucose 6-Phosphate on the Nucleotide Site of Glycogen Phosphorylase b. J. Biol. Chem. 1970, 245, 3115–3121. [Google Scholar] [CrossRef] [PubMed]
- Segel, I.H. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems; Wiley: New York, NY, USA, 1975. [Google Scholar]
- Yoshino, M. A Graphical Method for Determining Inhibition Parameters for Partial and Complete Inhibitors. Biochem. J. 1987, 248, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, B. A Guide to the Michaelis–Menten Equation: Steady State and Beyond. FEBS J. 2022, 289, 6086–6098. [Google Scholar] [CrossRef]
- Fontes, R.; Ribeiro, J.M.; Sillero, A. A Tridimensional Representation of Enzyme Inhibition Useful for Diagnostic Purposes. J. Enzym. Inhib. 1994, 8, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Baici, A. The Specific Velocity Plot. A Graphical Method for Determining Inhibition Parameters for Both Linear and Hyperbolic Enzyme Inhibitors. Eur. J. Biochem. 1981, 119, 9–14. [Google Scholar] [CrossRef]
- Balestri, F.; Cappiello, M.; Moschini, R.; Mura, U.; Del-Corso, A. Models of Enzyme Inhibition and Apparent Dissociation Constants from Kinetic Analysis to Study the Differential Inhibition of Aldose Reductase. J. Enzym. Inhib. Med. Chem. 2022, 37, 1426–1436. [Google Scholar] [CrossRef]
- Copeland, R.A. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, 3rd ed.; Wiley: Hoboken, NJ, USA, 2023. [Google Scholar]
- Cheng, Y.-C.; Prusoff, W.H. Relationship between the Inhibition Constant (KI) and the Concentration of Inhibitor Which Causes 50 per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Kazakova, R.R.; Masson, P. Quantitative Measurements of Pharmacological and Toxicological Activity of Molecules. Chemistry 2022, 4, 1466–1474. [Google Scholar] [CrossRef]
- Gelpi, J.L.; Aviles, J.J.; Busquets, M.; Imperial, S.; Mazo, A.; Cortes, A.; Halsall, D.J.; Holbrook, J.J. A Theoretical Approach to the Discrimination and Characterization of the Different Classes of Reversible Inhibitors. J. Chem. Educ. 1993, 70, 805. [Google Scholar] [CrossRef]
- Dixon, M. The Determination of Enzyme Inhibitor Constants. Biochem. J. 1953, 55, 170–171. [Google Scholar] [CrossRef] [PubMed]
- Mukhametgalieva, A.R.; Lushchekina, S.V.; Aglyamova, A.R.; Masson, P. Steady-State Kinetic Analysis of Human Cholinesterases over Wide Concentration Ranges of Competing Substrates. Biochim. Biophys. Acta BBA—Proteins Proteom. 2022, 1870, 140733. [Google Scholar] [CrossRef] [PubMed]
- Goličnik, M.; Masson, P. Time-course of enzyme-catalyzed competing substrate degradation for michaelian behavior and for enzymes showing activation/inhibition by excess substrate. Chem Biol Interact. 2019, 309, 108704. [Google Scholar] [CrossRef] [PubMed]
- Mukhametgalieva, A.R.; Nemtarev, A.V.; Sykaev, V.V.; Pashirova, T.N.; Masson, P. Activation/Inhibition of Cholinesterases by Excess Substrate: Interpretation of the Phenomenological b Factor in Steady-State Rate Equation. Int. J. Mol. Sci. 2023, 24, 10472. [Google Scholar] [CrossRef] [PubMed]
- Mukhametgalieva, A.R.; Aglyamova, A.R.; Lushchekina, S.V.; Goličnik, M.; Masson, P. Time-Course of Human Cholinesterases-Catalyzed Competing Substrate Kinetics. Chem. Biol. Interact. 2019, 310, 108702. [Google Scholar] [CrossRef] [PubMed]
- Whiteley, C.G. Mechanistic and Kinetic Studies of Inhibition of Enzymes. Cell Biochem. Biophys. 2000, 33, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, M.; Murakami, K. Analysis of the Substrate Inhibition of Complete and Partial Types. SpringerPlus 2015, 4, 292. [Google Scholar] [CrossRef]
- Grant, G.A. The Many Faces of Partial Inhibition: Revealing Imposters with Graphical Analysis. Arch. Biochem. Biophys. 2018, 653, 10–23. [Google Scholar] [CrossRef]
- Berman, H.A.; Leonard, K. Ligand Exclusion on Acetylcholinesterase. Biochemistry 1990, 29, 10640–10649. [Google Scholar] [CrossRef] [PubMed]
- Cornish-Bowden, A. Fundamentals of Enzyme Kinetics, 4th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2012. [Google Scholar]
- Reed, M.C.; Lieb, A.; Nijhout, H.F. The Biological Significance of Substrate Inhibition: A Mechanism with Diverse Functions. BioEssays 2010, 32, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Wu, B. Substrate Inhibition Kinetics in Drug Metabolism Reactions. Drug Metab. Rev. 2011, 43, 440–456. [Google Scholar] [CrossRef] [PubMed]
- Silman, I. The Multiple Biological Roles of the Cholinesterases. Prog. Biophys. Mol. Biol. 2021, 162, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Roche, T.E.; Baker, J.C.; Yan, X.; Hiromasa, Y.; Gong, X.; Peng, T.; Dong, J.; Turkan, A.; Kasten, S.A. Distinct Regulatory Properties of Pyruvate Dehydrogenase Kinase and Phosphatase Isoforms. In Progress in Nucleic Acid Research and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2001; Volume 70, pp. 33–75. [Google Scholar] [CrossRef]
- Kokkonen, P.; Beier, A.; Mazurenko, S.; Damborsky, J.; Bednar, D.; Prokop, Z. Substrate Inhibition by the Blockage of Product Release and Its Control by Tunnel Engineering. RSC Chem. Biol. 2021, 2, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, P.; Van Der Veken, B.; Van Der Veken, P.; Pintelon, I.; Roosens, L.; Adriaenssens, E.; Timmerman, V.; Guns, P.-J.; De Meyer, G.R.Y.; Martinet, W. Partial Inhibition of Glycolysis Reduces Atherogenesis Independent of Intraplaque Neovascularization in Mice. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1168–1181. [Google Scholar] [CrossRef] [PubMed]
- Eddleston, M. Novel Clinical Toxicology and Pharmacology of Organophosphorus Insecticide Self-Poisoning. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 341–360. [Google Scholar] [CrossRef]
- Masson, P.; Froment, M.-T.; Gillon, E.; Nachon, F.; Lockridge, O.; Schopfer, L.M. Kinetic Analysis of Effector Modulation of Butyrylcholinesterase-Catalysed Hydrolysis of Acetanilides and Homologous Esters: Modulation of Butyrylcholinesterase Catalytic Activitiy. FEBS J. 2008, 275, 2617–2631. [Google Scholar] [CrossRef]
- González Tanarro, C.M.; Gütschow, M. Hyperbolic Mixed-Type Inhibition of Acetylcholinesterase by Tetracyclic Thienopyrimidines. J. Enzym. Inhib. Med. Chem. 2011, 26, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Shahrivar-Gargari, M.; Hamzeh-Mivehroud, M.; Hemmati, S.; Shahbazi Mojarrad, J.; Notash, B.; Tüylü Küçükkılınç, T.; Ayazgök, B.; Dastmalchi, S. Design, Synthesis, and Biological Evaluation of Novel Indanone-Based Hybrids as Multifunctional Cholinesterase Inhibitors for Alzheimer’s Disease. J. Mol. Struct. 2021, 1229, 129787. [Google Scholar] [CrossRef]
- Jaswal, J.; Lopaschuk, G. Partial Inhibition of Fatty Acid β-Oxidation with Trimetazidine—A Novel Approach to the Treatment of Ischemic Heart Disease. Arch. Med. Sci. Special Issue 2007, 3, S1–S9. [Google Scholar]
- Hutzler, J.M.; Tracy, T.S. Atypical Kinetic Profiles in Drug Metabolism Reactions. Drug Metab. Dispos. 2002, 30, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Locke, S.; Naidoo, V.; Hassan, I.; Duncan, N. Effect of Cytochrome P450 Inhibition on Toxicity of Diclofenac in Chickens: Unravelling Toxicity in Gyps Vultures. Onderstepoort J. Vet. Res. 2022, 89, 1978. [Google Scholar] [CrossRef] [PubMed]
- Verespy Iii, S.; Mehta, A.Y.; Afosah, D.; Al-Horani, R.A.; Desai, U.R. Allosteric Partial Inhibition of Monomeric Proteases. Sulfated Coumarins Induce Regulation, Not Just Inhibition, of Thrombin. Sci. Rep. 2016, 6, 24043. [Google Scholar] [CrossRef]
- Nihei, K.; Kubo, I. Benzonitriles as Tyrosinase Inhibitors with Hyperbolic Inhibition Manner. Int. J. Biol. Macromol. 2019, 133, 929–932. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Duan, S.X.; Harmatz, J.S.; Wei, Z.; Singleton, C.A.; Greenblatt, D.J. Mechanism of Dasabuvir Inhibition of Acetaminophen Glucuronidation. J. Pharm. Pharmacol. 2022, 74, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Abou El-Magd, R.; Park, H.; Kawazoe, T.; Iwana, S.; Ono, K.; Chung, S.; Miyano, M.; Yorita, K.; Sakai, T.; Fukui, K. The Effect of Risperidone on D-Amino Acid Oxidase Activity as a Hypothesis for a Novel Mechanism of Action in the Treatment of Schizophrenia. J. Psychopharmacol. 2010, 24, 1055–1067. [Google Scholar] [CrossRef]
- Westley, A.M.; Westley, J. Enzyme Inhibition in Open Systems. Superiority of Uncompetitive Agents. J. Biol. Chem. 1996, 271, 5347–5352. [Google Scholar] [CrossRef]
- Facchetti, G.; Altafini, C. Partial Inhibition and Bilevel Optimization in Flux Balance Analysis. BMC Bioinform. 2013, 14, 344. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Inhibition | Intercept on Ordinate | Intercept on Abscissa | Slope | Replot of Slope vs. [S] |
|---|---|---|---|---|
| general case (mixed type) | Equation (6b) | −β/αKi | Equation (6c) | hyperbolic |
| competitive | Equation (7b) | −1/αKi | Equation (7c) | linear |
| noncompetitive | β/(1 − β) | −β/Ki | Ki/(1 − β) | constant ∀ [S] |
| uncompetitive | Equation (9b) | −β/αKi | Equation (9c) | hyperbolic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masson, P.; Mukhametgalieva, A.R. Partial Reversible Inhibition of Enzymes and Its Metabolic and Pharmaco-Toxicological Implications. Int. J. Mol. Sci. 2023, 24, 12973. https://doi.org/10.3390/ijms241612973
Masson P, Mukhametgalieva AR. Partial Reversible Inhibition of Enzymes and Its Metabolic and Pharmaco-Toxicological Implications. International Journal of Molecular Sciences. 2023; 24(16):12973. https://doi.org/10.3390/ijms241612973
Chicago/Turabian StyleMasson, Patrick, and Aliya R. Mukhametgalieva. 2023. "Partial Reversible Inhibition of Enzymes and Its Metabolic and Pharmaco-Toxicological Implications" International Journal of Molecular Sciences 24, no. 16: 12973. https://doi.org/10.3390/ijms241612973