
SARS-CoV-2 Spike Proteins and Cell–Cell Communication Induce P-Selectin and Markers of Endothelial Injury, NETosis, and Inflammation in Human Lung Microvascular Endothelial Cells and Neutrophils: Implications for the Pathogenesis of COVID-19 Coagulopathy
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Exposure of HLMEC or Neutrophils to S-Proteins and Endothelial–Neutrophil Interactions Increased P-Selectin Transcription
2.2. Exposure of HLMEC to S-Proteins Induced P-Selectin Expression
2.3. Delta Variant S-Proteins Induced Higher P-Selectin Transcription and Expression Than the Wuhan Variant
2.4. Exposure of Human Neutrophils and HLMEC to S-Proteins and Neutrophil–Endothelial Interactions Induces Histone H3 Citrullination
2.5. Exposure of HLMEC and Neutrophils to S-Proteins and Neutrophil–Endothelial Interactions Increased vWF Expression
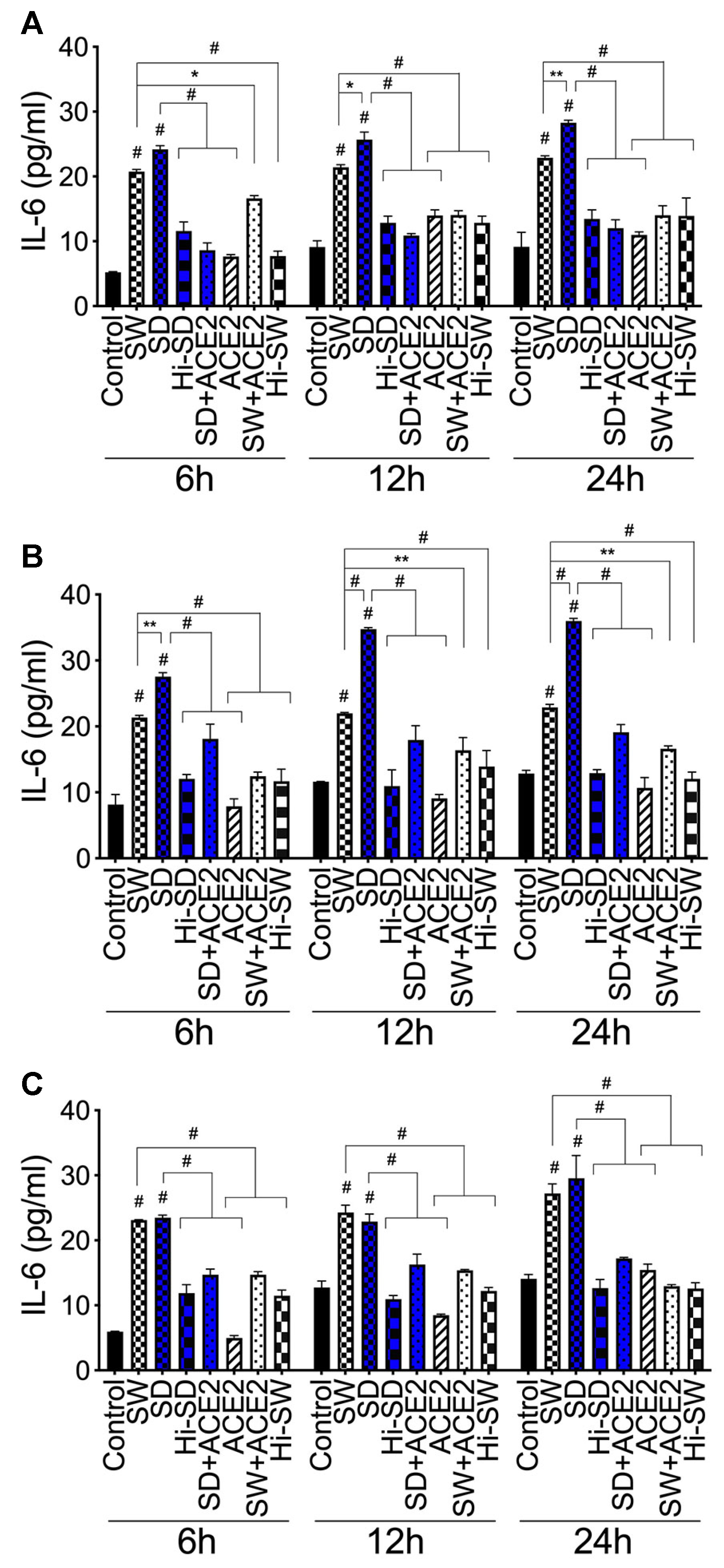
2.6. Exposure of HLMEC and Neutrophils to S-Proteins and Endothelial-Neutrophil Interactions Increased IL-6 Expression
2.7. rTFPI Blocked S-Protein-Induced Citrullination of Histone H3, Expression and Secretion of vWF and IL-6
2.8. DTNB Blocked S-Protein-Induced Citrullination of Histone H3, Expression and Secretion of vWF and IL-6
2.9. Thrombomodulin Blocked S-Protein-Induced Citrullination of Histone H3 and Blocked vWF and IL-6 Expression and Secretion
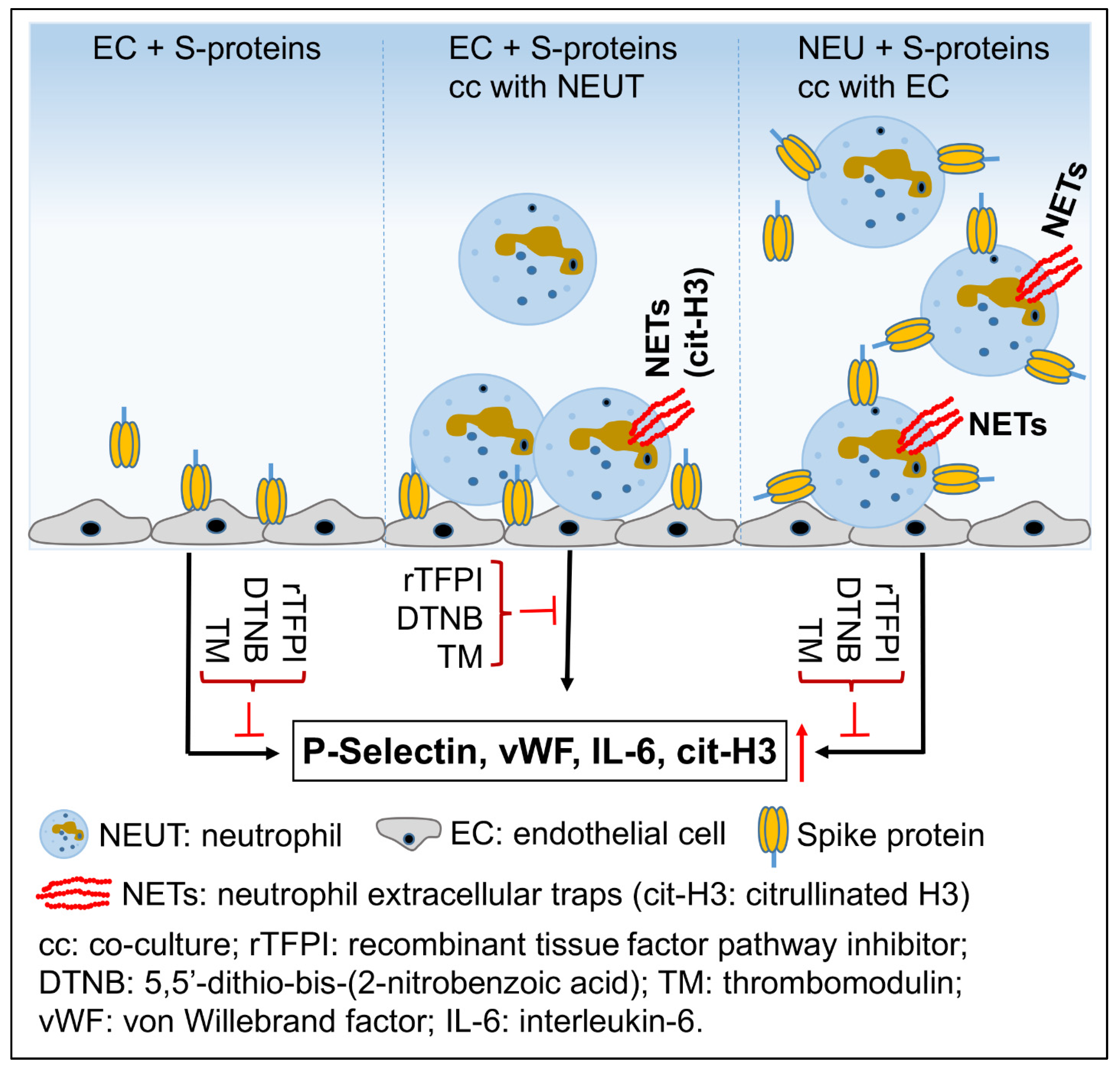
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. HLMEC and Neutrophils
4.3. Cell Treatment and Endothelial–Neutrophil Co-Culture
4.4. RNA Isolation and Real-Time PCR
4.5. Human vWF, cit-H3, and IL-6 ELISA
4.6. Immunofluorescence Analysis
4.7. Western Blot Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DTNB | 5:5′-dithio-bis-(2-nitrobenzoic acid) |
| TFPI | Tissue factor pathway inhibitor |
| rTFPI | Recombinant tissue factor pathway inhibitor |
| BDCA3/TM | Thrombomodulin |
| vWF | von Willebrand factor |
| IL-6 | Interleukin-6 |
| Cit-H3 | Citrulinated histone 3 |
| dsDNA | Double strand DNA |
| NETs | Neutrophils extracellular traps |
| MPO | Myeloperoxidase |
| TF | Tissue factor |
| F-V | Factor-V |
| F-VIII | Factor-VIII |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus-2 |
| COVID-19 | Coronavirus disease 2019 |
| ACE2 | Angiotensin-converting enzyme-2 |
| rhACE2 | Recombinant human ACE2 |
| S-proteins | Spike proteins |
| SW | Spike protein: Wuhan variant |
| SD | Spike protein: delta variant |
| Hi | Heat-inactivated |
| ELISA | Enzyme-linked immunosorbent assay |
| PCR | Polymerase chain reaction |
| cDNA | Complementary DNA |
| GAPDH | Glyceraldehyde-3-Phosphate Dehydrogenase |
References
- JHU. Coronavirus Resource Center: COVID-19 in the USA. 2023. Available online: https://coronavirus.jhu.edu/ (accessed on 24 April 2023).
- CDC. United States COVID-19 Cases and Deaths by State; US Center for Disease Control and Prevention: Atlanta, GA, USA, 2023. Available online: https://www.cdc.gov/covid-data-tracker/#cases (accessed on 24 April 2023).
- WHO. Coronavirus Disease (COVID-19) Pandemic; World Health Organization: Geneva, Switzerland, 2023; Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 24 April 2023).
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- O’Sullivan, J.M.; Gonagle, D.M.; Ward, S.E.; Preston, R.J.S.; O’Donnell, J.S. Endothelial cells orchestrate COVID-19 coagulopathy. Lancet Haematol. 2020, 7, e553–e555. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnoor, M.; Alcaide, P.; Voisin, M.B.; van Buul, J.D. Crossing the Vascular Wall: Common and Unique Mechanisms Exploited by Different Leukocyte Subsets during Extravasation. Mediat. Inflamm. 2015, 2015, 946509. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.K.; Dorosky, D.; Sharma, P.; Abbasi, S.A.; Dye, J.M.; Kranz, D.M.; Herbert, A.S.; Procko, E. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science 2020, 369, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- McKenna, E.; Wubben, R.; Isaza-Correa, J.M.; Melo, A.M.; Mhaonaigh, A.U.; Conlon, N.; O’Donnell, J.S.; Ni Cheallaigh, C.; Hurley, T.; Stevenson, N.J.; et al. Neutrophils in COVID-19: Not Innocent Bystanders. Front. Immunol. 2022, 13, 864387. [Google Scholar] [CrossRef]
- Calvert, B.A.; Quiroz, E.J.; Lorenzana, Z.; Doan, N.; Kim, S.; Senger, C.N.; Anders, J.J.; Wallace, W.D.; Salomon, M.P.; Henley, J.; et al. Neutrophilic inflammation promotes SARS-CoV-2 infectivity and augments the inflammatory responses in airway epithelial cells. Front. Immunol. 2023, 14, 1112870. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Kusunoki, A.; Machida, T.; Hirafuji, M. Angiotensin II reduces membranous angiotensin-converting enzyme 2 in pressurized human aortic endothelial cells. J. Renin Angiotensin Aldosterone Syst. 2009, 10, 210–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhao, Z.; Wang, Y.; Zhou, Y.; Ma, Y.; Zuo, W. Single-Cell RNA Expression Profiling of ACE2, the Receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020, 202, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Morigi, M.; Galbusera, M.; Pezzotta, A.; Gastoldi, S.; Imberti, B.; Perna, A.; Ruggenenti, P.; Donadelli, R.; Benigni, A.; et al. SARS-CoV-2 Spike Protein 1 Activates Microvascular Endothelial Cells and Complement System Leading to Platelet Aggregation. Front. Immunol. 2022, 13, 827146. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Pal, A.C.; Gagnon, J.; Timalsina, S.; Singh, P.; Vydyam, P.; Munshi, M.; Chiu, J.E.; Renard, I.; Harden, C.A.; et al. Evidence for SARS-CoV-2 Spike Protein in the Urine of COVID-19 Patients. Kidney360 2021, 2, 924–936. [Google Scholar] [CrossRef]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetite, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [Green Version]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Zuo, Y.; Zuo, M.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; Shi, H.; Woodard, W.; Lezak, S.P.; Lugogo, N.L.; Knight, J.S.; et al. Neutrophil extracellular traps and thrombosis in COVID-19. J. Thromb. Thrombolysis 2020, 51, 446–453. [Google Scholar] [CrossRef]
- Baugh, R.J.; Broze, G.J., Jr.; Krishnaswamy, S. Regulation of extrinsic pathway factor Xa formation by tissue factor pathway inhibitor. J. Biol. Chem. 1998, 273, 4378–4386. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, M.S.; Birktoft, J.J.; Steer, S.A.; Bajaj, S.P. Structure and biology of tissue factor pathway inhibitor. Thromb. Haemost. 2001, 86, 959–972. [Google Scholar] [PubMed]
- Lwaleed, B.A.; Bass, P.S. Tissue factor pathway inhibitor: Structure, biology and involvement in disease. J. Pathol. 2006, 208, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Maroney, S.A.; Hansen, K.G.; Mast, A.E. Cellular expression and biological activities of alternatively spliced forms of tissue factor pathway inhibitor. Curr. Opin. Hematol. 2013, 20, 403–409. [Google Scholar] [CrossRef] [Green Version]
- Loghmani, H.; Conway, E.M. Exploring traditional and nontraditional roles for thrombomodulin. Blood 2018, 132, 148–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Mauracher, L.M.; Posch, F.; Martinod, K.; Grilz, E.; Daullary, T.; Hell, L.; Brostjan, C.; Zielinski, C.; Ay, C.; Wagner, D.D.; et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J. Thromb. Haemost. 2018, 16, 508–518. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Zhang, Z.; Li, X.; Dong, G.; Zhang, M.; Xu, Z.; Yang, J. Neutrophil Extracellular Traps: Signaling Properties and Disease Relevance. Mediat. Inflamm. 2020, 2020, 9254087. [Google Scholar] [CrossRef]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Quincy Brown, J.; Vander Heide, R.S. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir. Med. 2020, 8, 681–686. [Google Scholar] [CrossRef]
- Barton, L.M.; Duval, E.J.; Stroberg, E.; Ghosh, S.; Mukhopadhyay, S. COVID-19 Autopsies, Oklahoma, USA. Am. J. Clin. Pathol. 2020, 153, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.W. Current Understanding in Neutrophil Differentiation and Heterogeneity. Immune Netw. 2017, 17, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Jover, E.; Matilla, L.; Garaikoetxea, M.; Fernandez-Celis, A.; Muntendam, P.; Jaisser, F.; Rossignol, P.; Lopez-Andres, N. Beneficial Effects of Mineralocorticoid Receptor Pathway Blockade against Endothelial Inflammation Induced by SARS-CoV-2 Spike Protein. Biomedicines 2021, 9, 639. [Google Scholar] [CrossRef] [PubMed]
- Patra, T.; Meyer, K.; Geerling, L.; Isbell, T.S.; Hoft, D.F.; Brien, J.; Pinto, A.K.; Ray, R.B.; Ray, R. SARS-CoV-2 spike protein promotes IL-6 trans-signaling by activation of angiotensin II receptor signaling in epithelial cells. PLoS Pathog. 2020, 16, e1009128. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-kappaB pathway. Elife 2021, 10, e68563. [Google Scholar] [CrossRef]
- Pantazi, I.; Al-Qahtani, A.A.; Alhamlan, F.S.; Alothaid, H.; Matou-Nasri, S.; Sourvinos, G.; Vergadi, E.; Tsatsanis, C. SARS-CoV-2/ACE2 Interaction Suppresses IRAK-M Expression and Promotes Pro-Inflammatory Cytokine Production in Macrophages. Front. Immunol. 2021, 12, 683800. [Google Scholar] [CrossRef]
- Cao, X.; Tian, Y.; Nguyen, V.; Zhang, Y.; Gao, C.; Yin, R.; Carver, W.; Fan, D.; Albrecht, H.; Cui, T.; et al. Spike protein of SARS-CoV-2 activates macrophages and contributes to induction of acute lung inflammation in male mice. FASEB J. 2021, 35, e21801. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Kizaki, T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon 2021, 7, e06187. [Google Scholar] [CrossRef]
- Masso-Silva, J.A.; Moshensky, A.; Lam, M.T.Y.; Odish, M.F.; Patel, A.; Xu, L.; Hansen, E.; Trescott, S.; Nguyen, C.; Kim, R.; et al. Increased Peripheral Blood Neutrophil Activation Phenotypes and Neutrophil Extracellular Trap Formation in Critically Ill Coronavirus Disease 2019 (COVID-19) Patients: A Case Series and Review of the Literature. Clin. Infect. Dis. 2022, 74, 479–489. [Google Scholar] [CrossRef]
- Patel, K.D.; Cuvelier, S.L.; Wiehler, S. Selectins: Critical mediators of leukocyte recruitment. Semin. Immunol. 2002, 14, 73–81. [Google Scholar] [CrossRef] [Green Version]
- McEver, R.P. Selectins: Initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res. 2015, 107, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Lenting, P.J.; Christophe, O.D.; Denis, C.V. von Willebrand factor biosynthesis, secretion, and clearance: Connecting the far ends. Blood 2015, 125, 2019–2028. [Google Scholar] [CrossRef] [Green Version]
- Vischer, U.M. von Willebrand factor, endothelial dysfunction, and cardiovascular disease. J. Thromb. Haemost. 2006, 4, 1186–1193. [Google Scholar] [CrossRef]
- Chauhan, A.K.; Kisucka, J.; Lamb, C.B.; Bergmeier, W.; Wagner, D.D. von Willebrand factor and factor VIII are independently required to form stable occlusive thrombi in injured veins. Blood 2007, 109, 2424–2429. [Google Scholar] [CrossRef] [Green Version]
- Bhargavan, B.; Kanmogne, G.D. SARS-CoV-2 Spike Proteins and Cell-Cell Communication Inhibits TFPI and Induces Thrombogenic Factors in Human Lung Microvascular Endothelial Cells and Neutrophils: Implications for COVID-19 Coagulopathy Pathogenesis. Int. J. Mol. Sci. 2022, 23, 10436. [Google Scholar] [CrossRef] [PubMed]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef] [PubMed]
- Pine, A.B.; Meizlish, M.L.; Goshua, G.; Chang, C.H.; Zhang, H.; Bishai, J.; Bahel, P.; Patel, A.; Gbyli, R.; Kwan, J.M.; et al. Circulating markers of angiogenesis and endotheliopathy in COVID-19. Pulm. Circ. 2020, 10, 2045894020966547. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Connors, J.M.; Levy, J.H. The coagulopathy, endotheliopathy, and vasculitis of COVID-19. Inflamm. Res. 2020, 69, 1181–1189. [Google Scholar] [CrossRef]
- Kusunoki, Y.; Nakazawa, D.; Shida, H.; Hattanda, F.; Miyoshi, A.; Masuda, S.; Nishio, S.; Tomaru, U.; Atsumi, T.; Ishizu, A. Peptidylarginine Deiminase Inhibitor Suppresses Neutrophil Extracellular Trap Formation and MPO-ANCA Production. Front. Immunol. 2016, 7, 227. [Google Scholar] [CrossRef] [Green Version]
- Rohrbach, A.S.; Slade, D.J.; Thompson, P.R.; Mowen, K.A. Activation of PAD4 in NET formation. Front. Immunol. 2012, 3, 360. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.L.; Wagner, D.D. Peptidylarginine deiminase 4: A nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB J. 2018, 32, 6358–6370. [Google Scholar] [CrossRef]
- Folco, E.J.; Mawson, T.L.; Vromman, A.; Bernardes-Souza, B.; Franck, G.; Persson, O.; Nakamura, M.; Newton, G.; Luscinskas, F.W.; Libby, P. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1alpha and Cathepsin G. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1901–1912. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed]
- Thalin, C.; Hisada, Y.; Lundstrom, S.; Mackman, N.; Wallen, H. Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1724–1738. [Google Scholar] [CrossRef] [PubMed]
- Longstaff, C.; Varju, I.; Sotonyi, P.; Szabo, L.; Krumrey, M.; Hoell, A.; Bota, A.; Varga, Z.; Komorowicz, E.; Kolev, K. Mechanical stability and fibrinolytic resistance of clots containing fibrin, DNA, and histones. J. Biol. Chem. 2013, 288, 6946–6956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varju, I.; Longstaff, C.; Szabo, L.; Farkas, A.Z.; Varga-Szabo, V.J.; Tanka-Salamon, A.; Machovich, R.; Kolev, K. DNA, histones and neutrophil extracellular traps exert anti-fibrinolytic effects in a plasma environment. Thromb. Haemost. 2015, 113, 1289–1298. [Google Scholar] [CrossRef]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef]
- Carmona-Rivera, C.; Zhao, W.; Yalavarthi, S.; Kaplan, M.J. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann. Rheum. Dis. 2015, 74, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M. NETs Activate Pulmonary Arterial Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2035–2037. [Google Scholar] [CrossRef] [Green Version]
- Dubey, A.; Choudhary, S.; Kumar, P.; Tomar, S. Emerging SARS-CoV-2 Variants: Genetic Variability and Clinical Implications. Curr. Microbiol. 2021, 79, 20. [Google Scholar] [CrossRef]
- Boehm, E.; Kronig, I.; Neher, R.A.; Eckerle, I.; Vetter, P.; Kaiser, L.; Geneva Centre for Emerging Viral, D. Novel SARS-CoV-2 variants: The pandemics within the pandemic. Clin. Microbiol. Infect. 2021, 27, 1109–1117. [Google Scholar] [CrossRef]
- Singh, J.; Pandit, P.; McArthur, A.G.; Banerjee, A.; Mossman, K. Evolutionary trajectory of SARS-CoV-2 and emerging variants. Virol. J. 2021, 18, 166. [Google Scholar] [CrossRef]
- Broze, G.J., Jr.; Girard, T.J. Tissue factor pathway inhibitor: Structure-function. Front. Biosci. 2012, 17, 262–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellery, P.E.; Adams, M.J. Tissue factor pathway inhibitor: Then and now. Semin. Thromb. Hemost. 2014, 40, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Mast, A.E. Tissue Factor Pathway Inhibitor: Multiple Anticoagulant Activities for a Single Protein. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.M. Tissue factor de-encryption, thrombus formation, and thiol-disulfide exchange. Semin. Thromb. Hemost. 2013, 39, 40–47. [Google Scholar] [CrossRef]
- Chen, V.M.; Ahamed, J.; Versteeg, H.H.; Berndt, M.C.; Ruf, W.; Hogg, P.J. Evidence for activation of tissue factor by an allosteric disulfide bond. Biochemistry 2006, 45, 12020–12028. [Google Scholar] [CrossRef]
- Prasad, R.; Banerjee, S.; Sen, P. Contribution of allosteric disulfide in the structural regulation of membrane-bound tissue factor-factor VIIa binary complex. J. Biomol. Struct. Dyn. 2019, 37, 3707–3720. [Google Scholar] [CrossRef]
- Winther, J.R.; Thorpe, C. Quantification of thiols and disulfides. Biochim. Biophys. Acta 2014, 1840, 838–846. [Google Scholar] [CrossRef] [Green Version]
- Khanna, K.; Raymond, W.; Jin, J.; Charbit, A.R.; Gitlin, I.; Tang, M.; Werts, A.D.; Barrett, E.G.; Cox, J.M.; Birch, S.M.; et al. Thiol drugs decrease SARS-CoV-2 lung injury in vivo and disrupt SARS-CoV-2 spike complex binding to ACE2 in vitro. bioRxiv 2021. [Google Scholar] [CrossRef]
- Khanna, K.; Raymond, W.W.; Jin, J.; Charbit, A.R.; Gitlin, I.; Tang, M.; Werts, A.D.; Barrett, E.G.; Cox, J.M.; Birch, S.M.; et al. Exploring antiviral and anti-inflammatory effects of thiol drugs in COVID-19. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 323, L372–L389. [Google Scholar] [CrossRef]
- Wu, X.; Xiang, M.; Jing, H.; Wang, C.; Novakovic, V.A.; Shi, J. Damage to endothelial barriers and its contribution to long COVID. Angiogenesis 2023, 1–18. [Google Scholar] [CrossRef]
- Liu, R.; Pan, J.; Zhang, C.; Sun, X. Cardiovascular Complications of COVID-19 Vaccines. Front. Cardiovasc. Med. 2022, 9, 840929. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, S.R.; Sogaard, O.S.; Tolstrup, M.; Staerke, N.B.; Lundgren, J.; Ostergaard, L.; Hvas, A.M. Inflammation and Platelet Activation After COVID-19 Vaccines—Possible Mechanisms Behind Vaccine-Induced Immune Thrombocytopenia and Thrombosis. Front. Immunol. 2021, 12, 779453. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C.; Hawley, H.B. Vaccine-Associated Thrombocytopenia and Thrombosis: Venous Endotheliopathy Leading to Venous Combined Micro-Macrothrombosis. Medicina 2021, 57, 1163. [Google Scholar] [CrossRef]
- Colunga Biancatelli, R.M.L.; Solopov, P.A.; Sharlow, E.R.; Lazo, J.S.; Marik, P.E.; Catravas, J.D. The SARS-CoV-2 spike protein subunit S1 induces COVID-19-like acute lung injury in Kappa18-hACE2 transgenic mice and barrier dysfunction in human endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L477–L484. [Google Scholar] [CrossRef] [PubMed]
- Kanmogne, G.D.; Kennedy, R.C.; Grammas, P. Analysis of human lung endothelial cells for susceptibility to HIV type 1 infection, coreceptor expression, and cytotoxicity of gp120 protein. AIDS Res. Hum. Retroviruses 2001, 17, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Kanmogne, G.D.; Primeaux, C.; Grammas, P. Induction of apoptosis and endothelin-1 secretion in primary human lung endothelial cells by HIV-1 gp120 proteins. Biochem. Biophys. Res. Commun. 2005, 333, 1107–1115. [Google Scholar] [CrossRef]
- Li, H.; Singh, S.; Potula, R.; Persidsky, Y.; Kanmogne, G.D. Dysregulation of claudin-5 in HIV-induced interstitial pneumonitis and lung vascular injury. Protective role of peroxisome proliferator-activated receptor-gamma. Am. J. Respir. Crit. Care Med. 2014, 190, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Woollard, S.M.; Li, H.; Singh, S.; Yu, F.; Kanmogne, G.D. HIV-1 induces cytoskeletal alterations and Rac1 activation during monocyte-blood-brain barrier interactions: Modulatory role of CCR5. Retrovirology 2014, 11, 20. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.Y.; Tjonnfjord, G.E.; Kanse, S.M.; Dahm, A.E.A.; Iversen, N.; Myklebust, C.F.; Sun, L.; Jiang, Z.X.; Ueland, T.; Campbell, J.J.; et al. Tissue factor pathway inhibitor upregulates CXCR7 expression and enhances CXCL12-mediated migration in chronic lymphocytic leukemia. Sci. Rep. 2021, 11, 5127. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Yang, B.; Gendelman, H.E.; Persidsky, Y.; Kanmogne, G.D. STAT1 signaling modulates HIV-1-induced inflammatory responses and leukocyte transmigration across the blood-brain barrier. Blood 2008, 111, 2062–2072. [Google Scholar] [CrossRef] [Green Version]
- Bhargavan, B.; Kanmogne, G.D. Toll-Like Receptor-3 Mediates HIV-1-Induced Interleukin-6 Expression in the Human Brain Endothelium via TAK1 and JNK Pathways: Implications for Viral Neuropathogenesis. Mol. Neurobiol. 2018, 55, 5976–5992. [Google Scholar] [CrossRef] [PubMed]
- Bhargavan, B.; Woollard, S.M.; McMillan, J.E.; Kanmogne, G.D. CCR5 antagonist reduces HIV-induced amyloidogenesis, tau pathology, neurodegeneration, and blood-brain barrier alterations in HIV-infected hu-PBL-NSG mice. Mol. Neurodegener. 2021, 16, 78. [Google Scholar] [CrossRef] [PubMed]
- Bhargavan, B.; Woollard, S.M.; Kanmogne, G.D. Toll-like receptor-3 mediates HIV-1 transactivation via NFκB and JNK pathways and histone acetylation, but prolonged activation suppresses Tat and HIV-1 replication. Cell. Signal. 2016, 28, 7–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhargavan, B.; Kanmogne, G.D. SARS-CoV-2 Spike Proteins and Cell–Cell Communication Induce P-Selectin and Markers of Endothelial Injury, NETosis, and Inflammation in Human Lung Microvascular Endothelial Cells and Neutrophils: Implications for the Pathogenesis of COVID-19 Coagulopathy. Int. J. Mol. Sci. 2023, 24, 12585. https://doi.org/10.3390/ijms241612585
Bhargavan B, Kanmogne GD. SARS-CoV-2 Spike Proteins and Cell–Cell Communication Induce P-Selectin and Markers of Endothelial Injury, NETosis, and Inflammation in Human Lung Microvascular Endothelial Cells and Neutrophils: Implications for the Pathogenesis of COVID-19 Coagulopathy. International Journal of Molecular Sciences. 2023; 24(16):12585. https://doi.org/10.3390/ijms241612585
Chicago/Turabian StyleBhargavan, Biju, and Georgette D. Kanmogne. 2023. "SARS-CoV-2 Spike Proteins and Cell–Cell Communication Induce P-Selectin and Markers of Endothelial Injury, NETosis, and Inflammation in Human Lung Microvascular Endothelial Cells and Neutrophils: Implications for the Pathogenesis of COVID-19 Coagulopathy" International Journal of Molecular Sciences 24, no. 16: 12585. https://doi.org/10.3390/ijms241612585