
CD155 and Its Receptors as Targets for Cancer Therapy


Abstract
:1. Introduction
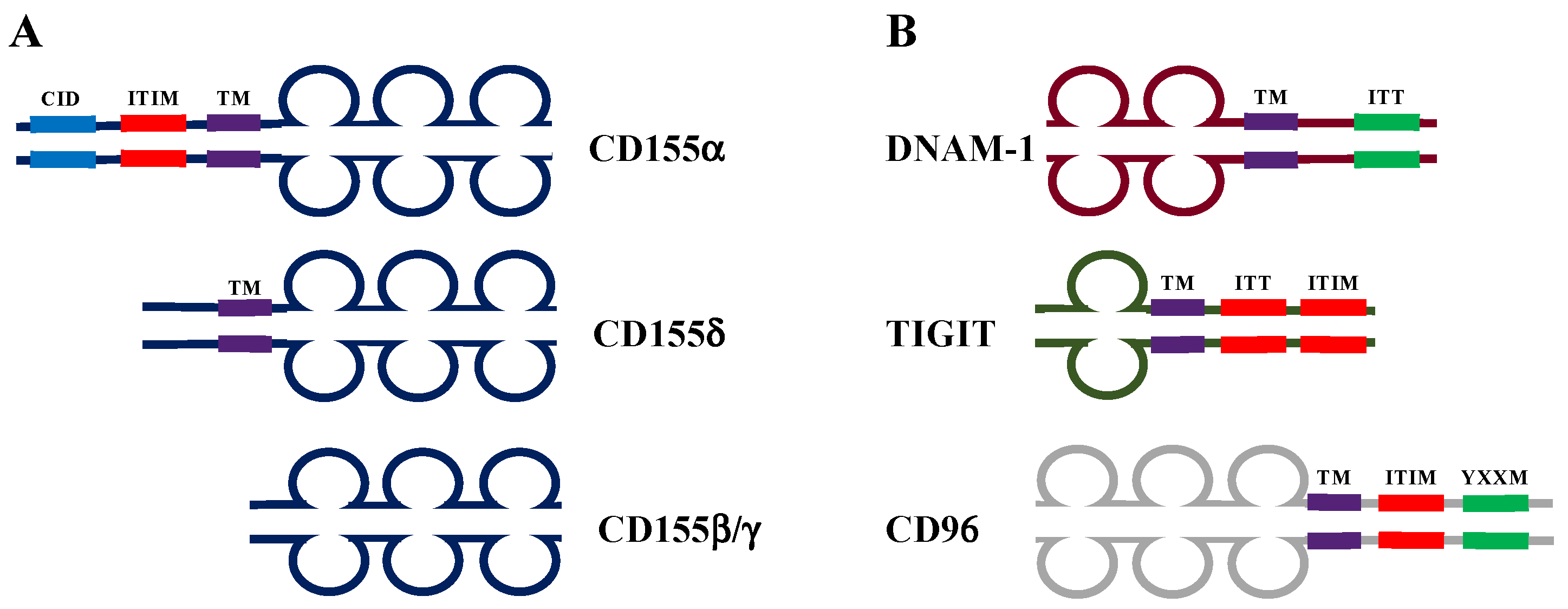
2. The Complex Role of CD155 in Tumor Progression
2.1. Regulation of CD155 Expression and Function in the Tumor Microenvironment
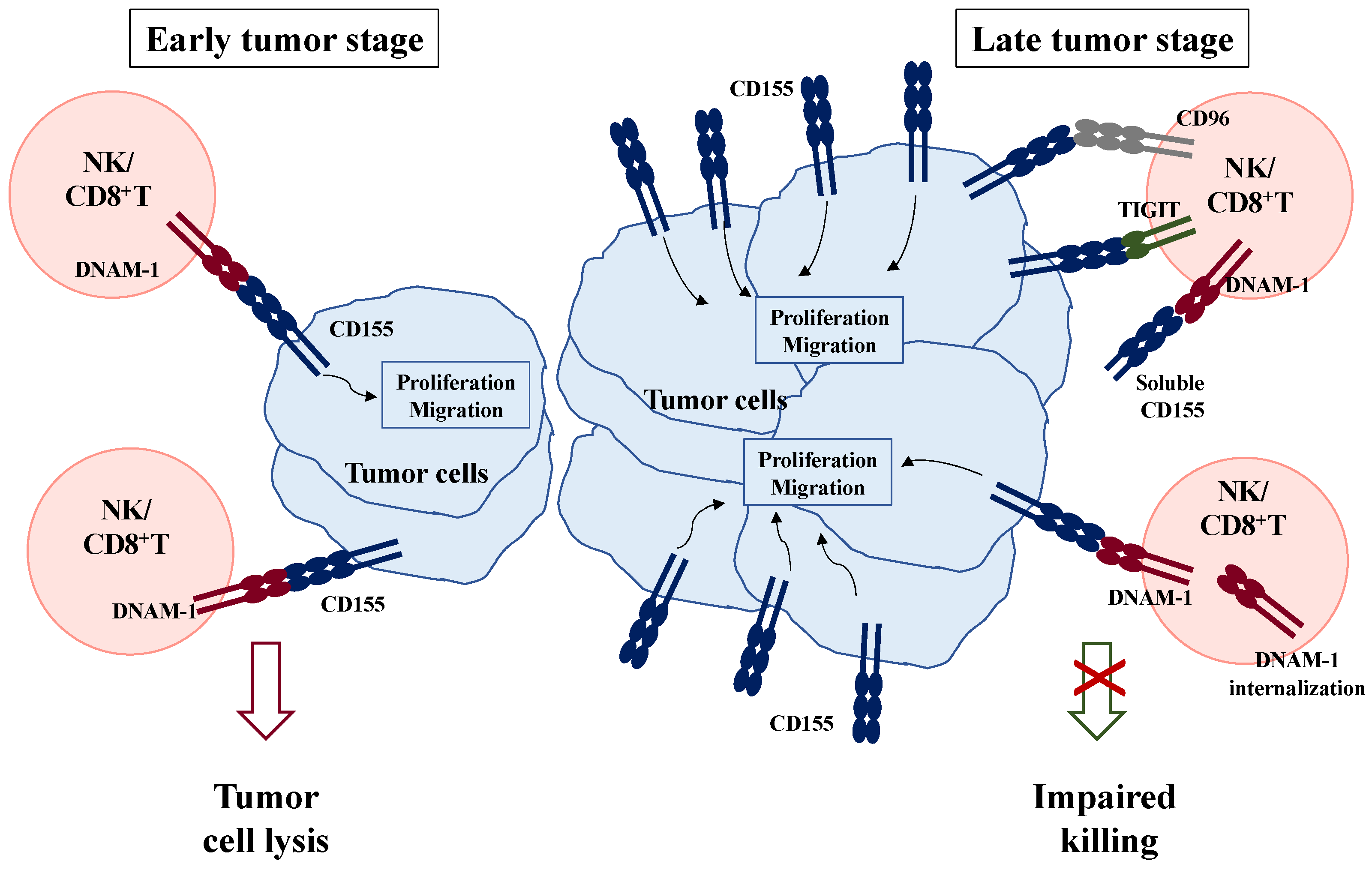
2.2. CD155: A Double-Edged Sword in Cancer Immune Surveillance
3. Current Anti-Cancer Strategies Targeting CD155 and Its Receptors
3.1. Oncolytic Viruses Targeting CD155
3.2. DNAM-1 Chimeric-Receptor-Based Therapies
3.3. Monoclonal Antibodies Targeting CD155 Inhibitory Receptors
4. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mendelsohn, C.L.; Wimmer, E.; Racaniello, V.R. Cellular receptor for poliovirus: Molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 1989, 56, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.; Colonna, M. The role of NK cell recognition of nectin and nectin-like proteins in tumor immunosurveillance. Semin. Cancer Biol. 2006, 16, 359–366. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Madore, J.; Li, X.Y.; Smyth, M.J. Tumor intrinsic and extrinsic immune functions of CD155. Semin. Cancer Biol. 2020, 65, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Welker, R.; Mueller, S.; Linehan, M.; Nomoto, A.; Wimmer, E. Immunofluorescence analysis of poliovirus receptor expression in Peyer’s patches of humans, primates, and CD155 transgenic mice: Implications for poliovirus infection. J. Infect. Dis. 2002, 186, 585–592. [Google Scholar] [CrossRef]
- Maier, M.K.; Seth, S.; Czeloth, N.; Qiu, Q.; Ravens, I.; Kremmer, E.; Ebel, M.; Müller, W.; Pabst, O.; Förster, R.; et al. The adhesion receptor CD155 determines the magnitude of humoral immune responses against orally ingested antigens. Eur. J. Immunol. 2007, 37, 2214–2225. [Google Scholar] [CrossRef]
- Escalante, N.K.; von Rossum, A.; Lee, M.; Choy, J.C. CD155 on human vascular endothelial cells attenuates the acquisition of effector functions in CD8 T cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1177–1184. [Google Scholar] [CrossRef]
- Nakai, R.; Maniwa, Y.; Tanaka, Y.; Nishio, W.; Yoshimura, M.; Okita, Y.; Ohbayashi, C.; Satoh, N.; Ogita, H.; Takai, Y.; et al. Overexpression of Necl-5 correlates with unfavorable prognosis in patients with lung adenocarcinoma. Cancer Sci. 2010, 101, 1326–1330. [Google Scholar] [CrossRef] [PubMed]
- Bevelacqua, V.; Bevelacqua, Y.; Candido, S.; Skarmoutsou, E.; Amoroso, A.; Guarneri, C.; Strazzanti, A.; Gangemi, P.; Mazzarino, M.C.; D’Amico, F.; et al. Nectin like-5 overexpression correlates with the malignant phenotype in cutaneous melanoma. Oncotarget 2012, 3, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, S.; Matsumine, A.; Toyoda, H.; Niimi, R.; Iino, T.; Sudo, A. Prognostic significance of CD155 mRNA expression in soft tissue sarcomas. Oncol. Lett. 2013, 5, 1771–1776. [Google Scholar] [CrossRef]
- Nishiwada, S.; Sho, M.; Yasuda, S.; Shimada, K.; Yamato, I.; Akahori, T.; Kinoshita, S.; Nagai, M.; Konishi, N.; Nakajima, Y. Clinical significance of CD155 expression in human pancreatic cancer. Anticancer Res. 2015, 35, 2287–2297, Erratum in Anticancer Res. 2015, 35, 4371. [Google Scholar]
- Zhuo, B.; Li, Y.; Gu, F.; Li, Z.; Sun, Q.; Shi, Y.; Shen, Y.; Zhang, F.; Wang, R.; Wang, X. Overexpression of CD155 relates to metastasis and invasion in osteosarcoma. Oncol. Lett. 2018, 15, 7312–7318. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Zhou, Q.; Song, Q.K.; Wang, R.B.; Lyu, S.; Guan, X.; Zhao, Y.J.; Wu, J.P. Overexpression of an Immune Checkpoint (CD155) in Breast Cancer Associated with Prognostic Significance and Exhausted Tumor-Infiltrating Lymphocytes: A Cohort Study. J. Immunol. Res. 2020, 2020, 3948928. [Google Scholar] [CrossRef] [PubMed]
- Murakami, D.; Matsuda, K.; Iwamoto, H.; Mitani, Y.; Mizumoto, Y.; Nakamura, Y.; Matsuzaki, I.; Iwamoto, R.; Takahashi, Y.; Kojima, F.; et al. Prognostic value of CD155/TIGIT expression in patients with colorectal cancer. PLoS ONE 2022, 17, e0265908. [Google Scholar] [CrossRef]
- Lee, B.H.; Kim, J.H.; Kang, K.W.; Lee, S.R.; Park, Y.; Sung, H.J.; Kim, B.S. PVR (CD155) Expression as a Potential Prognostic Marker in Multiple Myeloma. Biomedicines 2022, 10, 1099. [Google Scholar] [CrossRef] [PubMed]
- Boissière-Michot, F.; Chateau, M.C.; Thézenas, S.; Guiu, S.; Bobrie, A.; Jacot, W. Correlation of the TIGIT-PVR immune checkpoint axis with clinicopathological features in triple-negative breast cancer. Front. Immunol. 2022, 13, 1058424. [Google Scholar] [CrossRef] [PubMed]
- Kučan Brlić, P.; Lenac Roviš, T.; Cinamon, G.; Tsukerman, P.; Mandelboim, O.; Jonjić, S. Targeting PVR (CD155) and its receptors in anti-tumor therapy. Cell Mol. Immunol. 2019, 16, 40–52. [Google Scholar] [CrossRef]
- Molfetta, R.; Zitti, B.; Lecce, M.; Milito, N.D.; Stabile, H.; Fionda, C.; Cippitelli, M.; Gismondi, A.; Santoni, A.; Paolini, R. CD155: A Multi-Functional Molecule in Tumor Progression. Int. J. Mol. Sci. 2020, 21, 922. [Google Scholar] [CrossRef]
- De Andrade, L.F.; Smyth, M.J.; Martinet, L. DNAM-1 control of natural killer cells functions through nectin and nectin-like proteins. Immunol. Cell Biol. 2014, 92, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Martinet, L.; Smyth, M.J. Balancing natural killer cell activation through paired receptors. Nat. Rev. Immunol. 2015, 15, 243–254. [Google Scholar] [CrossRef]
- Rikitake, Y.; Mandai, K.; Takai, Y. The role of nectins in different types of cell-cell adhesion. J. Cell Sci. 2012, 125 Pt 16, 3713–3722. [Google Scholar] [CrossRef]
- Solecki, D.J.; Gromeier, M.; Mueller, S.; Bernhardt, G.; Wimmer, E. Expression of the human poliovirus receptor/CD155 gene is activated by sonic hedgehog. J. Biol. Chem. 2002, 277, 25697–25702. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Irie, K.; Okamoto, R.; Ikeda, W.; Takai, Y. Transcriptional activation of the mouse Necl-5/Tage4/PVR/CD155 gene by fibroblast growth factor or oncogenic Ras through the Raf-MEK-ERK-AP-1 pathway. Oncogene 2005, 24, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; Di Gialleonardo, V.; Cippitelli, M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009, 113, 3503–3511. [Google Scholar] [CrossRef] [PubMed]
- Fionda, C.; Abruzzese, M.P.; Zingoni, A.; Soriani, A.; Ricci, B.; Molfetta, R.; Paolini, R.; Santoni, A.; Cippitelli, M. Nitric oxide donors increase PVR/CD155 DNAM-1 ligand expression in multiple myeloma cells: Role of DNA damage response activation. BMC Cancer 2015, 15, 17. [Google Scholar] [CrossRef]
- Soriani, A.; Borrelli, C.; Ricci, B.; Molfetta, R.; Zingoni, A.; Fionda, C.; Carnevale, S.; Abruzzese, M.P.; Petrucci, M.T.; Ricciardi, M.R.; et al. p38 MAPK differentially controls NK activating ligands at transcriptional and post-transcriptional level on multiple myeloma cells. Oncoimmunology 2016, 6, e1264564. [Google Scholar] [CrossRef]
- Pende, D.; Castriconi, R.; Romagnani, P.; Spaggiari, G.M.; Marcenaro, S.; Dondero, A.; Lazzeri, E.; Lasagni, L.; Martini, S.; Rivera, P.; et al. Expression of the DNAM-1 ligands, Nectin-2 (CD112) and poliovirus receptor (CD155), on dendritic cells: Relevance for natural killer-dendritic cell interaction. Blood 2006, 107, 2030–2036. [Google Scholar] [CrossRef]
- Kamran, N.; Takai, Y.; Miyoshi, J.; Biswas, S.K.; Wong, J.S.; Gasser, S. Toll-like receptor ligands induce expression of the costimulatory molecule CD155 on antigen-presenting cells. PLoS ONE 2013, 8, e54406. [Google Scholar] [CrossRef]
- Mekhloufi, A.; Kosta, A.; Stabile, H.; Molfetta, R.; Zingoni, A.; Soriani, A.; Cippitelli, M.; Paolini, R.; Gismondi, A.; Ricciardi, M.R.; et al. Bone Marrow Stromal Cell-Derived IL-8 Upregulates PVR Expression on Multiple Myeloma Cells via NF-kB Transcription Factor. Cancers 2020, 12, 440. [Google Scholar] [CrossRef]
- Brandetti, E.; Focaccetti, C.; Pezzolo, A.; Ognibene, M.; Folgiero, V.; Cotugno, N.; Benvenuto, M.; Palma, P.; Manzari, V.; Rossi, P.; et al. Enhancement of Neuroblastoma NK-Cell-Mediated Lysis through NF-kB p65 Subunit-Induced Expression of FAS and PVR, the Loss of Which Is Associated with Poor Patient Outcome. Cancers 2021, 13, 4368. [Google Scholar] [CrossRef]
- Briukhovetska, D.; Suarez-Gosalvez, J.; Voigt, C.; Markota, A.; Giannou, A.D.; Schübel, M.; Jobst, J.; Zhang, T.; Dörr, J.; Märkl, F.; et al. T cell-derived interleukin-22 drives the expression of CD155 by cancer cells to suppress NK cell function and promote metastasis. Immunity 2023, 56, 143–161.e11. [Google Scholar] [CrossRef]
- Koike, S.; Horie, H.; Ise, I.; Okitsu, A.; Yoshida, M.; Iizuka, N.; Takeuchi, K.; Takegami, T.; Nomoto, A. The poliovirus receptor protein is produced both as membrane-bound and secreted forms. EMBO J. 1990, 9, 3217–3224. [Google Scholar] [CrossRef] [PubMed]
- Baury, B.; Masson, D.; McDermott, B.M., Jr.; Jarry, A.; Blottière, H.M.; Blanchardie, P.; Laboisse, C.L.; Lustenberger, P.; Racaniello, V.R.; Denis, M.G. Identification of secreted CD155 isoforms. Biochem. Biophys. Res. Commun. 2003, 309, 175–182. [Google Scholar] [CrossRef]
- Ohka, S.; Ohno, H.; Tohyama, K.; Nomoto, A. Basolateral sorting of human poliovirus receptor alpha involves an interaction with the mu1B subunit of the clathrin adaptor complex in polarized epithelial cells. Biochem. Biophys. Res. Commun. 2001, 287, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Ohka, S.; Nomoto, A. Ligand stimulation of CD155alpha inhibits cell adhesion and enhances cell migration in fibroblasts. Biochem. Biophys. Res. Commun. 2004, 319, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Molfetta, R.; Zingoni, A.; Santoni, A.; Paolini, R. Post-translational Mechanisms Regulating NK Cell Activating Receptors and Their Ligands in Cancer: Potential Targets for Therapeutic Intervention. Front. Immunol. 2019, 10, 2557. [Google Scholar] [CrossRef]
- Molfetta, R.; Petillo, S.; Cippitelli, M.; Paolini, R. SUMOylation and related post-translational modifications in natural killer cell anti-cancer responses. Front. Cell Dev. Biol. 2023, 11, 1213114. [Google Scholar] [CrossRef]
- Gong, J.; Fang, L.; Liu, R.; Wang, Y.; Xing, J.; Chen, Y.; Zhuang, R.; Zhang, Y.; Zhang, C.; Yang, A.; et al. UPR decreases CD226 ligand CD155 expression and sensitivity to NK cell-mediated cytotoxicity in hepatoma cells. Eur. J. Immunol. 2014, 44, 3758–3767. [Google Scholar] [CrossRef]
- Zitti, B.; Molfetta, R.; Fionda, C.; Quatrini, L.; Stabile, H.; Lecce, M.; de Turris, V.; Ricciardi, M.R.; Petrucci, M.T.; Cippitelli, M.; et al. Innate immune activating ligand SUMOylation affects tumor cell recognition by NK cells. Sci. Rep. 2017, 7, 10445. [Google Scholar] [CrossRef]
- Takai, Y.; Miyoshi, J.; Ikeda, W.; Ogita, H. Nectins and nectin-like molecules: Roles in contact inhibition of cell movement and proliferation. Nat. Rev. Mol. Cell Biol. 2008, 9, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Bowers, J.R.; Readler, J.M.; Sharma, P.; Excoffon, K.J.D.A. Poliovirus Receptor: More than a simple viral receptor. Virus Res. 2017, 242, 1–6. [Google Scholar] [CrossRef]
- Abe, A.; Fukui, H.; Fujii, S.; Kono, T.; Mukawa, K.; Yoshitake, N.; Sekikawa, A.; Ichikawa, K.; Tomita, S.; Yamagishi, H.; et al. Role of Necl-5 in the pathophysiology of colorectal lesions induced by dimethylhydrazine and/or dextran sodium sulphate. J. Pathol. 2009, 217, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Wang, B.; Gao, J.; Xin, N.; Wang, W.; Song, X.; Shao, Y.; Zhao, C. CD155 knockdown promotes apoptosis via AKT/Bcl-2/Bax in colon cancer cells. J. Cell Mol. Med. 2018, 22, 131–140. [Google Scholar] [CrossRef]
- Kakunaga, S.; Ikeda, W.; Shingai, T.; Fujito, T.; Yamada, A.; Minami, Y.; Imai, T.; Takai, Y. Enhancement of serum- and platelet-derived growth factor-induced cell proliferation by Necl-5/Tage4/poliovirus receptor/CD155 through the Ras-Raf-MEK-ERK signaling. J. Biol. Chem. 2004, 279, 36419–36425. [Google Scholar] [CrossRef] [PubMed]
- Kono, T.; Imai, Y.; Yasuda, S.; Ohmori, K.; Fukui, H.; Ichikawa, K.; Tomita, S.; Imura, J.; Kuroda, Y.; Ueda, Y.; et al. The CD155/poliovirus receptor enhances the proliferation of ras-mutated cells. Int. J. Cancer. 2008, 122, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, W.; Kakunaga, S.; Itoh, S.; Shingai, T.; Takekuni, K.; Satoh, K.; Inoue, Y.; Hamaguchi, A.; Morimoto, K.; Takeuchi, M.; et al. Tage4/Nectin-like molecule-5 heterophilically trans-interacts with cell adhesion molecule Nectin-3 and enhances cell migration. J. Biol. Chem. 2003, 278, 28167–28172. [Google Scholar] [CrossRef]
- Mueller, S.; Wimmer, E. Recruitment of nectin-3 to cell-cell junctions through trans-heterophilic interaction with CD155, a vitronectin and poliovirus receptor that localizes to alpha(v)beta3 integrin-containing membrane microdomains. J. Biol. Chem. 2003, 278, 31251–31260. [Google Scholar] [CrossRef]
- Lange, R.; Peng, X.; Wimmer, E.; Lipp, M.; Bernhardt, G. The poliovirus receptor CD155 mediates cell-to-matrix contacts by specifically binding to vitronectin. Virology 2001, 285, 218–227. [Google Scholar] [CrossRef]
- Fujito, T.; Ikeda, W.; Kakunaga, S.; Minami, Y.; Kajita, M.; Sakamoto, Y.; Monden, M.; Takai, Y. Inhibition of cell movement and proliferation by cell-cell contact-induced interaction of Necl-5 with nectin-3. J. Cell Biol. 2005, 171, 165–173. [Google Scholar] [CrossRef]
- Ikeda, W.; Kakunaga, S.; Takekuni, K.; Shingai, T.; Satoh, K.; Morimoto, K.; Takeuchi, M.; Imai, T.; Takai, Y. Nectin-like molecule-5/Tage4 enhances cell migration in an integrin-dependent, Nectin-3-independent manner. J. Biol. Chem. 2004, 279, 18015–18025. [Google Scholar] [CrossRef]
- Sloan, K.E.; Eustace, B.K.; Stewart, J.K.; Zehetmeier, C.; Torella, C.; Simeone, M.; Roy, J.E.; Unger, C.; Louis, D.N.; Ilag, L.L.; et al. CD155/PVR plays a key role in cell motility during tumor cell invasion and migration. BMC Cancer 2004, 4, 73. [Google Scholar] [CrossRef]
- Sloan, K.E.; Stewart, J.K.; Treloar, A.F.; Matthews, R.T.; Jay, D.G. CD155/PVR enhances glioma cell dispersal by regulating adhesion signaling and focal adhesion dynamics. Cancer Res. 2005, 65, 10930–10937. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, A.; Campbell, D.; Hannum, C.; Yssel, H.; Franz-Bacon, K.; McClanahan, T.; Kitamura, T.; Nicholl, J.; Sutherland, G.R.; Lanier, L.L.; et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity 1996, 4, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Iguchi-Manaka, A.; Kai, H.; Yamashita, Y.; Shibata, K.; Tahara-Hanaoka, S.; Honda, S.; Yasui, T.; Kikutani, H.; Shibuya, K.; Shibuya, A. Accelerated tumor growth in mice deficient in DNAM-1 receptor. J. Exp. Med. 2008, 205, 2959–2964. [Google Scholar] [CrossRef]
- Gilfillan, S.; Chan, C.J.; Cella, M.; Haynes, N.M.; Rapaport, A.S.; Boles, K.S.; Andrews, D.M.; Smyth, M.J.; Colonna, M. DNAM-1 promotes activation of cytotoxic lymphocytes by nonprofessional antigen-presenting cells and tumors. J. Exp. Med. 2008, 205, 2965–2973. [Google Scholar] [CrossRef] [PubMed]
- Tahara-Hanaoka, S.; Shibuya, K.; Kai, H.; Miyamoto, A.; Morikawa, Y.; Ohkochi, N.; Honda, S.; Shibuya, A. Tumor rejection by the poliovirus receptor family ligands of the DNAM-1 (CD226) receptor. Blood 2006, 107, 1491–1496. [Google Scholar] [CrossRef]
- Chan, C.J.; Andrews, D.M.; McLaughlin, N.M.; Yagita, H.; Gilfillan, S.; Colonna, M.; Smyth, M.J. DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J. Immunol. 2010, 184, 902–911. [Google Scholar] [CrossRef]
- Castriconi, R.; Dondero, A.; Corrias, M.V.; Lanino, E.; Pende, D.; Moretta, L.; Bottino, C.; Moretta, A. Natural killer cell-mediated killing of freshly isolated neuroblastoma cells: Critical role of DNAX accessory molecule-1-poliovirus receptor interaction. Cancer Res. 2004, 64, 9180–9184. [Google Scholar] [CrossRef]
- Pende, D.; Spaggiari, G.M.; Marcenaro, S.; Martini, S.; Rivera, P.; Capobianco, A.; Falco, M.; Lanino, E.; Pierri, I.; Zambello, R.; et al. Analysis of the receptor-ligand interactions in the natural killer-mediated lysis of freshly isolated myeloid or lymphoblastic leukemias: Evidence for the involvement of the Poliovirus receptor (CD155) and Nectin-2 (CD112). Blood 2005, 105, 2066–2073. [Google Scholar] [CrossRef]
- Carlsten, M.; Björkström, N.K.; Norell, H.; Bryceson, Y.; van Hall, T.; Baumann, B.C.; Hanson, M.; Schedvins, K.; Kiessling, R.; Ljunggren, H.G.; et al. DNAX accessory molecule-1 mediated recognition of freshly isolated ovarian carcinoma by resting natural killer cells. Cancer Res. 2007, 67, 1317–1325. [Google Scholar] [CrossRef]
- El-Sherbiny, Y.M.; Meade, J.L.; Holmes, T.D.; McGonagle, D.; Mackie, S.L.; Morgan, A.W.; Cook, G.; Feyler, S.; Richards, S.J.; Davies, F.E.; et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res. 2007, 67, 8444–8449. [Google Scholar] [CrossRef]
- Lakshmikanth, T.; Burke, S.; Ali, T.H.; Kimpfler, S.; Ursini, F.; Ruggeri, L.; Capanni, M.; Umansky, V.; Paschen, A.; Sucker, A.; et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J. Clin. Investig. 2009, 119, 1251–1263. [Google Scholar] [CrossRef] [PubMed]
- Iguchi-Manaka, A.; Okumura, G.; Kojima, H.; Cho, Y.; Hirochika, R.; Bando, H.; Sato, T.; Yoshikawa, H.; Hara, H.; Shibuya, A.; et al. Increased Soluble CD155 in the Serum of Cancer Patients. PLoS ONE 2016, 11, e0152982. [Google Scholar] [CrossRef] [PubMed]
- Okumura, G.; Iguchi-Manaka, A.; Murata, R.; Yamashita-Kanemaru, Y.; Shibuya, A.; Shibuya, K. Tumor-derived soluble CD155 inhibits DNAM-1-mediated antitumor activity of natural killer cells. J. Exp. Med. 2020, 217, 1. [Google Scholar] [CrossRef]
- Braun, M.; Aguilera, A.R.; Sundarrajan, A.; Corvino, D.; Stannard, K.; Krumeich, S.; Das, I.; Lima, L.G.; Meza Guzman, L.G.; Li, K.; et al. CD155 on Tumor Cells Drives Resistance to Immunotherapy by Inducing the Degradation of the Activating Receptor CD226 in CD8+ T Cells. Immunity 2020, 53, 805–823.e15. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, M.; Norell, H.; Bryceson, Y.T.; Poschke, I.; Schedvins, K.; Ljunggren, H.G.; Kiessling, R.; Malmberg, K.J. Primary human tumor cells expressing CD155 impair tumor targeting by down-regulating DNAM-1 on NK cells. J. Immunol. 2009, 183, 4921–4930. [Google Scholar] [CrossRef]
- Carlsten, M.; Baumann, B.C.; Simonsson, M.; Jädersten, M.; Forsblom, A.M.; Hammarstedt, C.; Bryceson, Y.T.; Ljunggren, H.G.; Hellström-Lindberg, E.; Malmberg, K.J. Reduced DNAM-1 expression on bone marrow NK cells associated with impaired killing of CD34+ blasts in myelodysplastic syndrome. Leukemia 2010, 24, 1607–1616. [Google Scholar] [CrossRef]
- Sanchez-Correa, B.; Gayoso, I.; Bergua, J.M.; Casado, J.G.; Morgado, S.; Solana, R.; Tarazona, R. Decreased expression of DNAM-1 on NK cells from acute myeloid leukemia patients. Immunol. Cell Biol. 2012, 90, 109–115. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Bi, J.; Tian, Z. NK Cell Dysfunction and Checkpoint Immunotherapy. Front. Immunol. 2019, 10, 1999. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, W.; Jankovic, V.; Golubov, J.; Poon, P.; Oswald, E.M.; Gurer, C.; Wei, J.; Ramos, I.; Wu, Q.; et al. Combination cancer immunotherapy targeting PD-1 and GITR can rescue CD8+ T cell dysfunction and maintain memory phenotype. Sci. Immunol. 2018, 3, eaat7061. [Google Scholar] [CrossRef]
- Banta, K.L.; Xu, X.; Chitre, A.S.; Au-Yeung, A.; Takahashi, C.; O’Gorman, W.E.; Wu, T.D.; Mittman, S.; Cubas, R.; Comps-Agrar, L.; et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8+ T cell responses. Immunity 2022, 55, 512–526.e9. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Huang, Q.; Huang, M.; Wen, H.; Lin, R.; Zheng, M.; Qu, K.; Li, K.; Wei, H.; Xiao, W.; et al. Human CD96 Correlates to Natural Killer Cell Exhaustion and Predicts the Prognosis of Human Hepatocellular Carcinoma. Hepatology 2019, 70, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.A.; Schirm, D.; Bendzick, L.; Hopps, R.; Selleck, C.; Hinderlie, P.; Felices, M.; Miller, J.S. Balanced engagement of activating and inhibitory receptors mitigates human NK cell exhaustion. JCI Insight 2022, 7, e150079. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Wherry, E.J. TIGIT and CD226: Tipping the balance between costimulatory and coinhibitory molecules to augment the cancer immunotherapy toolkit. Cancer Cell 2014, 26, 785–787. [Google Scholar] [CrossRef] [PubMed]
- Milito, N.D.; Zingoni, A.; Stabile, H.; Soriani, A.; Capuano, C.; Cippitelli, M.; Gismondi, A.; Santoni, A.; Paolini, R.; Molfetta, R. NKG2D engagement on human NK cells leads to DNAM-1 hypo-responsiveness through different converging mechanisms. Eur. J. Immunol. 2023, 53, e2250198. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef]
- Stanietsky, N.; Rovis, T.L.; Glasner, A.; Seidel, E.; Tsukerman, P.; Yamin, R.; Enk, J.; Jonjic, S.; Mandelboim, O. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with PVR. Eur. J. Immunol. 2013, 43, 2138–2150. [Google Scholar] [CrossRef]
- Lozano, E.; Dominguez-Villar, M.; Kuchroo, V.; Hafler, D.A. The TIGIT/CD226 axis regulates human T cell function. J. Immunol. 2012, 188, 3869–3875. [Google Scholar] [CrossRef]
- Harjunpää, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.J.; Martinet, L.; Gilfillan, S.; Souza-Fonseca-Guimaraes, F.; Chow, M.T.; Town, L.; Ritchie, D.S.; Colonna, M.; Andrews, D.M.; Smyth, M.J. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 2014, 15, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, H.; Ravens, I.; Papadogianni, G.; Bernhardt, G. Coming of Age: CD96 Emerges as Modulator of Immune Responses. Front. Immunol. 2018, 9, 1072. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Li, M.; Hu, D.; Li, C.; Ge, B.; Jin, B.; Fan, Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013, 20, 456–464. [Google Scholar] [CrossRef]
- Li, M.; Xia, P.; Du, Y.; Liu, S.; Huang, G.; Chen, J.; Zhang, H.; Hou, N.; Cheng, X.; Zhou, L.; et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-γ production of natural killer cells via β-arrestin 2-mediated negative signaling. J. Biol. Chem. 2014, 289, 17647–17657. [Google Scholar] [CrossRef] [PubMed]
- Kurtulus, S.; Sakuishi, K.; Ngiow, S.F.; Joller, N.; Tan, D.J.; Teng, M.W.; Smyth, M.J.; Kuchroo, V.K.; Anderson, A.C. TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Investig. 2015, 125, 4053–4062. [Google Scholar] [CrossRef]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Fuchs, A.; Cella, M.; Giurisato, E.; Shaw, A.S.; Colonna, M. Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155). J. Immunol. 2004, 172, 3994–3998. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.J.; Stannard, K.; Liu, J.; Allen, S.; Yong, M.C.; Mittal, D.; Aguilera, A.R.; Miles, J.J.; Lutzky, V.P.; de Andrade, L.F.; et al. Suppression of Metastases Using a New Lymphocyte Checkpoint Target for Cancer Immunotherapy. Cancer Discov. 2016, 6, 446–459. [Google Scholar] [CrossRef]
- Roman Aguilera, A.; Lutzky, V.P.; Mittal, D.; Li, X.Y.; Stannard, K.; Takeda, K.; Bernhardt, G.; Teng, M.W.L.; Dougall, W.C.; Smyth, M.J. CD96 targeted antibodies need not block CD96-CD155 interactions to promote NK cell anti-metastatic activity. Oncoimmunology 2018, 7, e1424677. [Google Scholar] [CrossRef]
- Mittal, D.; Lepletier, A.; Madore, J.; Aguilera, A.R.; Stannard, K.; Blake, S.J.; Whitehall, V.L.J.; Liu, C.; Bettington, M.L.; Takeda, K.; et al. CD96 Is an Immune Checkpoint That Regulates CD8+ T-cell Antitumor Function. Cancer Immunol. Res. 2019, 7, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Chiang, E.Y.; de Almeida, P.E.; de AlmeidaNagata, D.E.; Bowles, K.H.; Du, X.; Chitre, A.S. CD96 Functions as a Co-Stimulatory Receptor to Enhance CD8(+) T Cell Activation and Effector Responses. Eur. J. Immunol. 2020, 50, 891–902. [Google Scholar] [CrossRef]
- Rogel, A.; Ibrahim, F.M.; Thirdborough, S.M.; Renart-Depontieu, F.; Birts, C.N.; Buchan, S.L.; Preville, X.; King, E.V.; Al-Shamkhani, A. Fcγ receptor-mediated cross-linking codefines the immunostimulatory activity of anti-human CD96 antibodies. JCI Insight 2022, 7, e158444. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.; Seth, S.; Albrecht, J.; Maier, M.K.; du Pasquier, L.; Ravens, I.; Dreyer, L.; Burger, R.; Gramatzki, M.; Schwinzer, R.; et al. CD96 interaction with CD155 via its first Ig-like domain is modulated by alternative splicing or mutations in distal Ig-like domains. J. Biol. Chem. 2009, 284, 2235–2244. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Luo, C.; Liu, F.; Liu, Z.; Chen, F. CD96 Correlates With Immune Infiltration and Impacts Patient Prognosis: A Pan-Cancer Analysis. Front. Oncol. 2021, 11, 634617. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Hamad, A.; Yusubalieva, G.M.; Baklaushev, V.P.; Chumakov, P.M.; Lipatova, A.V. Recent Developments in Glioblastoma Therapy: Oncolytic Viruses and Emerging Future Strategies. Viruses 2023, 15, 547. [Google Scholar] [CrossRef]
- Merrill, M.K.; Bernhardt, G.; Sampson, J.H.; Wikstrand, C.J.; Bigner, D.D.; Gromeier, M. Poliovirus receptor CD155-targeted oncolysis of glioma. Neuro. Oncol. 2004, 6, 208–217. [Google Scholar] [CrossRef]
- Cello, J.; Toyoda, H.; Dejesus, N.; Dobrikova, E.Y.; Gromeier, M.; Wimmer, E. Growth phenotypes and biosafety profiles in poliovirus-receptor transgenic mice of recombinant oncolytic polio/human rhinoviruses. J. Med. Virol. 2008, 80, 352–359. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Herndon JE 2nd Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; Peters, K.B.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Thompson, E.M.; Landi, D.; Brown, M.C.; Friedman, H.S.; McLendon, R.; Herndon JE 2nd Buckley, E.; Bolognesi, D.P.; Lipp, E.; Schroeder, K.; Becher, O.J.; et al. Recombinant polio-rhinovirus immunotherapy for recurrent paediatric high-grade glioma: A phase 1b trial. Lancet Child. Adolesc. Health 2023, 7, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, H.; Yin, J.; Mueller, S.; Wimmer, E.; Cello, J. Oncolytic treatment and cure of neuroblastoma by a novel attenuated poliovirus in a novel poliovirus-susceptible animal model. Cancer Res. 2007, 67, 2857–2864. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, H.; Wimmer, E.; Cello, J. Oncolytic poliovirus therapy and immunization with poliovirus-infected cell lysate induces potent antitumor immunity against neuroblastoma in vivo. Int. J. Oncol. 2011, 38, 81–87. [Google Scholar] [PubMed]
- Holl, E.K.; Brown, M.C.; Boczkowski, D.; McNamara, M.A.; George, D.J.; Bigner, D.D.; Gromeier, M.; Nair, S.K. Recombinant oncolytic poliovirus, PVSRIPO, has potent cytotoxic and innate inflammatory effects, mediating therapy in human breast and prostate cancer xenograft models. Oncotarget 2016, 7, 79828–79841. [Google Scholar] [CrossRef]
- Atsumi, S.; Matsumine, A.; Toyoda, H.; Niimi, R.; Iino, T.; Nakamura, T.; Matsubara, T.; Asanuma, K.; Komada, Y.; Uchida, A.; et al. Oncolytic virotherapy for human bone and soft tissue sarcomas using live attenuated poliovirus. Int. J. Oncol. 2012, 41, 893–902. [Google Scholar] [CrossRef]
- Beasley, G.M.; Nair, S.K.; Farrow, N.E.; Landa, K.; Selim, M.A.; Wiggs, C.A.; Jung, S.H.; Bigner, D.D.; True Kelly, A.; Gromeier, M.; et al. Phase I trial of intratumoral PVSRIPO in patients with unresectable, treatment-refractory melanoma. J. Immunother. Cancer 2021, 9, e002203. [Google Scholar] [CrossRef]
- Brown, M.C.; Holl, E.K.; Boczkowski, D.; Dobrikova, E.; Mosaheb, M.; Chandramohan, V.; Bigner, D.D.; Gromeier, M.; Nair, S.K. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci. Transl. Med. 2017, 9, eaan4220. [Google Scholar] [CrossRef]
- Mosaheb, M.M.; Dobrikova, E.Y.; Brown, M.C.; Yang, Y.; Cable, J.; Okada, H.; Nair, S.K.; Bigner, D.D.; Ashley, D.M.; Gromeier, M. Genetically stable poliovirus vectors activate dendritic cells and prime antitumor CD8 T cell immunity. Nat. Commun. 2020, 11, 524. [Google Scholar] [CrossRef]
- Brown, M.C.; Mosaheb, M.M.; Mohme, M.; McKay, Z.P.; Holl, E.K.; Kastan, J.P.; Yang, Y.; Beasley, G.M.; Hwang, E.S.; Ashley, D.M.; et al. Viral infection of cells within the tumor microenvironment mediates antitumor immunotherapy via selective TBK1-IRF3 signaling. Nat. Commun. 2021, 12, 1858. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.H.; Oh, E.; Kim, Y.; Nam, D.H.; Jeon, S.Y.; Yu, J.H.; Minn, D. Recent Advances in CAR-Based Solid Tumor Immunotherapy. Cells 2023, 12, 1606. [Google Scholar] [CrossRef]
- Reymond, N.; Imbert, A.M.; Devilard, E.; Fabre, S.; Chabannon, C.; Xerri, L.; Farnarier, C.; Cantoni, C.; Bottino, C.; Moretta, A.; et al. DNAM-1 and PVR regulate monocyte migration through endothelial junctions. J. Exp. Med. 2004, 199, 1331–1341. [Google Scholar] [CrossRef]
- Wu, M.R.; Zhang, T.; Alcon, A.; Sentman, C.L. DNAM-1-based chimeric antigen receptors enhance T cell effector function and exhibit in vivo efficacy against melanoma. Cancer Immunol. Immunother. 2015, 64, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Kilgour, M.K.; Bastin, D.J.; Lee, S.H.; Ardolino, M.; McComb, S.; Visram, A. Advancements in CAR-NK therapy: Lessons to be learned from CAR-T therapy. Front. Immunol. 2023, 14, 1166038. [Google Scholar] [CrossRef] [PubMed]
- Focaccetti, C.; Benvenuto, M.; Pighi, C.; Vitelli, A.; Napolitano, F.; Cotugno, N.; Fruci, D.; Palma, P.; Rossi, P.; Bei, R.; et al. DNAM-1-chimeric receptor-engineered NK cells, combined with Nutlin-3a, more effectively fight neuroblastoma cells in vitro: A proof-of-concept study. Front. Immunol. 2022, 13, 886319. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.H.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Thibaudin, M.; Limagne, E.; Hampe, L.; Ballot, E.; Truntzer, C.; Ghiringhelli, F. Targeting PD-L1 and TIGIT could restore intratumoral CD8 T cell function in human colorectal cancer. Cancer Immunol. Immunother. 2022, 71, 2549–2563. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.C.; Abreu, D.R.; Hussein, M.; Cobo, M.; Patel, A.J.; Secen, N.; Lee, K.H.; Massuti, B.; Hiret, S.; Yang, J.C.H.; et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): Primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. 2022, 23, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Maurice-Dror, C.; Lee, D.H.; Kim, D.W.; Nagrial, A.; Voskoboynik, M.; Chung, H.C.; Mileham, K.; Vaishampayan, U.; Rasco, D.; et al. First-in-human phase 1 study of the anti-TIGIT antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer. Ann. Oncol. 2022, 33, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Florou, V.; Garrido-Laguna, I. Clinical Development of Anti-TIGIT Antibodies for Immunotherapy of Cancer. Curr. Oncol. Rep. 2022, 24, 1107–1112. [Google Scholar] [CrossRef]
- Mettu, N.B.; Ulahannan, S.V.; Bendell, J.C.; Garrido-Laguna, I.; Strickler, J.H.; Moore, K.N.; Stagg, R.; Kapoun, A.M.; Faoro, L.; Sharma, S. A Phase 1a/b Open-Label, Dose-Escalation Study of Etigilimab Alone or in Combination with Nivolumab in Patients with Locally Advanced or Metastatic Solid Tumors. Clin. Cancer Res. 2022, 28, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Kim, S.; St Jean, N.; O’Brien, S.; Lian, L.; Sun, J.; Verona, R.I.; Moon, E. Addition of anti-TIM3 or anti-TIGIT Antibodies to anti-PD1 Blockade Augments Human T cell Adoptive Cell Transfer. Oncoimmunology 2021, 10, 1873607. [Google Scholar] [CrossRef]
- Brooks, J.; Fleischmann-Mundt, B.; Woller, N.; Niemann, J.; Ribback, S.; Peters, K.; Demir, I.E.; Armbrecht, N.; Ceyhan, G.O.; Manns, M.P.; et al. Perioperative, Spatiotemporally Coordinated Activation of T and NK Cells Prevents Recurrence of Pancreatic Cancer. Cancer Res. 2018, 78, 475–488. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, C.; Qiu, J.; Qu, X.; Peng, J.; Lu, C.; Zhang, M.; Zhang, M.; Qi, X.; Li, G.; et al. Targeting CD96 overcomes PD-1 blockade resistance by enhancing CD8+ TIL function in cervical cancer. J. Immunother. Cancer 2022, 10, e003667. [Google Scholar] [CrossRef]
- Ozmadenci, D.; Shankara Narayanan, J.S.; Andrew, J.; Ojalill, M.; Barrie, A.M.; Jiang, S.; Iyer, S.; Chen, X.L.; Rose, M.; Estrada, V.; et al. Tumor FAK orchestrates immunosuppression in ovarian cancer via the CD155/TIGIT axis. Proc. Natl. Acad. Sci. USA 2022, 119, e2117065119. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Therapeutic Agent | Tumor | Clinical Phase | Identification Number |
|---|---|---|---|
| Tiragolumab | NSCLC | III | NCT04294810 |
| Vibostolimab | NSCLC Melanoma | I/II I/II | NCT04165070 NCT04305041 |
| Domvanalimab | NSCLC Metastatic NSLC | II III | NCT04262856 NCT04736173 |
| Etigilimab | Metastatic solid tumors | I/II | NCT04761198 |
| Ociperlimab | Cervical cancer NSCLC Esophageal Squamous Cell Carcinoma | II III II | NCT04693234 NCT04746924 NCT04732494 |
| EOS-448 | Advanced cancers | I | NCT04335253 |
| BMS-986207 | Multiple Myeloma Solid tumors | I/II | NCT04150965 NCT02913313 |
| ASP8374 | Solid tumors | I | NCT03260322 |
| COM902 | Advanced cancers | I | NCT04354246 |
| IBI939 | NSCLC | I | NCT04672369 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paolini, R.; Molfetta, R. CD155 and Its Receptors as Targets for Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 12958. https://doi.org/10.3390/ijms241612958
Paolini R, Molfetta R. CD155 and Its Receptors as Targets for Cancer Therapy. International Journal of Molecular Sciences. 2023; 24(16):12958. https://doi.org/10.3390/ijms241612958
Chicago/Turabian StylePaolini, Rossella, and Rosa Molfetta. 2023. "CD155 and Its Receptors as Targets for Cancer Therapy" International Journal of Molecular Sciences 24, no. 16: 12958. https://doi.org/10.3390/ijms241612958