
Transcriptional Response to Standard AML Drugs Identifies Synergistic Combinations
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. AML Cell Lines Exhibit Variable Response to Standard Drugs
2.2. Cytotoxic Response of AML Cells Correlates with the Transcription Response
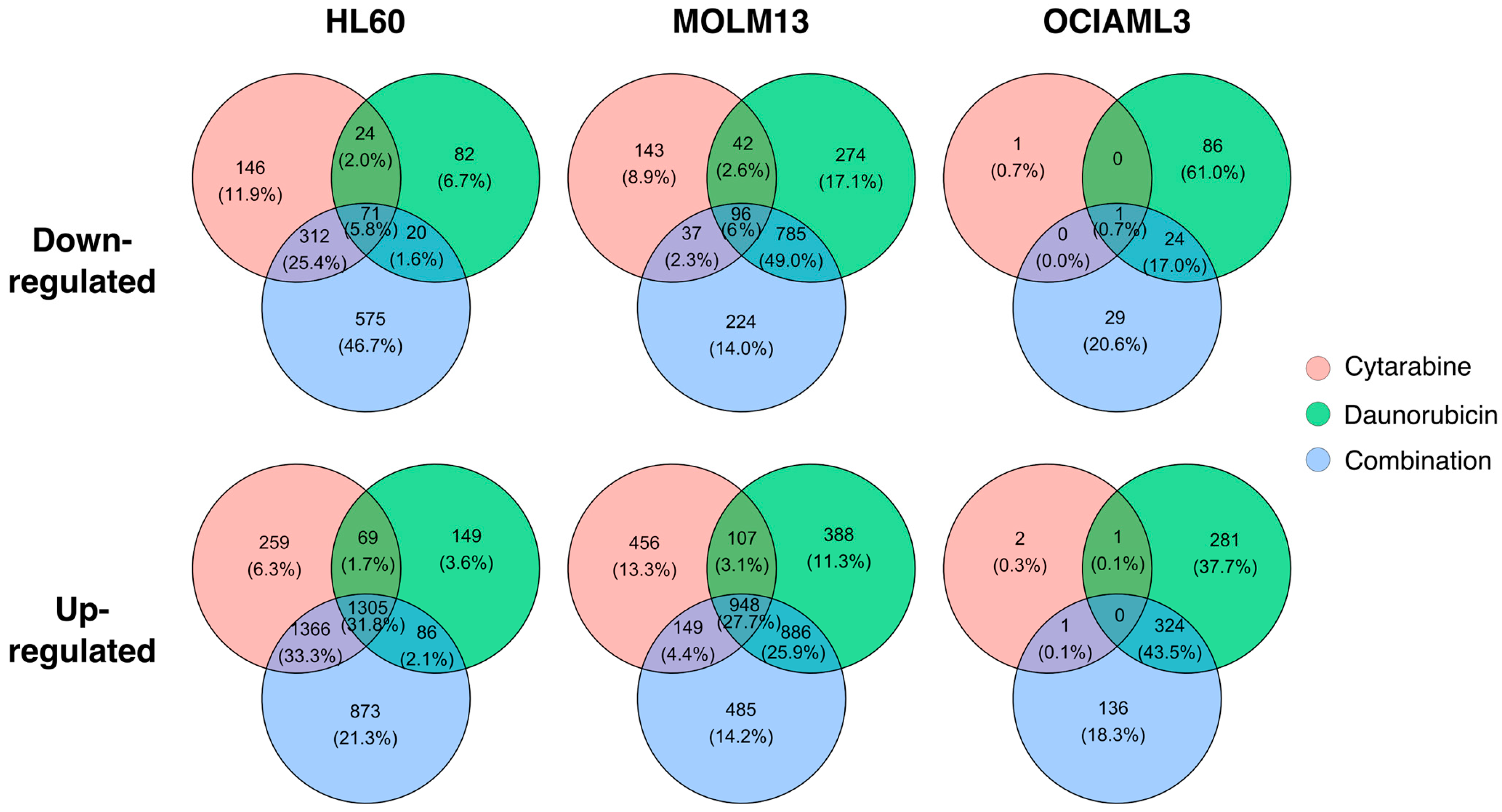
2.3. Transcription Response of AML Cells Vary between Individual Drugs and the Combination
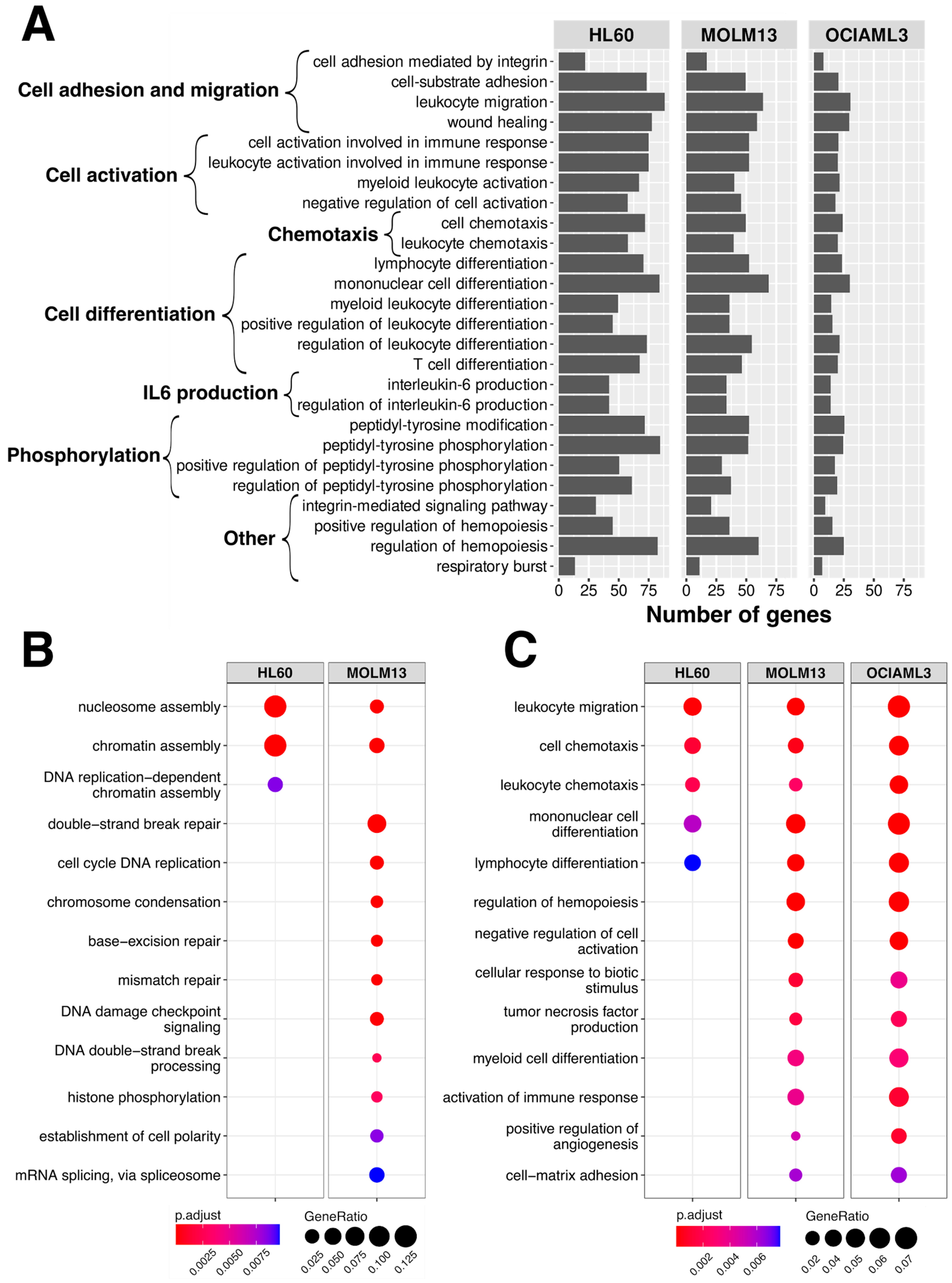
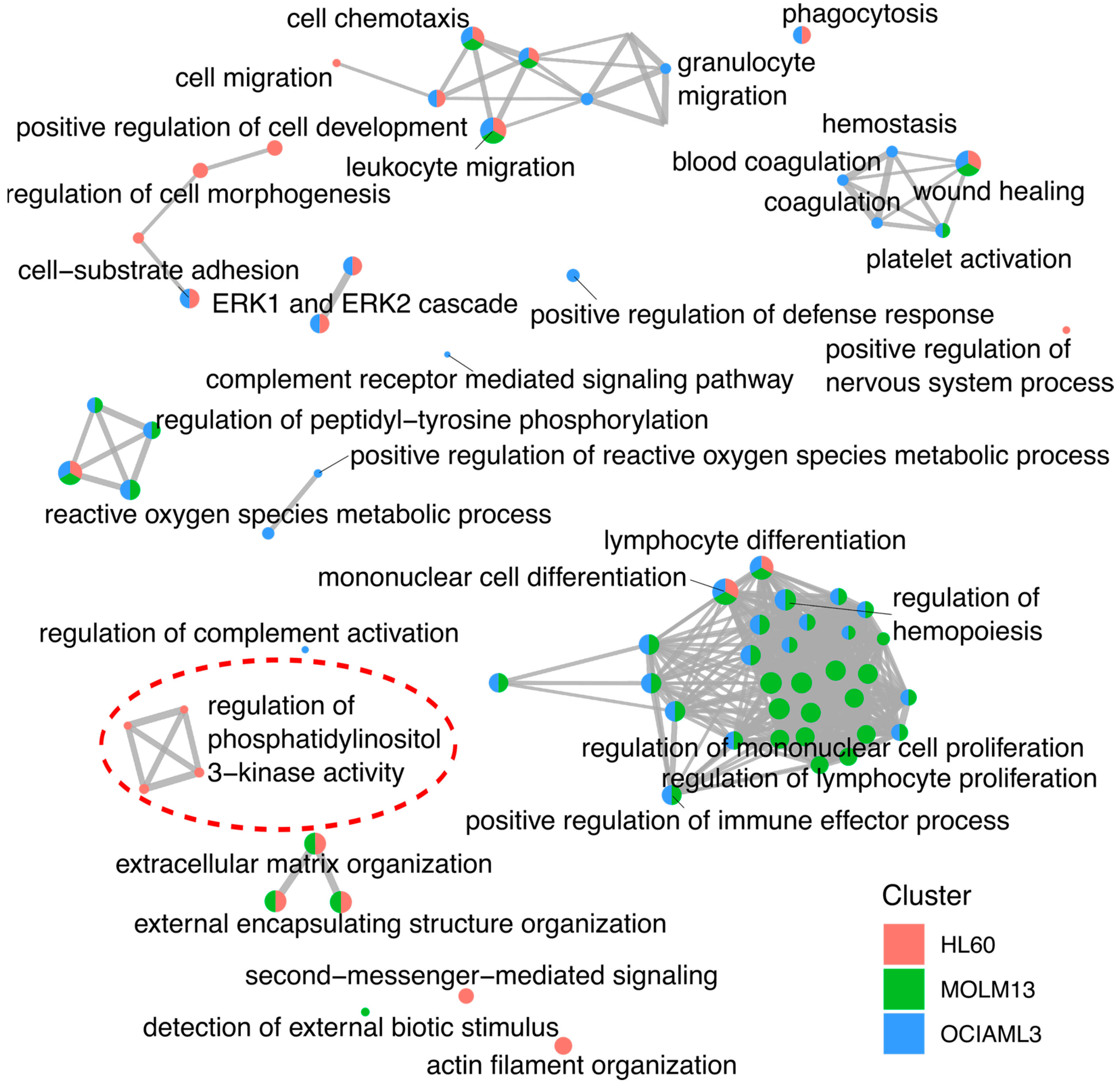
2.4. Pathway Annotation of the Gene Expression Changes
2.5. Gene Expression Changes Predict Daunorubicin Combination Partners
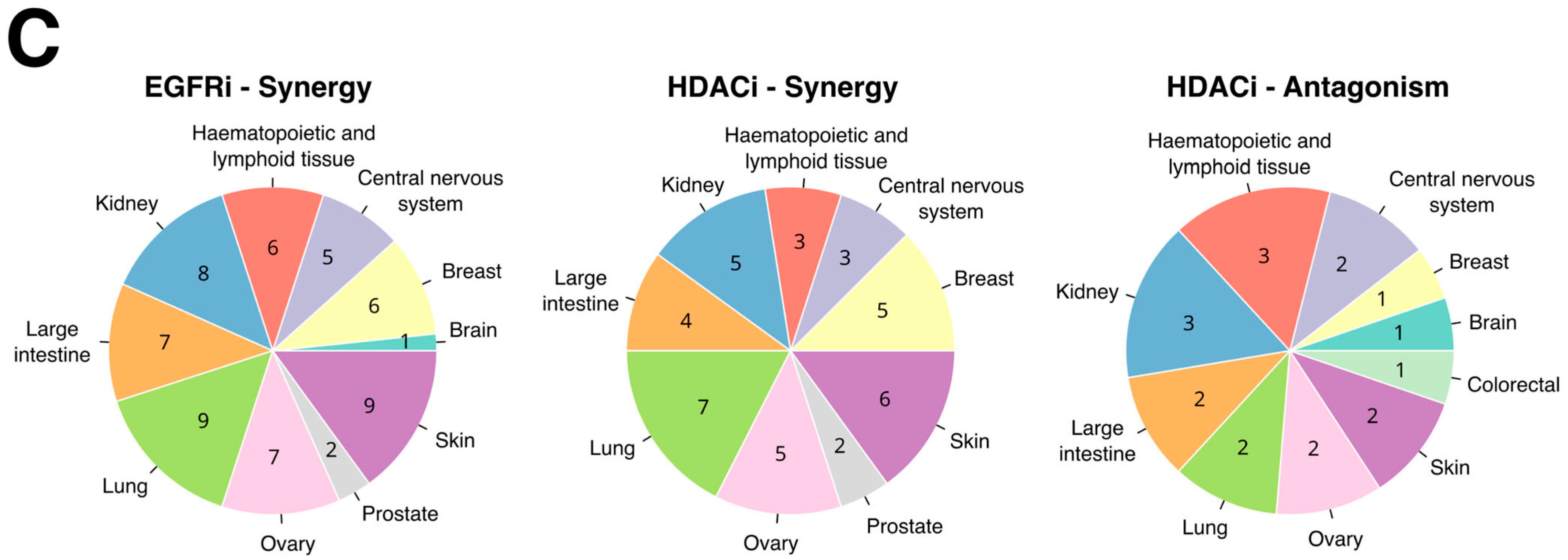
2.6. Predicted Combinations Exert Synergistic Response in AML Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Drugs Treatment
4.2. Primary Patient Samples
4.3. CellTiter-Glo Cell Toxicity Assay
4.4. RNA Extraction and Library Preparation
4.5. RNA-Seq and Downstream Analysis
4.6. Daunorubicin Combination Identification
4.7. Daunorubicin Combination Verification
4.8. Statistical Association of the Drug Synergy with Genomic Markers
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daver, N.; Wei, A.H.; Pollyea, D.A.; Fathi, A.T.; Vyas, P.; DiNardo, C.D. New Directions for Emerging Therapies in Acute Myeloid Leukemia: The next Chapter. Blood Cancer J. 2020, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, R.; Paterno, G.; De Bellis, E.; Mercante, L.; Buzzatti, E.; Esposito, F.; Del Principe, M.I.; Maurillo, L.; Buccisano, F.; Venditti, A. Therapeutic Choice in Older Patients with Acute Myeloid Leukemia: A Matter of Fitness. Cancers 2020, 12, 120. [Google Scholar] [CrossRef] [PubMed]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International Phase 3 Study of Azacitidine vs Conventional Care Regimens in Older Patients with Newly Diagnosed AML with >30% Blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef] [PubMed]
- De Lima, M.; Roboz, G.J.; Platzbecker, U.; Craddock, C.; Ossenkoppele, G. AML and the Art of Remission Maintenance. Blood Rev. 2021, 49, 100829. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Kadia, T.M.; DiNardo, C.D.; Welch, M.A.; Ravandi, F. Acute Myeloid Leukemia: Treatment and Research Outlook for 2021 and the MD Anderson Approach. Cancer 2021, 127, 1186–1207. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Löwenberg, B. Towards Precision Medicine for AML. Nat. Rev. Clin. Oncol. 2021, 18, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.-Y.; Li, J.-F.; Zhu, Y.-M.; Lin, X.-J.; Wen, L.-J.; Zhang, F.; Zhang, Y.-L.; Zhao, M.; Fang, H.; Wang, S.-Y.; et al. Transcriptome-Based Molecular Subtypes and Differentiation Hierarchies Improve the Classification Framework of Acute Myeloid Leukemia. Proc. Natl. Acad. Sci. USA 2022, 119, e2211429119. [Google Scholar] [CrossRef] [PubMed]
- Small, S.; Oh, T.S.; Platanias, L.C. Role of Biomarkers in the Management of Acute Myeloid Leukemia. Int. J. Mol. Sci. 2022, 23, 14543. [Google Scholar] [CrossRef]
- Walker, C.J.; Mrózek, K.; Ozer, H.G.; Nicolet, D.; Kohlschmidt, J.; Papaioannou, D.; Genutis, L.K.; Bill, M.; Powell, B.L.; Uy, G.L.; et al. Gene Expression Signature Predicts Relapse in Adult Patients with Cytogenetically Normal Acute Myeloid Leukemia. Blood Adv. 2021, 5, 1474–1482. [Google Scholar] [CrossRef]
- Zhu, Y.-M.; Wang, P.-P.; Huang, J.-Y.; Chen, Y.-S.; Chen, B.; Dai, Y.-J.; Yan, H.; Hu, Y.; Cheng, W.-Y.; Ma, T.-T.; et al. Gene Mutational Pattern and Expression Level in 560 Acute Myeloid Leukemia Patients and Their Clinical Relevance. J. Transl. Med. 2017, 15, 178. [Google Scholar] [CrossRef]
- Palve, V.; Liao, Y.; Remsing Rix, L.L.; Rix, U. Turning Liabilities into Opportunities: Off-Target Based Drug Repurposing in Cancer. Semin. Cancer Biol. 2021, 68, 209–229. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Feng, Q.; Kerchberger, V.E.; Nelson, S.D.; Chen, Q.; Li, B.; Edwards, T.L.; Cox, N.J.; Phillips, E.J.; Stein, C.M.; et al. Integrating Gene Expression and Clinical Data to Identify Drug Repurposing Candidates for Hyperlipidemia and Hypertension. Nat. Commun. 2022, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- More, P.; Goedtel-Armbrust, U.; Shah, V.; Mathaes, M.; Kindler, T.; Andrade-Navarro, M.A.; Wojnowski, L. Drivers of Topoisomerase II Poisoning Mimic and Complement Cytotoxicity in AML Cells. Oncotarget 2019, 10, 5298–5312. [Google Scholar] [CrossRef] [PubMed]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of Cancer Therapeutic Targets Using CRISPR-Cas9 Screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Silva, E.; Ideker, T. Transcriptional Responses to DNA Damage. DNA Repair 2019, 79, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Steurer, B.; Janssens, R.C.; Geijer, M.E.; Aprile-Garcia, F.; Geverts, B.; Theil, A.F.; Hummel, B.; van Royen, M.E.; Evers, B.; Bernards, R.; et al. DNA Damage-Induced Transcription Stress Triggers the Genome-Wide Degradation of Promoter-Bound Pol II. Nat. Commun. 2022, 13, 3624. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, L.H.; Svejstrup, J.Q. The Cellular Response to Transcription-Blocking DNA Damage. Trends Biochem. Sci. 2018, 43, 327–341. [Google Scholar] [CrossRef]
- Wei, F.; Hao, P.; Zhang, X.; Hu, H.; Jiang, D.; Yin, A.; Wen, L.; Zheng, L.; He, J.Z.; Mei, W.; et al. Etoposide-Induced DNA Damage Affects Multiple Cellular Pathways in Addition to DNA Damage Response. Oncotarget 2018, 9, 24122–24139. [Google Scholar] [CrossRef]
- Tomic, B.; Smoljo, T.; Lalic, H.; Dembitz, V.; Batinic, J.; Batinic, D.; Bedalov, A.; Visnjic, D. Cytarabine-Induced Differentiation of AML Cells Depends on Chk1 Activation and Shares the Mechanism with Inhibitors of DHODH and Pyrimidine Synthesis. Sci. Rep. 2022, 12, 11344. [Google Scholar] [CrossRef]
- Récher, C. Clinical Implications of Inflammation in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 623952. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The Complexity of NF-ΚB Signaling in Inflammation and Cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, I.; Sundkvist, M.; Björn, N.; Gréen, H.; Lotfi, K. Early Changes in Gene Expression Profiles in AML Patients during Induction Chemotherapy. BMC Genom. 2022, 23, 752. [Google Scholar] [CrossRef] [PubMed]
- Abuetabh, Y.; Wu, H.H.; Chai, C.; Al Yousef, H.; Persad, S.; Sergi, C.M.; Leng, R. DNA Damage Response Revisited: The P53 Family and Its Regulators Provide Endless Cancer Therapy Opportunities. Exp. Mol. Med. 2022, 54, 1658–1669. [Google Scholar] [CrossRef] [PubMed]
- Drosten, M.; Sum, E.Y.M.; Lechuga, C.G.; Simón-Carrasco, L.; Jacob, H.K.C.; García-Medina, R.; Huang, S.; Beijersbergen, R.L.; Bernards, R.; Barbacid, M. Loss of P53 Induces Cell Proliferation via Ras-Independent Activation of the Raf/Mek/Erk Signaling Pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 15155–15160. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.K.; Toker, A. The Phosphoinositide 3-Kinase Pathway and Therapy Resistance in Cancer. F1000Prime Rep. 2015, 7, 13. [Google Scholar] [CrossRef]
- Abourehab, M.A.S.; Alqahtani, A.M.; Youssif, B.G.M.; Gouda, A.M. Globally Approved EGFR Inhibitors: Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism. Molecules 2021, 26, 6677. [Google Scholar] [CrossRef]
- de Almeida, L.Y.; Rego, E.M. Is the EGFR Pathway Relevant for the Pathogenesis but Not for Treatment of Acute Myeloid Leukemia? J. Cancer Metastasis Treat. 2021, 7, 57. [Google Scholar] [CrossRef]
- Nath, S.; Bhattacharyya, J.; Sarma, P.P.; Saxena, R.; Sazawal, S.; Barman, M.P.; Saikia, K.K. The Prognostic Impact of Epidermal Growth Factor Receptor (EGFR) in Patients with Acute Myeloid Leukaemia. Indian J. Hematol. Blood Transfus. 2020, 36, 749–753. [Google Scholar] [CrossRef]
- Dalle, I.A.; Cortes, J.E.; Pinnamaneni, P.; Lamothe, B.; Duque, A.D.; Randhawa, J.; Pemmaraju, N.; Jabbour, E.; Ferrajoli, A.; Wierda, W.G.; et al. A Pilot Phase II Study of Erlotinib for the Treatment of Patients with Relapsed/Refractory Acute Myeloid Leukemia. Acta Haematol. 2018, 140, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Machado, C.B.; de Pinho Pessoa, F.M.C.; da Silva, E.L.; da Costa Pantoja, L.; Ribeiro, R.M.; de Moraes Filho, M.O.; de Moraes, M.E.A.; Montenegro, R.C.; Burbano, R.M.R.; Khayat, A.S.; et al. Kinase Inhibition in Relapsed/Refractory Leukemia and Lymphoma Settings: Recent Prospects into Clinical Investigations. Pharmaceutics 2021, 13, 1604. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Penninger, J.M. Mitogen-Activated Protein Kinases in Apoptosis Regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Jia, Y.; Wang, Y.; Cai, X. Dysregulated MAPK Signaling Pathway in Acute Myeloid Leukemia with RUNX1 Mutations. Expert Rev. Hematol. 2022, 15, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Marampon, F.; Ciccarelli, C.; Zani, B.M. Biological Rationale for Targeting MEK/ERK Pathways in Anti-Cancer Therapy and to Potentiate Tumour Responses to Radiation. Int. J. Mol. Sci. 2019, 20, 2530. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Moffat, J.; Schaefer, G.; Chan, J.; Wang, X.; Orr, C.; Cheng, J.; Hunsaker, T.; Shao, L.; Wang, S.J.; et al. Combined MEK and ERK Inhibition Overcomes Therapy-Mediated Pathway Reactivation in RAS Mutant Tumors. PLoS ONE 2017, 12, e0185862. [Google Scholar] [CrossRef] [PubMed]
- Laurent, G.; Jaffrézou, J.-P. Signaling Pathways Activated by Daunorubicin. Blood 2001, 98, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S. Combination Therapies with Kinase Inhibitors for Acute Myeloid Leukemia Treatment. Hematol. Rep. 2023, 15, 331–346. [Google Scholar] [CrossRef]
- Jain, N.; Curran, E.; Iyengar, N.M.; Diaz-Flores, E.; Kunnavakkam, R.; Popplewell, L.; Kirschbaum, M.H.; Karrison, T.; Erba, H.P.; Green, M.; et al. Phase II Study of the Oral MEK Inhibitor Selumetinib in Advanced Acute Myelogenous Leukemia: A University of Chicago Phase II Consortium Trial. Clin. Cancer Res. 2014, 20, 490–498. [Google Scholar] [CrossRef]
- Maiti, A.; Naqvi, K.; Kadia, T.M.; Borthakur, G.; Takahashi, K.; Bose, P.; Daver, N.G.; Patel, A.; Alvarado, Y.; Ohanian, M.; et al. Phase II Trial of MEK Inhibitor Binimetinib (MEK162) in RAS-Mutant Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2019, 19, 142–148.e1. [Google Scholar] [CrossRef]
- Desikan, S.P.; Ravandi, F.; Pemmaraju, N.; Konopleva, M.; Loghavi, S.; Jabbour, E.J.; Daver, N.; Jain, N.; Chien, K.S.; Maiti, A.; et al. A Phase II Study of Azacitidine, Venetoclax, and Trametinib in Relapsed or Refractory Acute Myeloid Leukemia Harboring RAS Pathway-Activating Mutations. Acta Haematol. 2022, 145, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Kayser, S.; Levis, M.J. The Clinical Impact of the Molecular Landscape of Acute Myeloid Leukemia. Haematologica 2023, 108, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Poreba, E.; Durzynska, J. Nuclear Localization and Actions of the Insulin-like Growth Factor 1 (IGF-1) System Components: Transcriptional Regulation and DNA Damage Response. Mutat. Res. Rev. Mutat. Res. 2020, 784, 108307. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, Y.; Xiong, J.; Lan, B.; Wang, X.; Liu, J.; Lin, J.; Fei, Z.; Zheng, X.; Chen, C. Role of PRKDC in Cancer Initiation, Progression, and Treatment. Cancer Cell Int. 2021, 21, 563. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Kizawa, R.; Yoshida, A.; Koizumi, R.; Motohashi, S.; Shimoyama, Y.; Hannya, Y.; Yoshida, S.; Oikawa, T.; Shimoda, M.; et al. Extracellular PKCδ Signals to Epidermal Growth Factor Receptor for Tumor Proliferation in Liver Cancer Cells. Cancer Sci. 2022, 113, 2378–2385. [Google Scholar] [CrossRef]
- Holbeck, S.L.; Camalier, R.; Crowell, J.A.; Govindharajulu, J.P.; Hollingshead, M.; Anderson, L.W.; Polley, E.; Rubinstein, L.; Srivastava, A.; Wilsker, D.; et al. The National Cancer Institute ALMANAC: A Comprehensive Screening Resource for the Detection of Anticancer Drug Pairs with Enhanced Therapeutic Activity. Cancer Res. 2017, 77, 3564–3576. [Google Scholar] [CrossRef] [PubMed]
- Zagidullin, B.; Aldahdooh, J.; Zheng, S.; Wang, W.; Wang, Y.; Saad, J.; Malyutina, A.; Jafari, M.; Tanoli, Z.; Pessia, A.; et al. DrugComb: An Integrative Cancer Drug Combination Data Portal. Nucleic Acids Res. 2019, 47, W43–W51. [Google Scholar] [CrossRef] [PubMed]
- Pabst, C.; Krosl, J.; Fares, I.; Boucher, G.; Ruel, R.; Marinier, A.; Lemieux, S.; Hébert, J.; Sauvageau, G. Identification of Small Molecules That Support Human Leukemia Stem Cell Activity Ex Vivo. Nat. Methods 2014, 11, 436–442. [Google Scholar] [CrossRef]
- Andrews, S. FASTQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 3 June 2022).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- R Core Team R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022.
- Love, M.I.; Soneson, C.; Hickey, P.F.; Johnson, L.K.; Pierce, N.T.; Shepherd, L.; Morgan, M.; Patro, R. Tximeta: Reference Sequence Checksums for Provenance Identification in RNA-Seq. PLoS Comput. Biol. 2020, 16, e1007664. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. G:Profiler: A Web Server for Functional Enrichment Analysis and Conversions of Gene Lists (2019 Update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 3.0: An Interactive Analysis and Consensus Interpretation of Multi-Drug Synergies across Multiple Samples. Nucleic Acids Res. 2022, 50, W739–W743. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic Identification of Genomic Markers of Drug Sensitivity in Cancer Cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef]
- Van der Meer, D.; Barthorpe, S.; Yang, W.; Lightfoot, H.; Hall, C.; Gilbert, J.; Francies, H.E.; Garnett, M.J. Cell Model Passports—A Hub for Clinical, Genetic and Functional Datasets of Preclinical Cancer Models. Nucleic Acids Res. 2019, 47, D923–D929. [Google Scholar] [CrossRef]
- Cokelaer, T.; Chen, E.; Iorio, F.; Menden, M.P.; Lightfoot, H.; Saez-Rodriguez, J.; Garnett, M.J. GDSCTools for Mining Pharmacogenomic Interactions in Cancer. Bioinformatics 2018, 34, 1226–1228. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. R Package Version 1.14.0. 2022. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 28 June 2023).






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
More, P.; Ngaffo, J.A.M.; Goedtel-Armbrust, U.; Hähnel, P.S.; Hartwig, U.F.; Kindler, T.; Wojnowski, L. Transcriptional Response to Standard AML Drugs Identifies Synergistic Combinations. Int. J. Mol. Sci. 2023, 24, 12926. https://doi.org/10.3390/ijms241612926
More P, Ngaffo JAM, Goedtel-Armbrust U, Hähnel PS, Hartwig UF, Kindler T, Wojnowski L. Transcriptional Response to Standard AML Drugs Identifies Synergistic Combinations. International Journal of Molecular Sciences. 2023; 24(16):12926. https://doi.org/10.3390/ijms241612926
Chicago/Turabian StyleMore, Piyush, Joëlle Aurelie Mekontso Ngaffo, Ute Goedtel-Armbrust, Patricia S. Hähnel, Udo F. Hartwig, Thomas Kindler, and Leszek Wojnowski. 2023. "Transcriptional Response to Standard AML Drugs Identifies Synergistic Combinations" International Journal of Molecular Sciences 24, no. 16: 12926. https://doi.org/10.3390/ijms241612926