
Molecular Mechanism of a FRET Biosensor for the Cardiac Ryanodine Receptor Pathologically Leaky State
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
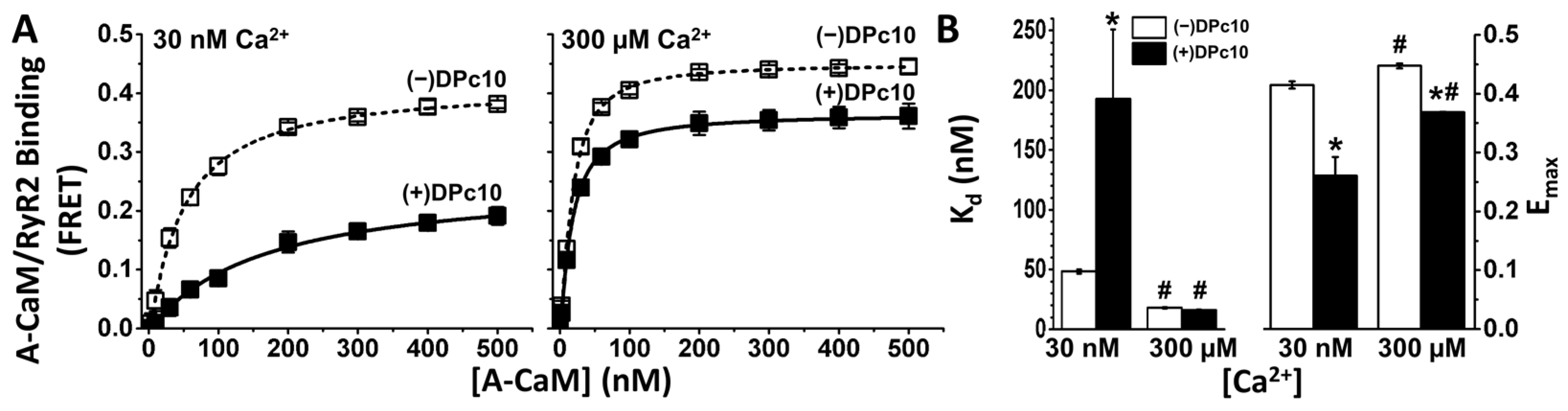
2.1. Effect of DPc10 on CaM/RyR2 Binding
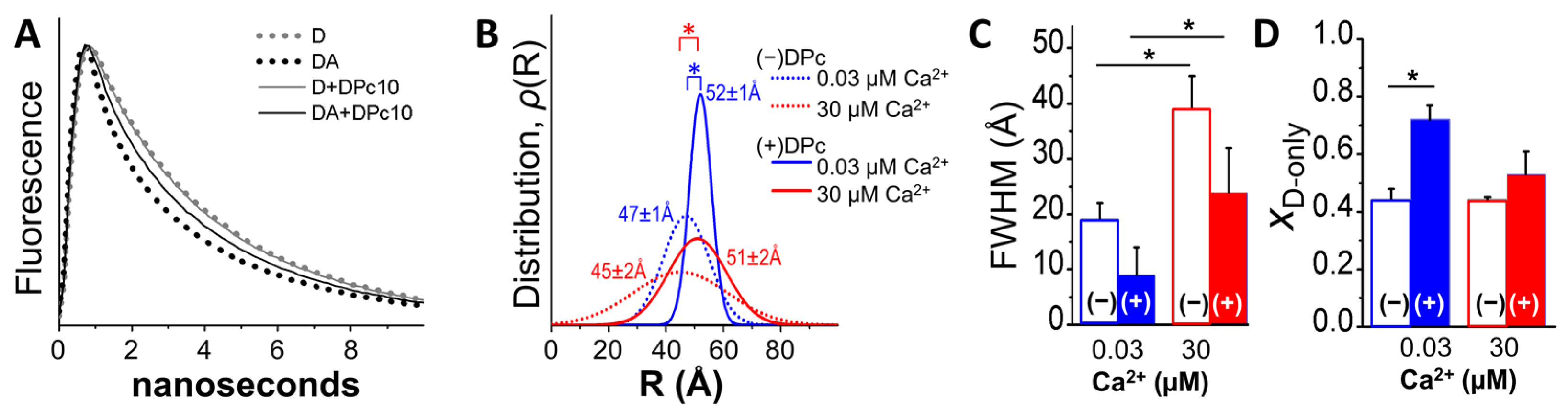
2.2. Fluorescence Lifetime (FLT)-Detected FRET Resolves DPc10 Effects on CaM/RyR2 Structure
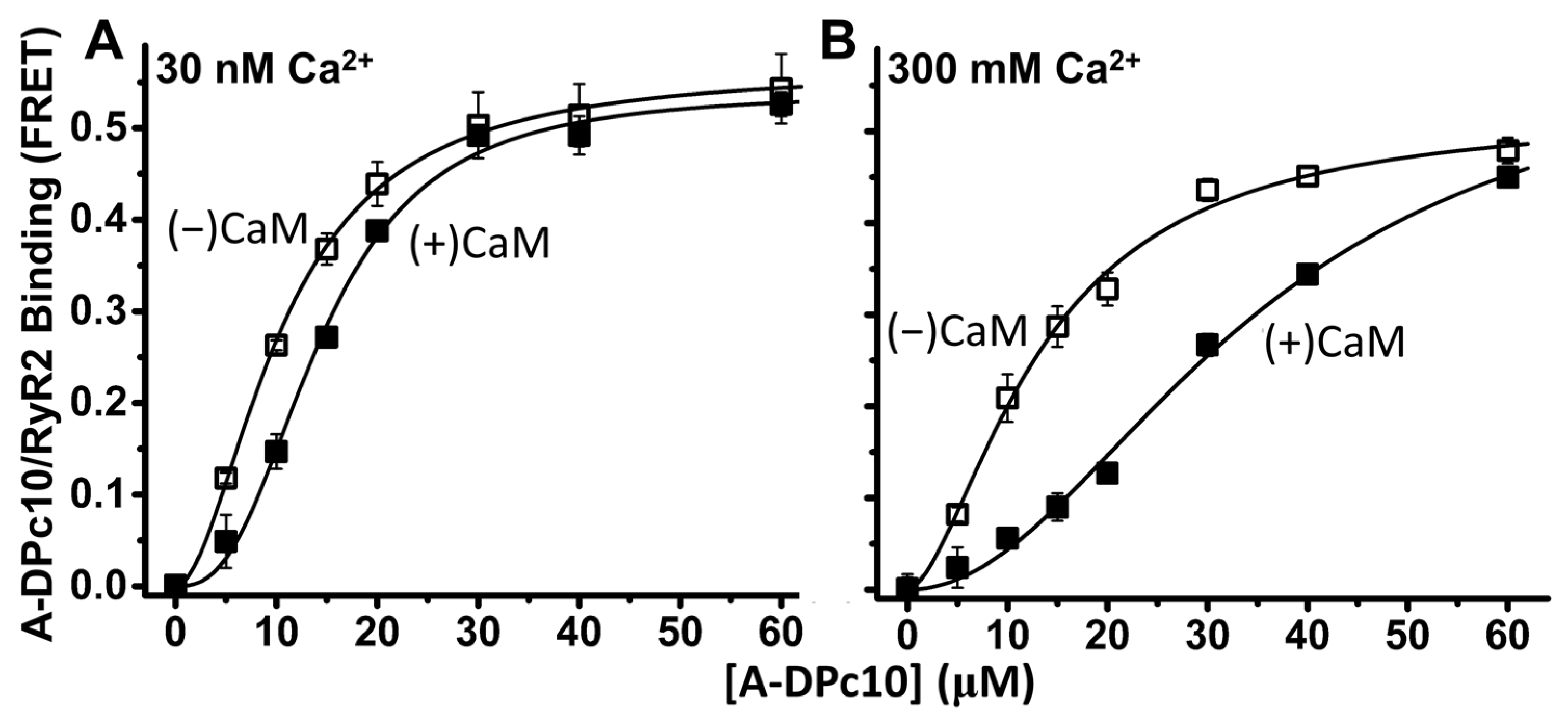
2.3. Effect of CaM on DPc10 Binding to RyR2
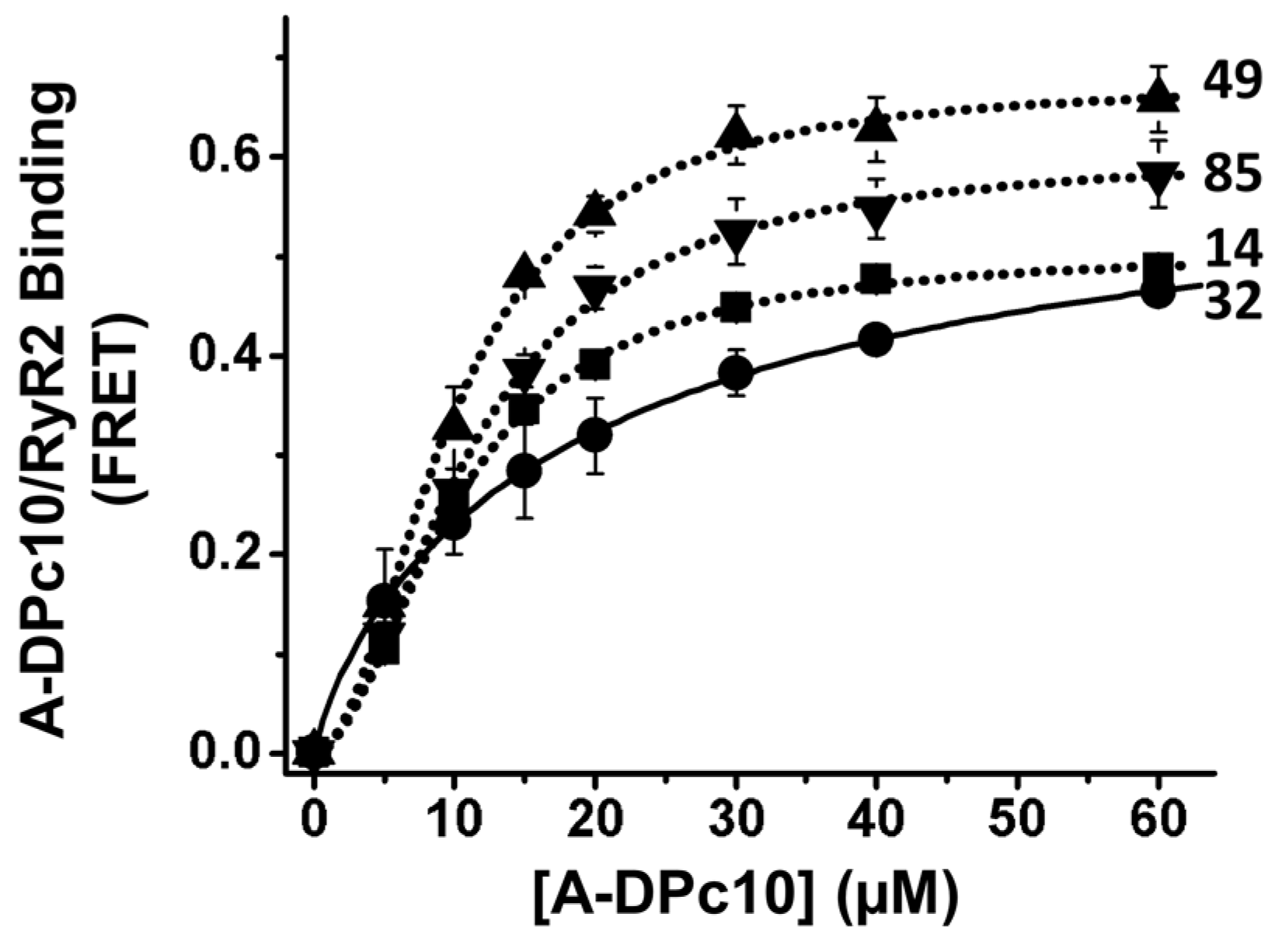
2.4. Effect of FKBP12.6 on DPc10/RyR2 Binding
3. Discussion
3.1. Ca2+ Effect on A-CaM and A-DPc10 Binding
3.2. FKBP- and CaM-Dependent Cooperativity of DPc10 Binding to RyR2
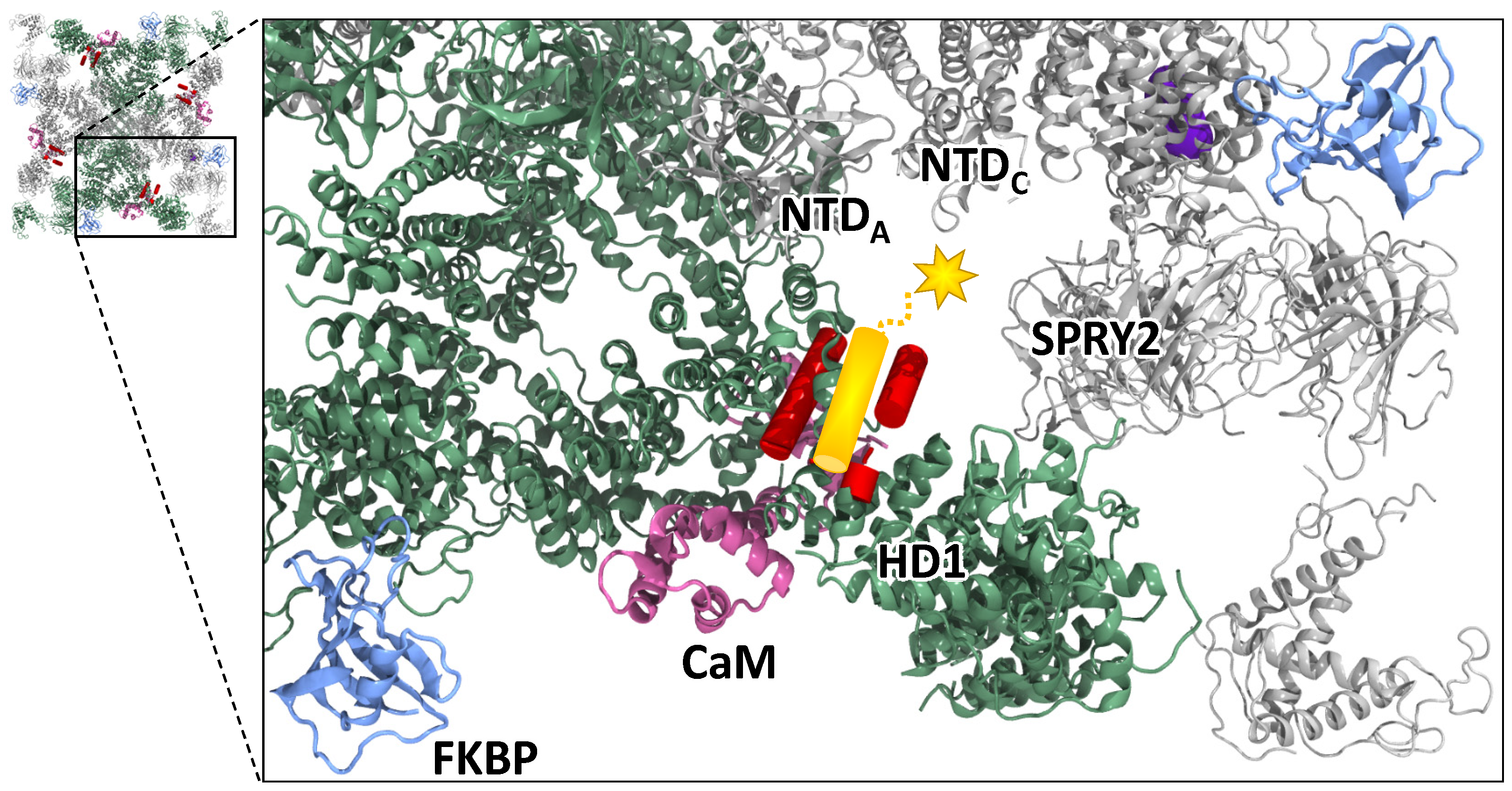
3.3. Revised DPc10 Binding Location and Effect on RyR2 Function
4. Materials and Methods
4.1. Materials
4.2. FRET Measurements
4.3. Model-Based Fitting of Time-Resolved FRET Data
4.4. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bers, D.M. Cardiac sarcoplasmic reticulum calcium leak: Basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 2014, 76, 107–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Petegem, F. Ryanodine receptors: Allosteric ion channel giants. J. Mol. Biol. 2015, 427, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Balshaw, D.M.; Xu, L.; Yamaguchi, N.; Pasek, D.A.; Meissner, G. Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). J. Biol. Chem. 2001, 276, 20144–20153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchinoumi, H.; Yang, Y.; Oda, T.; Li, N.; Alsina, K.M.; Puglisi, J.L.; Chen-Izu, Y.; Cornea, R.L.; Wehrens, X.H.; Bers, D.M. CaMKII-dependent phosphorylation of RyR2 promotes targetable pathological RyR2 conformational shift. J. Mol. Cell. Cardiol. 2016, 98, 62–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Guo, T.; Oda, T.; Chakraborty, A.; Chen, L.; Uchinoumi, H.; Knowlton, A.A.; Fruen, B.R.; Cornea, R.L.; Meissner, G.; et al. Cardiac myocyte Z-line calmodulin is mainly RyR2-bound, and reduction is arrhythmogenic and occurs in heart failure. Circ. Res. 2014, 114, 295–306. [Google Scholar] [CrossRef]
- Ono, M.; Yano, M.; Hino, A.; Suetomi, T.; Xu, X.; Susa, T.; Uchinoumi, H.; Tateishi, H.; Oda, T.; Okuda, S.; et al. Dissociation of calmodulin from cardiac ryanodine receptor causes aberrant Ca2+ release in heart failure. Cardiovasc. Res. 2010, 87, 609–617. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Yano, M.; Uchinoumi, H.; Hino, A.; Suetomi, T.; Ono, M.; Tateishi, H.; Oda, T.; Okuda, S.; Doi, M.; et al. Defective calmodulin binding to the cardiac ryanodine receptor plays a key role in CPVT-associated channel dysfunction. Biochem. Biophys. Res. Commun. 2010, 394, 660–666. [Google Scholar] [CrossRef] [Green Version]
- Oda, T.; Yang, Y.; Uchinoumi, H.; Thomas, D.D.; Chen-Izu, Y.; Kato, T.; Yamamoto, T.; Yano, M.; Cornea, R.L.; Bers, D.M. Oxidation of ryanodine receptor (RyR) and calmodulin enhance Ca release and pathologically alter, RyR structure and calmodulin affinity. J. Mol. Cell. Cardiol. 2015, 85, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, M.; Yamamoto, T.; Nishimura, S.; Kato, T.; Murakami, W.; Hino, A.; Ono, M.; Tateishi, H.; Oda, T.; Okuda, S.; et al. Enhanced binding of calmodulin to RyR2 corrects arrhythmogenic channel disorder in CPVT-associated myocytes. Biochem. Biophys. Res. Commun. 2014, 448, 1–7. [Google Scholar] [CrossRef]
- Brillantes, A.B.; Ondrias, K.; Scott, A.; Kobrinsky, E.; Ondriasova, E.; Moschella, M.C.; Jayaraman, T.; Landers, M.; Ehrlich, B.E.; Marks, A.R. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell 1994, 77, 513–523. [Google Scholar] [CrossRef]
- Ahern, G.P.; Junankar, P.R.; Dulhunty, A.F. Subconductance states in single-channel activity of skeletal muscle ryanodine receptors after removal of FKBP12. Biophys. J. 1997, 72, 146–162. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Tu, Y.; Rappel, W.J.; Levine, H. Excitation-contraction coupling gain and cooperativity of the cardiac ryanodine receptor: A modeling approach. Biophys. J. 2005, 89, 3017–3025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Shen, H.; Wu, J.; Guo, W.; Pan, X.; Wang, R.; Chen, S.R.; Yan, N. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 2016, 354, aah5324. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Cornea, R.L.; Huke, S.; Camors, E.; Yang, Y.; Picht, E.; Fruen, B.R.; Bers, D.M. Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks. Circ. Res. 2010, 106, 1743–1752. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Yano, M.; Yamamoto, T.; Tokuhisa, T.; Okuda, S.; Doi, M.; Ohkusa, T.; Ikeda, Y.; Kobayashi, S.; Ikemoto, N.; et al. Defective regulation of interdomain interactions within the ryanodine receptor plays a key role in the pathogenesis of heart failure. Circulation 2005, 111, 3400–3410. [Google Scholar] [CrossRef] [Green Version]
- Laver, D.R.; Honen, B.N.; Lamb, G.D.; Ikemoto, N. A domain peptide of the cardiac ryanodine receptor regulates channel sensitivity to luminal Ca2+ via cytoplasmic Ca2+ sites. Eur. Biophys. J. 2008, 37, 455–467. [Google Scholar] [CrossRef]
- Yang, Z.; Ikemoto, N.; Lamb, G.D.; Steele, D.S. The RyR2 central domain peptide DPc10 lowers the threshold for spontaneous Ca2+ release in permeabilized cardiomyocytes. Cardiovasc. Res. 2006, 70, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Rebbeck, R.; Ginsburg, K.S.; Ko, C.Y.; Fasoli, A.; Rusch, K.; Cai, G.F.; Dong, X.; Thomas, D.D.; Bers, D.M.; Cornea, R.L. Synergistic FRET assays for drug discovery targeting RyR2 channels. J. Mol. Cell. Cardiol. 2022, 168, 13–23. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ikemoto, N. Peptide probe study of the critical regulatory domain of the cardiac ryanodine receptor. Biochem. Biophys. Res. Commun. 2002, 291, 1102–1108. [Google Scholar] [CrossRef]
- Oda, T.; Yang, Y.; Nitu, F.R.; Svensson, B.; Lu, X.; Fruen, B.R.; Cornea, R.L.; Bers, D.M. In cardiomyocytes, binding of unzipping peptide activates ryanodine receptor 2 and reciprocally inhibits calmodulin binding. Circ. Res. 2013, 112, 487–497. [Google Scholar] [CrossRef]
- Hamada, T.; Gangopadhyay, J.P.; Mandl, A.; Erhardt, P.; Ikemoto, N. Defective regulation of the ryanodine receptor induces hypertrophy in cardiomyocytes. Biochem. Biophys. Res. Commun. 2009, 380, 493–497. [Google Scholar] [CrossRef] [Green Version]
- Marks, A.R. Targeting ryanodine receptors to treat human diseases. J. Clin. Investig. 2023, 133, e162891. [Google Scholar] [CrossRef]
- Cornea, R.L.; Nitu, F.; Gruber, S.; Kohler, K.; Satzer, M.; Thomas, D.D.; Fruen, B.R. FRET-based mapping of calmodulin bound to the RyR1 Ca2+ release channel. Proc. Natl. Acad. Sci. USA 2009, 106, 6128–6133. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, R.T.; Nitu, F.R.; Rohde, D.; Most, P.; Bers, D.M.; Thomas, D.D.; Cornea, R.L. S100A1 Protein Does Not Compete with Calmodulin for Ryanodine Receptor Binding but Structurally Alters the Ryanodine Receptor.Calmodulin Complex. J. Biol. Chem. 2016, 291, 15896–15907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, B.; Oda, T.; Nitu, F.R.; Yang, Y.; Cornea, I.; Chen-Izu, Y.; Fessenden, J.D.; Bers, D.M.; Thomas, D.D.; Cornea, R.L. FRET-based trilateration of probes bound within functional ryanodine receptors. Biophys. J. 2014, 107, 2037–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Fruen, B.R.; Nitu, F.R.; Nguyen, T.D.; Yang, Y.; Cornea, R.L.; Bers, D.M. FRET detection of calmodulin binding to the cardiac RyR2 calcium release channel. Biophys. J. 2011, 101, 2170–2177. [Google Scholar] [CrossRef] [Green Version]
- Cornea, R.L.; Nitu, F.R.; Samso, M.; Thomas, D.D.; Fruen, B.R. Mapping the ryanodine receptor FK506-binding protein subunit using fluorescence resonance energy transfer. J. Biol. Chem. 2010, 285, 19219–19226. [Google Scholar] [CrossRef] [Green Version]
- Uchinoumi, H.; Yano, M.; Suetomi, T.; Ono, M.; Xu, X.; Tateishi, H.; Oda, T.; Okuda, S.; Doi, M.; Kobayashi, S.; et al. Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ. Res. 2010, 106, 1413–1424. [Google Scholar] [CrossRef]
- Gong, D.; Chi, X.; Wei, J.; Zhou, G.; Huang, G.; Zhang, L.; Wang, R.; Lei, J.; Chen, S.R.W.; Yan, N. Modulation of cardiac ryanodine receptor 2 by calmodulin. Nature 2019, 572, 347–351. [Google Scholar] [CrossRef]
- Dhindwal, S.; Lobo, J.; Cabra, V.; Santiago, D.J.; Nayak, A.R.; Dryden, K.; Samso, M. A cryo-EM-based model of phosphorylation- and FKBP12.6-mediated allosterism of the cardiac ryanodine receptor. Sci. Signal. 2017, 10, veaai8842. [Google Scholar] [CrossRef]
- Fruen, B.R.; Balog, E.M.; Schafer, J.; Nitu, F.R.; Thomas, D.D.; Cornea, R.L. Direct detection of calmodulin tuning by ryanodine receptor channel targets using a Ca2+-sensitive acrylodan-labeled calmodulin. Biochemistry 2005, 44, 278–284. [Google Scholar] [CrossRef]
- Sondergaard, M.T.; Liu, Y.; Larsen, K.T.; Nani, A.; Tian, X.; Holt, C.; Wang, R.; Wimmer, R.; Van Petegem, F.; Fill, M.; et al. The Arrhythmogenic Calmodulin p.Phe142Leu Mutation Impairs C-domain Ca2+ Binding but Not Calmodulin-dependent Inhibition of the Cardiac Ryanodine Receptor. J. Biol. Chem. 2017, 292, 1385–1395. [Google Scholar] [CrossRef] [Green Version]
- Sondergaard, M.T.; Tian, X.; Liu, Y.; Wang, R.; Chazin, W.J.; Chen, S.R.; Overgaard, M.T. Arrhythmogenic Calmodulin Mutations Affect the Activation and Termination of Cardiac Ryanodine Receptor-mediated Ca2+ Release. J. Biol. Chem. 2015, 290, 26151–26162. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Fruen, B.R.; Black, D.J.; Bloomquist, R.A.; Bardy, J.M.; Johnson, J.D.; Louis, C.F.; Balog, E.M. Regulation of the RYR1 and RYR2 Ca2+ release channel isoforms by Ca2+- insensitive mutants of calmodulin. Biochemistry 2003, 42, 2740–2747. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ikemoto, N. Spectroscopic monitoring of local conformational changes during the intramolecular domain-domain interaction of the ryanodine receptor. Biochemistry 2002, 41, 1492–1501. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Thomas, D.D. Time-resolved FRET reveals the structural mechanism of SERCA-PLB regulation. Biochem. Biophys. Res. Commun. 2014, 449, 196–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guhathakurta, P.; Prochniewicz, E.; Thomas, D.D. Amplitude of the actomyosin power stroke depends strongly on the isoform of the myosin essential light chain. Proc. Natl. Acad. Sci. USA 2015, 112, 4660–4665. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 30 nM Ca2+ | 300 μM Ca2+ | |||||
|---|---|---|---|---|---|---|
| KD (µM) | Emax | nH | KD (µM) | Emax | nH | |
| (−) CaM | 10.4 (0.3) * | 0.57 (0.05) | 2.0 (0.1) * | 13.2 (1.3) * | 0.54 (0.03) | 1.8 (0.1) |
| (+) CaM | 14.6 (0.1) * | 0.54 (0.03) | 2.6 (0.3) * | 33.9 (3.8) * | 0.57 (0.06) | 2.3 (0.4) |
| D-x-FKBP 2 | KD (µM) | Emax | nH |
|---|---|---|---|
| 14 | 10.0 (0.2) | 0.51 (0.01) | 1.9 (0.07) |
| 32 | 13.1 (0.3) | 0.55 (0.04) | 1.3 (0.03) |
| 49 | 9.9 (0.4) | 0.67 (0.02) | 2.0 (0.15) |
| 85 | 10.4 (0.3) | 0.57 (0.05) | 2.0 (0.10) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Svensson, B.; Nitu, F.R.; Rebbeck, R.T.; McGurran, L.M.; Oda, T.; Thomas, D.D.; Bers, D.M.; Cornea, R.L. Molecular Mechanism of a FRET Biosensor for the Cardiac Ryanodine Receptor Pathologically Leaky State. Int. J. Mol. Sci. 2023, 24, 12547. https://doi.org/10.3390/ijms241612547
Svensson B, Nitu FR, Rebbeck RT, McGurran LM, Oda T, Thomas DD, Bers DM, Cornea RL. Molecular Mechanism of a FRET Biosensor for the Cardiac Ryanodine Receptor Pathologically Leaky State. International Journal of Molecular Sciences. 2023; 24(16):12547. https://doi.org/10.3390/ijms241612547
Chicago/Turabian StyleSvensson, Bengt, Florentin R. Nitu, Robyn T. Rebbeck, Lindsey M. McGurran, Tetsuro Oda, David D. Thomas, Donald M. Bers, and Razvan L. Cornea. 2023. "Molecular Mechanism of a FRET Biosensor for the Cardiac Ryanodine Receptor Pathologically Leaky State" International Journal of Molecular Sciences 24, no. 16: 12547. https://doi.org/10.3390/ijms241612547