
Brain Mitochondrial Bioenergetics in Genetic Neurodevelopmental Disorders: Focus on Down, Rett and Fragile X Syndromes

Abstract
:1. Introduction
2. Mitochondrial Energy Metabolism in the Brain: Role of Neurons and Glia
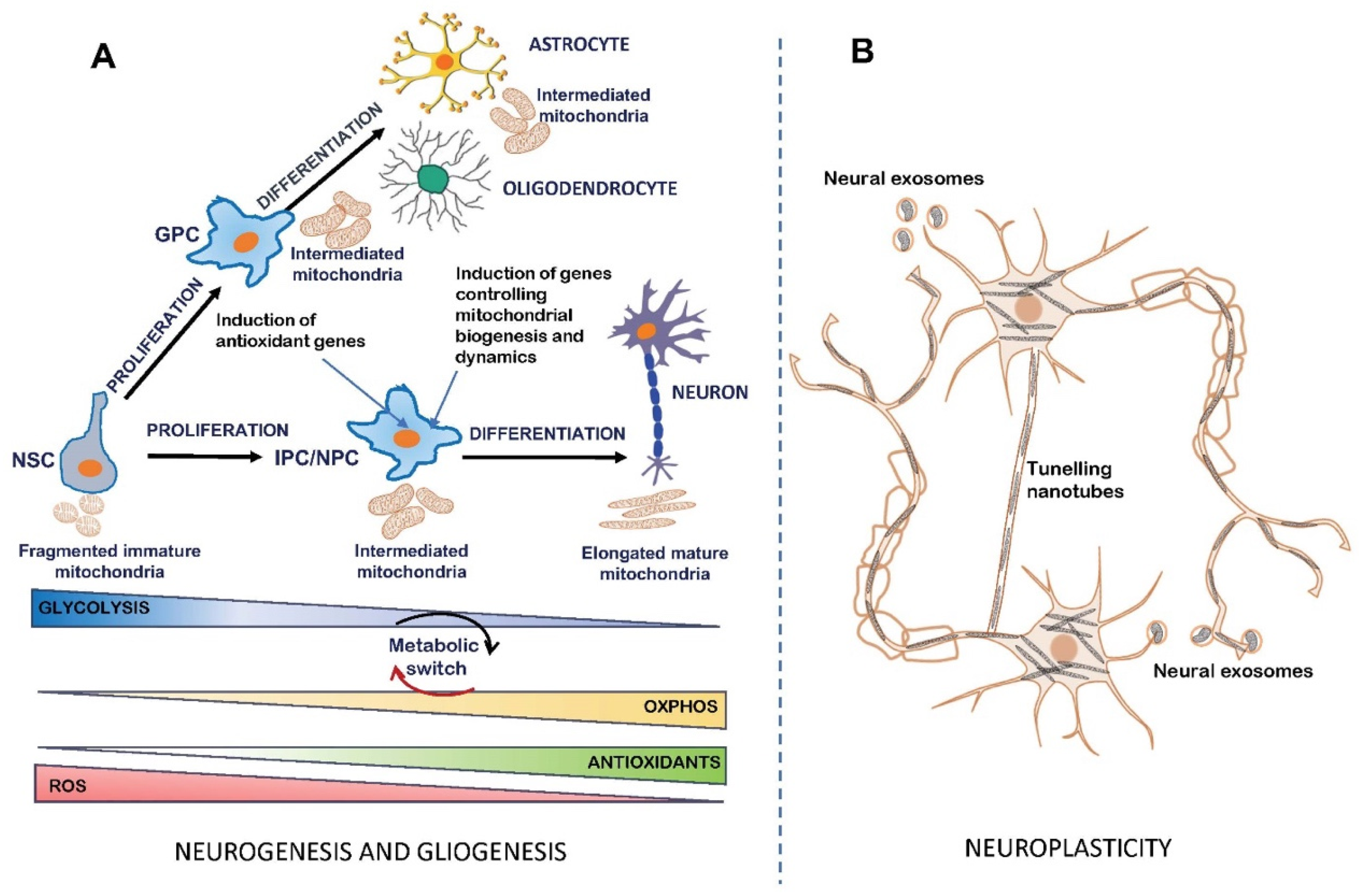
3. Mitochondrial Bioenergetics in the Regulation of Neurogenesis

4. Critical Roles of Brain Mitochondria as Master Regulators of Neuronal Plasticity and Central Hubs of Synaptic Modulation
5. Defective Brain Mitochondrial Bioenergetics in Intellectual Disability Diseases: The Case of Particular Genetic Neurodevelopmental Diseases
5.1. Down Syndrome
5.2. Rett Syndrome
5.3. Fragile X Syndrome

{kind=link}
{kind=link}
| Genetic NDDs | Neurobiological Alterations | Mitochondrial Phenotype | Drugs Targeting Mitochondrial Signaling |
|---|---|---|---|
| Down syndrome | Defects in foetal and adult hippocampal neurogenesis; Reduced granule neurons and dendritic spine density; Impaired neuro-differentiation; Impaired neurotransmission; Axonogenesis and dendritic abnormalities; Astrocyte-mediated synaptic dysfunction; Reduced brain volume; Neurodegeneration. (For refs., see [16,17,18,124,125,126,127,128,129]). | Deficit in MRC complex I, ATP synthase, ANT and ADK activities [130,131]; Deficit in mitochondrial ATP production [131]; Reduced mitochondrial membrane potential [130]; Mitochondrial ROS overproduction [130]; Altered mitochondrial morphology, dynamics and biogenesis [127,134]. | EGCG targets: DYRK1A; APP; cAMP/PKA; Sirt1/PGC-1α; AMPK [132,151,152] Resveratrol targets: RCAN1; miR-155; Sirt1/PGC-1α; AMPK [132,154] 7,8-DHF targets: TrkB receptor; MAPKs/CREB; PGC-1α [123] Metformin targets: NRIP1; PGC-1α/PPARGC1A [163] |
| Rett syndrome | Neurological cognitive and motor regression; Failure in the maturation of brain circuits; Reduced dendritic arborization; Reduced brain volume. (For refs., see [175,176,177,178]). | Down regulation of MRC complex II, complex III and complex IV expression [19,185]; Deficit in complex I, II and ATP synthase activity [19,187]. Reduced OXPHOS capacity [19,185,187]; Mitochondrial ROS overproduction [19,187]; Altered mitochondrial morphology and dynamics [193]; Reduced mitophagy [194]. | Metformin targets: PGC-1α/TFAM; PGC-1α/NRF2; PINK1 [197,198] Bacterial Toxin CNF1 targets: Rho GTPases; mTOR. [185,197] LP-211 targets: 5-HT7R; Rho GTPases [187,199] |
| Fragile X syndrome | Abnormalities in dendritic spine shape; Altered neural differentiation and migration; Defective cortical maturation and synaptic plasticity. (For refs., see [200,201,202]). | Up regulation of MRC complexes expression and activities [21]; Reduced OXPHOS [21,213]; ROS overproduction [21]; Mitochondrial proton leak [212]. | Mitochondrial CoQ target: Mitochondrial proton leak block [214] Dexpramipexole target: ATP synthase modulation [213] |
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Picard, M.; Wallace, D.C.; Burelle, Y. The rise of mitochondria in medicine. Mitochondrion 2016, 30, 105–116. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Makinde, E.; Ma, L.; Mellick, G.D.; Feng, Y. Mitochondrial Modulators: The Defender. Biomolecules 2023, 13, 226. [Google Scholar] [CrossRef]
- Valenti, D.; Vacca, R.A.; Moro, L.; Atlante, A. Mitochondria Can Cross Cell Boundaries: An Overview of the Biological Relevance, Pathophysiological Implications and Therapeutic Perspectives of Intercellular Mitochondrial Transfer. Int. J. Mol. Sci. 2021, 22, 8312. [Google Scholar] [CrossRef]
- Go, Y.M.; Fernandes, J.; Hu, X.; Uppal, K.; Jones, D.P. Mitochondrial network responses in oxidative physiology and disease. Free Radic. Biol. Med. 2018, 116, 31–40. [Google Scholar] [CrossRef]
- Pei, L.; Wallace, D.C. Mitochondrial etiology of neuropsychiatric disorders. Biol. Psychiat. 2018, 83, 722–730. [Google Scholar] [CrossRef]
- Kohshour, M.O.; Gonçalves, V.F. Mitochondrial genetics in mental disorders: The bioenergy viewpoint. Eur. Neuropsychopharmacol. 2023, 67, 80–82. [Google Scholar] [CrossRef]
- Bülow, P.; Patgiri, A.; Faundez, V. Mitochondrial protein synthesis and the bioenergetic cost of neurodevelopment. iScience 2022, 25, 104920. [Google Scholar] [CrossRef]
- Oyarzábal, A.; Musokhranova, U.; Barros, L.; García-Cazorla, A. Energy metabolism in childhood neurodevelopmental disorders. EBioMedicine 2021, 69, 103474. [Google Scholar] [CrossRef]
- Weber, B.; Barros, L.F. The astrocyte: Powerhouse and recycling center. Cold Spring Harb. Perspect. Biol. 2015, 7, a020396. [Google Scholar] [CrossRef] [Green Version]
- Kundakovic, M.; Champagne, F.A. Early life experience, epigenetics, and the developing brain. Neuropsychopharmacology 2015, 40, 141–153. [Google Scholar] [CrossRef] [Green Version]
- Andrade-Talavera, Y.; Pérez-Rodríguez, M.; Prius-Mengual, J.; Rodríguez-Moreno, A. Neuronal and astrocyte determinants of critical periods of plasticity. Trends Neurosci. 2023, 16, 566–580. [Google Scholar] [CrossRef]
- Morris-Rosendahl, D.J. Neurodevelopmental disorders-the history and future of a diagnostic concept. Dialogues Clin Neurosci 2020, 22, 65–72. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders DSM5; American Psychiatric Association Publishing: Arlington, VA, USA, 2013. [Google Scholar]
- Ortiz-González, X.R. Mitochondrial Dysfunction: A Common Denominator in Neurodevelopmental Disorders? Dev. Neurosci. 2021, 43, 222–229. [Google Scholar] [CrossRef]
- Valenti, D.; de Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46 Pt 2, 202–217. [Google Scholar] [CrossRef]
- Valenti, D.; Braidy, N.; De Rasmo, D.; Signorile, A.; Rossi, L.; Atanasov, A.G.; Volpicella, M.; Henrion-Caude, A.; Nabavi, S.M.; Vacca, R.A. Mitochondria as pharmacological targets in Down syndrome. Free Radic. Biol. Med. 2018, 114, 69–83. [Google Scholar] [CrossRef]
- Vacca, R.A.; Bawari, S.; Valenti, D.; Tewari, D.; Nabavi, S.F.; Shirooie, S.; Sah, A.N.; Volpicella, M.; Braidy, N.; Nabavi, S.M. Down syndrome: Neurobiological alterations and therapeutic targets. Neurosci. Biobehav. Rev. 2019, 98, 234–255. [Google Scholar] [CrossRef]
- De Filippis, B.; Valenti, D.; de Bari, L.; De Rasmo, D.; Musto, M.; Fabbri, A.; Ricceri, L.; Fiorentini, C.; Laviola, G.; Vacca, R.A. Mitochondrial free radical overproduction due to respiratory chain impairment in the brain of a mouse model of Rett syndrome: Protective effect of CNF1. Free Radic. Biol. Med. 2015, 83, 167–177. [Google Scholar] [CrossRef]
- Müller, M. Disturbed redox homeostasis and oxidative stress: Potential players in the developmental regression in Rett syndrome. Neurosci. Biobehav. Rev. 2019, 98, 154–163. [Google Scholar] [CrossRef]
- D’Antoni, S.; de Bari, L.; Valenti, D.; Borro, M.; Bonaccorso, C.M.; Simmaco, M.; Vacca, R.A.; Catania, M.V. Aberrant mitochondrial bioenergetics in the cerebral cortex of the Fmr1 knockout mouse model of fragile X syndrome. Biol. Chem. 2020, 401, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Napoli, E.; Kim, K.; McLennan, Y.A.; Hagerman, R.J.; Giulivi, C. Brain Atrophy and White Matter Damage Linked to Peripheral Bioenergetic Deficits in the Neurodegenerative Disease FXTAS. Int. J. Mol. Sci. 2021, 22, 9171. [Google Scholar] [CrossRef]
- von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S.J. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef] [Green Version]
- Verkhratskiĭ, A.N.; Butt, A. Glial Physiology and Pathophysiology; Wiley-Blackwell: Chichester, UK; Hoboken, NJ, USA, 2013; Volume XXXI, p. 527. [Google Scholar]
- Barres, B.A. The mystery and magic of glia: A perspective on their roles in health and disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Fields, R.D.; Woo, D.H.; Basser, P.J. Glial regulation of the neuronal connectome through local and long-distant communication. Neuron 2015, 86, 374–386. [Google Scholar] [CrossRef] [Green Version]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in physiology and disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic regulation of microglia. Glia 2018, 66, 1200–1212. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G. JMicroglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.J.; Attwell, D. The energetics of CNS white matter. J. Neurosci. 2012, 32, 356–371. [Google Scholar] [CrossRef] [Green Version]
- Rangaraju, V.; Lewis, T.L.; Hirabayashi, Y.; Bergami, M.; Motori, E.; Cartoni, R.; Kwon, S.-K.; Courchet, J. Pleiotropic mitochondria: The infuence of mitochondria on neuronal development and disease. J. Neurosci. 2019, 39, 8200–8208. [Google Scholar] [CrossRef] [Green Version]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [Green Version]
- Supplie, L.M.; Düking, T.; Campbell, G.; Diaz, F.; Moraes, C.T.; Götz, M.; Hamprecht, B.; Boretius, S.; Mahad, D.; Nave, K.A. Respiration-deficient astrocytes survive as glycolytic cells in vivo. J. Neurosci. 2017, 37, 4231–4242. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Gutierrez, C.; Bonora, N.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Fernandez, E.; Josephine, C.; Bonvento, G.; Enriquez, J.A.; Almeida, A.; et al. Astrocytic mitochondrial ROS modulate brain metabolism and mouse behaviour. Nat. Metab. 2019, 1, 201–211. [Google Scholar] [CrossRef]
- Beard, E.; Lengacher, S.; Dias, S.; Magistretti, P.J.; Finsterwald, C. Astrocytes as Key Regulators of Brain Energy Metabolism: New Therapeutic Perspectives. Front. Physiol. 2022, 12, 825816. [Google Scholar] [CrossRef]
- Clarke, L.E.; Barres, B.A. Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci. 2013, 14, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.X. Antioxidant defence of the brain: A role for astrocytes. Can. J. Physiol. Pharmacol. 1997, 75, 1149–1163. [Google Scholar] [CrossRef]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef] [Green Version]
- Nehlig, A. Brain uptake and metabolism of ketone bodies in animal models. Prostaglandins Leukot. Essent. Fatty Acids 2004, 70, 265–275. [Google Scholar] [CrossRef]
- Magistretti, P.J. Brain energy metabolism. In Fundamental Neuroscience; Squire, L.R., Berg, D., Bloom, F.E., du Lac, S., Ghosh, A., Spitzer, N.C., Eds.; Academic Press: San Diego, CA, USA, 2008; pp. 271–293. [Google Scholar]
- van Hall, G.; Stromstad, M.; Rasmussen, P.; Jans, O.; Zaar, M.; Gam, C.; Quistorff, B.; Secher, N.H.; Nielsen, H.B. Blood lactate is an important energy source for the human brain. J. Cereb. Blood Flow Metab. 2009, 29, 1121–1129. [Google Scholar] [CrossRef] [Green Version]
- Zielke, H.R.; Zielke, C.L.; Baab, P.J. Direct measurement of oxidative metabolism in the living brain by microdialysis: A review. J. Neurochem. 2009, 109 (Suppl. S1), 24–29. [Google Scholar] [CrossRef] [Green Version]
- Magistretti, P.J.; Pellerin, L.; Rothman, D.L.; Shulman, R.G. Energy on demand. Science 1999, 283, 496–497. [Google Scholar] [CrossRef]
- Baltan, S. Can lactate serve as an energy substrate for axons in good times and in bad, in sickness and in health? Metab. Brain Dis. 2015, 30, 25–30. [Google Scholar] [CrossRef]
- Castillo, X.; Rosafio, K.; Wyss, M.T.; Drandarov, K.; Buck, A.; Pellerin, L.; Weber, B.; Hirt, L. A probable dual mode of action for both L- and D-lactate neuroprotection in cerebral ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Mächler, P.; Wyss, M.T.; Elsayed, M.; Stobart, J.; Gutierrez, R.; von Faber-Castell, A.; Kaelin, V.; Zuend, M.; San Martín, A.; Romero-Gómez, I.; et al. In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell Metab. 2016, 23, 94–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Omuro, H.; Liu, Y.F.; Soya, M.; Shima, T.; McEwen, B.S.; Soya, H. Astrocytic glycogen-derived lactate fuels the brain during exhaustive exercise to maintain endurance capacity. Proc. Natl. Acad. Sci. USA 2017, 114, 6358–6363. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic endproduct to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Wyss, M.T.; Jolivet, R.; Buck, A.; Magistretti, P.J.; Weber, B. In vivo evidence for lactate as a neuronal energy source. J. Neurosci. 2011, 31, 7477–7485. [Google Scholar] [CrossRef] [Green Version]
- Sobieski, C.; Warikoo, N.; Shu, H.J.; Mennerick, S. Ambient but not local lactate underlies neuronal tolerance to prolonged glucose deprivation. PLoS ONE 2018, 13, e0195520. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Magistretti, P.J. Sweet sixteen for ANLS. J. Cereb. Blood Flow. Metab. 2012, 32, 1152–1166. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Sorg, O.; Yu, N.; Martin, J.L.; Pellerin, L. Neurotransmitters regulate energy metabolism in astrocytes: Implications for the metabolic trafficking between neural cells. Dev. Neurosci. 1993, 15, 306–312. [Google Scholar] [CrossRef]
- Brown, A.M. Brain glycogen re-awakened. J. Neurochem. 2004, 89, 537–552. [Google Scholar] [CrossRef]
- Sickmann, H.M.; Walls, A.B.; Schousboe, A.; Bouman, S.D.; Waagepetersen, H.S. Functional significance of brain glycogen in sustaining glutamatergic neurotransmission. J. Neurochem. 2009, 109 (Suppl. S1), 80–86. [Google Scholar] [CrossRef]
- Walls, A.B.; Heimbürger, C.M.; Bouman, S.D.; Schousboe, A.; Waagepetersen, H.S. Robust glycogen shunt activity in astrocytes: Effects of glutamatergic and adrenergic agents. Neuroscience 2009, 158, 284–292. [Google Scholar] [CrossRef]
- Dringen, R.; Gebhardt, R.; Hamprecht, B. Glycogen in astrocytes: Possible function as lactate supply for neighboring cells. Brain Res. 1993, 623, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Bonvento, G.; Bolaños, J.P. Astrocyte-neuron metabolic cooperation shapes brain activity. Cell Metab. 2021, 33, 1546–1564. [Google Scholar] [CrossRef]
- Gage, F.H. Mammalian neural stem cells. Science 2000, 287, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Nuebel, E.; Daley, G.Q.; Koehler, C.M.; Teitell, M.A. Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell 2012, 11, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Cerdeño, V.; Noctor, S.C. Neural progenitor cell erminology. Front. Neuroanat. 2018, 12, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lui, J.H.; Hansen, D.V.; Kriegstein, A.R. Development and evolution of the human neocortex. Cell 2011, 146, 18–36. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, P.S.; Perfilieva, E.; Björk-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef]
- Gage, F.H. Adult neurogenesis in mammals. Science 2019, 364, 827–828. [Google Scholar] [CrossRef]
- Bond, A.M.; Ming, G.L.; Song, H. Adult Mammalian Neural Stem Cells and Neurogenesis: Five Decades Later. Cell Stem Cell 2015, 17, 385–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempermann, G.; Song, H.; Gage, F.H. Neurogenesis in the Adult Hippocampus. Cold Spring Harb Perspect Biol. 2015, 5, a018812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, G.L.; Song, H. Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [Green Version]
- Sorrells, S.F.; Paredes, M.F.; Cebrian-Silla, A.; Sandoval, K.; Qi, D.; Kelley, K.W.; James, D.; Mayer, S.; Chang, J.; Auguste, K.I.; et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 2018, 555, 377–381. [Google Scholar] [CrossRef]
- Boldrini, M.; Fulmore, C.A.; Tartt, A.N.; Simeon, L.R.; Pavlova, I.; Poposka, V.; Rosoklija, G.B.; Stankov, A.; Arango, V.; Dwork, A.J.; et al. Human hippocampal neurogenesis persists throughout aging. Cell Stem Cell 2018, 22, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Tobin, M.K.; Musaraca, K.; Disouky, A.; Shetti, A.; Bheri, A.; Honer, W.G.; Kim, N.; Dawe, R.J.; Bennett, D.A.; Arfanakis, K.; et al. Human hippocampal neurogenesis persists in aged adults and Alzheimer’s disease patients. Cell Stem Cell 2019, 24, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Jurkowski, M.P.; Bettio, L.; Woo, E.K.; Patten, A.; Yau, S.Y.; Gil-Mohapel, J. Beyond the Hippocampus and the SVZ: Adult Neurogenesis Throughout the Brain. Front. Cell Neurosci. 2020, 14, 576444. [Google Scholar] [CrossRef]
- Khacho, M.; Slack, R.S. Mitochondrial activity in the regulation of stem cell self-renewal and differentiation. Curr. Opin. Cell Biol. 2017, 49, 1–8. [Google Scholar] [CrossRef]
- Sun, X.; St John, J.C. The role of the mtDNA set point in differentiation, development and tumorigenesis. Biochem J. 2016, 473, 2955–2971. [Google Scholar] [CrossRef]
- Beckervordersandforth, R.; Häberle, B.M.; Lie, D.C. Metabolic regulation of adult stem cell-derived neurons. Front. Biol. 2015, 10, 107–116. [Google Scholar] [CrossRef]
- Wanet, A.; Arnould, T.; Najimi, M.; Renard, P. Connecting mitochondria, metabolism, and stem cell fate. Stem Cells Dev. 2015, 24, 1957–1971. [Google Scholar] [CrossRef] [Green Version]
- Ottoboni, L.; Merlini, A.; Martino, G. Neural stem cell plasticity: Advantages in therapy for the injured central nervous system. Front. Cell Dev. Biol. 2017, 5, 52. [Google Scholar] [CrossRef] [Green Version]
- Colangelo, A.M.; Cirillo, G.; Alberghina, L.; Papa, M.; Westerhoff, H.V. Neural plasticity and adult neurogenesis: The deep biology perspective. Neural Regen. Res. 2019, 14, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Pontzer, H.; Brown, M.H.; Raichlen, D.A.; Dunsworth, H.; Hare, B.; Walker, K.; Luke, A.; Dugas, L.R.; Durazo-Arvizu, R.; Schoeller, D.; et al. Metabolic acceleration and the evolution of human brain size and life history. Nature 2016, 533, 390–392. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.; Jimenez-Blasco, D.; Bolaños, J.P. Cross-talk between energy and redox metabolism in astrocyte-neuron functional cooperation. Essays Biochem. 2023, 67, 17–26. [Google Scholar] [CrossRef]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife 2016, 5, e13374. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.C.; Keeney, P.M.; Bennett, J.P., Jr. Differentiation of Human Neural Stem Cells into Motor Neurons Stimulates Mitochondrial Biogenesis and Decreases Glycolytic Flux. Stem Cells Dev. 2015, 24, 1984–1994. [Google Scholar] [CrossRef] [Green Version]
- Agostini, M.; Romeo, F.; Inoue, S.; Niklison-Chirou, M.V.; Elia, A.J.; Dinsdale, D.; Morone, N.; Knight, R.A.; Mak, T.W.; Melino, G. Metabolic reprogramming during neuronal differentiation. Cell Death Differ. 2016, 23, 1502–1514. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.S.; Vieira, H.L.A. Role of cell metabolism and mitochondrial function during adult neurogenesis. Neurochem. Res. 2017, 42, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Mils, V.; Bosch, S.; Roy, J.; Bel-Vialar, S.; Belenguer, P.; Pituello, F.; Miquel, M.C. Mitochondrial reshaping accompanies neural differentiation in the developing spinal cord. PLoS ONE 2015, 10, e0128130. [Google Scholar] [CrossRef] [Green Version]
- Khacho, M.; Harris, R.; Slack, R.S. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat. Rev. Neurosci. 2019, 20, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Baum, T.; Gama, V. Dynamic properties of mitochondria during human corticogenesis. Development 2021, 148, dev194183. [Google Scholar] [CrossRef] [PubMed]
- Iwata, R.; Casimir, P.; Vanderhaeghen, P. Mitochondrial dynamics in postmitotic cells regulate neurogenesis. Science 2020, 369, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Beckervordersandforth, R. Mitochondrial metabolism-mediated regulation of adult neurogenesis. Brain Plast. 2017, 3, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Audano, M.; Pedretti, S.; Crestani, M.; Caruso, D.; De Fabiani, E.; Mitro, N. Mitochondrial dysfunction increases fatty acid β-oxidation and translates into impaired neuroblast maturation. FEBS Lett. 2019, 593, 3173–3189. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misgeld, T.; Schwarz, T.L. Mitostasis in neurons: Maintaining mitochondria in an extended cellular architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Okamoto, K.-I.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Devine, M.; Kittler, J. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef]
- Ertük, A.; Wang, Y.; Sheng, M. Local pruning of dendrites and spines by caspase-3 dependent and proteasome-limited mechanisms. J. Neurosci. 2014, 34, 1672–1688. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.J.; Pekkurnaz, G. Powerhouse of the mind: Mitochondrial plasticity at the synapse. Curr. Opin. Neurobiol. 2019, 57, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.C.; Bartol, T.M.; Phan, S.; Bushong, E.A.; Perkins, G.; Sejnowski, T.J.; Ellisman, M.H.; Skupin, A. Mitochondrial morphology provides a mechanism for energy bufering at synapses. Sci. Rep. 2019, 9, 18306. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Hou, Y.; Mattson, M.P. Mitochondria and neuroplasticity. ASN Neuro. 2010, 2, e00045. [Google Scholar] [CrossRef] [PubMed]
- Haydon, P.G.; Carmignoto, G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 2006, 86, 1009–1031. [Google Scholar] [CrossRef] [Green Version]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, R.G. NMDA receptors and memory encoding. Neuropharmacology 2013, 74, 32–40. [Google Scholar] [CrossRef]
- Mattson, M.P.; Liu, D. Mitochondrial potassium channels and uncoupling proteins in synaptic plasticity and neuronal cell death. Biochem. Biophys. Res. Commun. 2003, 304, 539–549. [Google Scholar] [CrossRef]
- Wieraszko, A. Changes in the hippocampal slices energy metabolism following stimulation and long-term potentiation of Schaffer collaterals-pyramidal cell synapses tested with the 2-deoxyglucose technique. Brain Res. 1982, 237, 449–457. [Google Scholar] [CrossRef]
- Stanton, P.K.; Schanne, F.A. Hippocampal long-term potentiation increases mitochondrial calcium pump activity in rat. Brain Res. 1986, 382, 185–188. [Google Scholar] [CrossRef]
- Williams, J.M.; Thompson, V.L.; Mason-Parker, S.E.; Abraham, W.C.; Tate, W.P. Synaptic activity-dependent modulation of mitochondrial gene expression in the rat hippocampus. Brain Res. Mol. Brain Res. 1998, 60, 50–56. [Google Scholar] [CrossRef]
- Zinsmaier, K.E.; Babic, M.; Russo, G.J. Mitochondrial transport dynamics in axons and dendrites. Results Probl. Cell Differ. 2009, 48, 107–139. [Google Scholar] [CrossRef]
- Cameron, H.A.; Kaliszewski, C.K.; Greer, C.A. Organization of mitochondria in olfactory bulb granule cell dendritic spines. Synapse 1991, 8, 107–118. [Google Scholar] [CrossRef]
- Popov, V.; Medvedev, N.I.; Davies, H.A.; Stewart, M.G. Mitochondria form a filamentous reticular network in hippocampal dendrites but are present as discrete bodies in axons: A three-dimensional ultrastructural study. Comp. Neurol. J. 2005, 492, 50–65. [Google Scholar] [CrossRef]
- Liesa, M.; Palacìn, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef] [Green Version]
- Macaskill, A.F.; Rinholm, J.E.; Twelvetrees, A.E.; Arancibia-Carcamo, I.L.; Muir, J.; Fransson, A.; Aspenstrom, P.; Attwell, D.; Kittler, J.T. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 2009, 61, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Rangaraju, V.; Lauterbach, M.; Schuman, E.M. Spatially Stable Mitochondrial Compartments Fuel Local Translation during Plasticity. Cell 2019, 176, 73–84.e15. [Google Scholar] [CrossRef] [Green Version]
- Kuzniewska, B.; Cysewski, D.; Wasilewski, M.; Sakowska, P.; Milek, J.; Kulinski, T.M.; Winiarski, M.; Kozielewicz, P.; Knapska, E.; Dadlez, M.; et al. Mitochondrial protein biogenesis in the synapse is supported by local translation. EMBO Rep. 2020, 21, e48882. [Google Scholar] [CrossRef]
- Cheng, A.; Wan, R.; Yang, J.L.; Kamimura, N.; Son, T.G.; Ouyang, X.; Luo, Y.; Okun, E.; Mattson, M.P. Involvement of PGC-1alpha in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 2012, 3, 1250. [Google Scholar] [CrossRef] [Green Version]
- Muller, M.; Mironov, S.; Ivannikov, M.; Schmidt, J.; Richter, D. Mitochondrial organization and motility probed by two-photon microscopy in cultured mouse brainstem neurons. Exp. Cell Res. 2004, 303, 114–127. [Google Scholar] [CrossRef]
- Seager, R.; Lee, L.; Henley, J.M.; Wilkinson, K.A. Mechanisms and roles of mitochondrial localisation and dynamics in neuronal function. Neuronal Signal 2020, 4, NS20200008. [Google Scholar] [CrossRef]
- Ligon, L.A.; Steward, O. Movement of mitochondria in theaxons and dendrites of cultured hippocampal neurons. J. Comp. Neurol. 2000, 427, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Wong, H.-T.C.; Pinter, K.; Mosqueda, N.; Beirl, A.; Lomash, R.M.; Won, S.; Kindt, K.S.; Drerup, C.M. Retrograde mitochondrial transport is essential for organelle distribution and health in zebrafish neurons. J. Neurosci. 2021, 41, 1371–1392. [Google Scholar] [CrossRef] [PubMed]
- Kruppa, A.J.; Buss, F. Motor proteins at the mitochondria-cytoskeleton interface. J. Cell Sci. 2021, 134, jcs226084. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xiong, G.J.; Huang, N.; Sheng, Z.H. The cross-talk of energy sensing and mitochondrial anchoring sustains synaptic efficacy by maintaining presynaptic metabolism. Nat. Metab. 2020, 2, 1077–1095. [Google Scholar] [CrossRef]
- Duarte, F.V.; Ciampi, D.; Duarte, C.B. Mitochondria as central hubs in synaptic modulation. Cell Mol. Life Sci. 2023, 80, 173. [Google Scholar] [CrossRef]
- Fecher, C.; Trovò, L.; Müller, S.A.; Snaidero, N.; Wettmarshausen, J.; Heink, S.; Ortiz, O.; Wagner, I.; Kühn, R.; Hartmann, J.; et al. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat. Neurosci. 2019, 22, 1731–1742. [Google Scholar] [CrossRef]
- Liu, S.; Akula, N.; Reardon, P.K.; Russ, J.; Torres, E.; Clasen, L.S.; Blumenthal, J.; Lalonde, F.; McMahon, F.J.; Szele, F.; et al. Aneuploidy effects on human gene expression across three cell types. Proc. Natl. Acad. Sci. USA 2023, 120, e2218478120. [Google Scholar] [CrossRef]
- Seol, S.; Kwon, J.; Kang, H.J. Cell type characterization of spatiotemporal gene co-expression modules in Down syndrome brain. iScience 2022, 26, 105884. [Google Scholar] [CrossRef]
- Vacca, R.A.; Augello, A.; Gallo, L.; Caggianese, G.; Malizia, V.; La Grutta, S.; Murero, M.; Valenti, D.; Tullo, A.; Balech, B.; et al. Serious Games in the new era of digital-health interventions: A narrative review of their therapeutic applications to manage neurobehavior in neurodevelopmental disorders. Neurosci. Biobehav. Rev. 2023, 149, 105156. [Google Scholar] [CrossRef]
- Valenti, D.; Stagni, F.; Emili, M.; Guidi, S.; Bartesaghi, R.; Vacca, R.A. Impaired Brain Mitochondrial Bioenergetics in the Ts65Dn Mouse Model of Down Syndrome Is Restored by Neonatal Treatment with the Polyphenol 7,8-Dihydroxyflavone. Antioxidants 2021, 11, 62. [Google Scholar] [CrossRef]
- Tan, K.L.; Lee, H.C.; Cheah, P.S.; Ling, K.H. Mitochondrial Dysfunction in Down Syndrome: From Pathology to Therapy. Neuroscience 2023, 511, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Prutton, K.M.; Marentette, J.O.; Maclean, K.N.; Roede, J.R. Characterization of mitochondrial and metabolic alterations induced by trisomy 21 during neural differentiation. Free Radic. Biol. Med. 2023, 196, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Sarver, D.C.; Xu, C.; Velez, L.M.; Aja, S.; Jaffe, A.E.; Seldin, M.M.; Reeves, R.H.; Wong, G.W. Dysregulated systemic metabolism in a Down syndrome mouse model. Mol. Metab. 2023, 68, 101666. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, B.B.; Kadam, N.N. Triplication of HSA21 on alterations in structure and function of mitochondria. Mitochondrion 2022, 65, 88–101. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef]
- Zamponi, E.; Helguera, P.R. The Shape of Mitochondrial Dysfunction in Down Syndrome. Dev. Neurobiol. 2019, 79, 613–621. [Google Scholar] [CrossRef]
- Valenti, D.; Manente, G.A.; Moro, L.; Marra, E.; Vacca, R.A. Deficit of complex I activity in human skin fibroblasts with chromosome 21 trisomy and overproduction of reactive oxygen species by mitochondria: Involvement of the cAMP/PKA signalling pathway. Biochem. J. 2011, 435, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Valenti, D.; Tullo, A.; Caratozzolo, M.F.; Merafina, R.S.; Scartezzini, P.; Marra, E.; Vacca, R.A. Impairment of F1Fo-ATPase, adenine nucleotide translocator and adenylate kinase causes mitochondrial energy deficit in human skin fibroblasts with chromosome 21 trisomy. Biochem. J. 2010, 431, 299–310. [Google Scholar] [CrossRef]
- Valenti, D.; de Bari, L.; de Rasmo, D.; Signorile, A.; Henrion-Caude, A.; Contestabile, A.; Vacca, R.A. The polyphenols resveratrol and epigallocatechin-3-gallate restore the severe impairment of mitochondria in hippocampal progenitor cells from a Down syndrome mouse model. Biochim. Biophys. Acta 2016, 1862, 1093–1104. [Google Scholar] [CrossRef]
- Mollo, N.; Cicatiello, R.; Aurilia, M.; Scognamiglio, R.; Genesio, R.; Charalambous, M.; Paladino, S.; Conti, A.; Nitsch, L.; Izzo, A. Targeting Mitochondrial Network Architecture in Down Syndrome and Aging. Int. J. Mol. Sci. 2020, 21, 3134. [Google Scholar] [CrossRef]
- Valenti, D.; Rossi, L.; Marzulli, D.; Bellomo, F.; De Rasmo, D.; Signorile, A.; Vacca, R.A. Inhibition of Drp1-mediated mitochondrial fission improves mitochondrial dynamics and bioenergetics stimulating neurogenesis in hippocampal progenitor cells from a Down syndrome mouse model. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3117–3127. [Google Scholar] [CrossRef] [PubMed]
- Urbano-Gámez, J.D.; Casañas, J.J.; Benito, I.; Montesinos, M.L. Prenatal treatment with rapamycin restores enhanced hippocampal mGluR-LTD and mushroom spine size in a Down’s syndrome mouse model. Mol. Brain 2021, 14, 84. [Google Scholar] [CrossRef]
- Perluigi, M.; Picca, A.; Montanari, E.; Calvani, R.; Marini, F.; Matassa, R.; Tramutola, A.; Villani, A.; Familiari, G.; Domenico, F.D.; et al. Aberrant crosstalk between insulin signaling and mTOR in young Down syndrome individuals revealed by neuronal-derived extracellular vesicles. Alzheimers Dement. 2022, 18, 1498–1510. [Google Scholar] [CrossRef] [PubMed]
- Dierssen, M.; Fructuoso, M.; Martínez de Lagrán, M.; Perluigi, M.; Barone, E. Down Syndrome Is a Metabolic Disease: Altered Insulin Signaling Mediates Peripheral and Brain Dysfunctions. Front. Neurosci. 2020, 14, 670. [Google Scholar] [CrossRef] [PubMed]
- Bayona-Bafaluy, M.P.; Garrido-Pérez, N.; Meade, P.; Iglesias, E.; Jiménez-Salvador, I.; Montoya, J.; Martínez-Cué, C.; Ruiz-Pesini, E. Down syndrome is an oxidative phosphorylation disorder. Redox Biol. 2021, 41, 101871. [Google Scholar] [CrossRef]
- Zampieri, B.L.; Costa, A.C.S. Evidence of Energy Metabolism Alterations in Cultured Neonatal Astrocytes Derived from the Ts65Dn Mouse Model of Down Syndrome. Brain Sci. 2022, 12, 83. [Google Scholar] [CrossRef] [PubMed]
- Mollo, N.; Esposito, M.; Aurilia, M.; Scognamiglio, R.; Accarino, R.; Bonfiglio, F.; Cicatiello, R.; Charalambous, M.; Procaccini, C.; Micillo, T.; et al. Human Trisomic iPSCs from Down Syndrome Fibroblasts Manifest Mitochondrial Alterations Early during Neuronal Differentiation. Biology 2021, 10, 609. [Google Scholar] [CrossRef]
- Contestabile, A.; Fila, T.; Ceccarelli, C.; Bonasoni, P.; Bonapace, L.; Santini, D.; Bartesaghi, R.; Ciani, E. Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with Down syndrome and in Ts65Dn mice. Hippocampus 2007, 17, 665–678. [Google Scholar] [CrossRef]
- Stagni, F.; Giacomini, A.; Emili, M.; Guidi, S.; Bartesaghi, R. Neurogenesis impairment: An early developmental defect in Down syndrome. Free Radic. Biol. Med. 2018, 114, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Uguagliati, B.; Stagni, F.; Emili, M.; Giacomini, A.; Russo, C.; Guidi, S.; Bartesaghi, R. Early Appearance of Dendritic Alterations in Neocortical Pyramidal Neurons of the Ts65Dn Model of Down Syndrome. Dev. Neurosci. 2022, 44, 23–38. [Google Scholar] [CrossRef]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508. [Google Scholar] [CrossRef]
- Lu, J.; Esposito, G.; Scuderi, C.; Steardo, L.; Delli-Bovi, L.C.; Hecht, J.L.; Dickinson, B.C.; Chang, C.J.; Mori, T.; Sheen, V. S100B and APP promote a gliocentric shift and impaired neurogenesis in Down syndrome neural progenitors. PLoS ONE 2011, 6, e22126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Acunzo, P.; Pérez-González, R.; Kim, Y.; Hargash, T.; Miller, C.; Alldred, M.J.; Erdjument-Bromage, H.; Penikalapati, S.C.; Pawlik, M.; Saito, M.; et al. Mitovesicles are a novel population of extracellular vesicles of mitochondrial origin altered in Down syndrome. Sci. Adv. 2021, 7, eabe5085. [Google Scholar] [CrossRef] [PubMed]
- Vacca, R.A.; Valenti, D.; Caccamese, S.; Daglia, M.; Braidy, N.; Nabavi, S.M. Plant polyphenols as natural drugs for the management of Down syndrome and related disorders. Neurosci. Biobehav. Rev. 2016, 71, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Noll, C.; Kandiah, J.; Moroy, G.; Gu, Y.; Dairou, J.; Janel, N. Catechins as a Potential Dietary Supplementation in Prevention of Comorbidities Linked with Down Syndrome. Nutrients 2022, 14, 2039. [Google Scholar] [CrossRef]
- Serra, D.; Almeida, L.M.; Dinis, T.C.P. Polyphenols in the management of brain disorders: Modulation of the microbiota-gut-brain axis. Adv. Food Nutr. Res. 2020, 91, 1–27. [Google Scholar] [CrossRef]
- Valenti, D.; De Rasmo, D.; Signorile, A.; Rossi, L.; de Bari, L.; Scala, I.; Granese, B.; Papa, S.; Vacca, R.A. Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with Down’s syndrome. Biochim. Biophys. Acta 2013, 1832, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Moroy, G.; Paul, J.L.; Rebillat, A.S.; Dierssen, M.; de la Torre, R.; Cieuta-Walti, C.; Dairou, J.; Janel, N. Molecular Rescue of Dyrk1A Overexpression Alterations in Mice with Fontup® Dietary Supplement: Role of Green Tea Catechins. Int. J. Mol. Sci. 2020, 21, 1404. [Google Scholar] [CrossRef] [Green Version]
- De Toma, I.; Ortega, M.; Aloy, P.; Sabidó, E.; Dierssen, M. DYRK1A Overexpression Alters Cognition and Neural-Related Proteomic Pathways in the Hippocampus That Are Rescued by Green Tea Extract and/or Environmental Enrichment. Front. Mol. Neurosci. 2019, 12, 272. [Google Scholar] [CrossRef] [PubMed]
- Raefsky, S.M.; Mattson, M.P. Adaptive responses of neuronal mitochondria to bioenergetic challenges: Roles in neuroplasticity and disease resistance. Free Radic. Biol. Med. 2017, 102, 203–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latruffe, N.; Lançon, A.; Frazzi, R.; Aires, V.; Delmas, D.; Michaille, J.J.; Djouadi, F.; Bastin, J.; Cherkaoui-Malki, M. Exploring new ways of regulation by resveratrol involving miRNAs, with emphasis on inflammation. Ann. N. Y. Acad. Sci. 2015, 1348, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Naomi, R.; Yazid, M.D.; Teoh, S.H.; Balan, S.S.; Shariff, H.; Kumar, J.; Bahari, H.; Embong, H. Dietary Polyphenols as a Protection against Cognitive Decline: Evidence from Animal Experiments; Mechanisms and Limitations. Antioxidants 2023, 12, 1054. [Google Scholar] [CrossRef] [PubMed]
- Sahadevan, R.; Singh, S.; Binoy, A.; Sadhukhan, S. Chemico-biological aspects of (-)-epigallocatechin-3-gallate (EGCG) to improve its stability, bioavailability and membrane permeability: Current status and future prospects. Crit. Rev. Food Sci. Nutr. 2022, 2, 1–30. [Google Scholar] [CrossRef]
- de la Torre, R.; de Sola, S.; Hernandez, G.; Farré, M.; Pujol, J.; Rodriguez, J.; Espadaler, J.M.; Langohr, K.; Cuenca-Royo, A.; Principe, A.; et al. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with Down’s syndrome (TESDAD): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Cieuta-Walti, C.; Cuenca-Royo, A.; Langohr, K.; Rakic, C.; López-Vílchez, M.Á.; Lirio, J.; González-Lamuño Leguina, D.; González, T.B.; García, J.G.; Roure, M.R.; et al. PERSEUS Study Group. Safety and preliminary efficacy on cognitive performance and adaptive functionality of epigallocatechin gallate (EGCG) in children with Down syndrome. A randomized phase Ib clinical trial (PERSEUS study). Genet. Med. 2022, 24, 2004–2013. [Google Scholar] [CrossRef]
- Andreu-Fernández, V.; Almeida Toledano, L.; Pizarro, N.; Navarro-Tapia, E.; Gómez-Roig, M.D.; de la Torre, R.; García-Algar, Ó. Bioavailability of Epigallocatechin Gallate Administered with Different Nutritional Strategies in Healthy Volunteers. Antioxidants 2020, 9, 440. [Google Scholar] [CrossRef]
- Vacca, R.A.; Valenti, D. Green tea EGCG plus fish oil omega-3 dietary supplements rescue mitochondrial dysfunctions and are safe in a Down’s syndrome child. Clin. Nutr. 2015, 34, 783–784. [Google Scholar] [CrossRef]
- Scala, I.; Valenti, D.; Scotto D’Aniello, V.; Marino, M.; Riccio, M.P.; Bravaccio, C.; Vacca, R.A.; Strisciuglio, P. Epigallocatechin-3-Gallate Plus Omega-3 Restores the Mitochondrial Complex I and FoF1-ATP Synthase Activities in PBMCs of Young Children with Down Syndrome: A Pilot Study of Safety and Efficacy. Antioxidants 2021, 10, 469. [Google Scholar] [CrossRef]
- Giunta, B.; Hou, H.; Zhu, Y.; Salemi, J.; Ruscin, A.; Shytle, R.D.; Tan, J. Fish oil enhances anti-amyloidogenic properties of green tea EGCG in Tg2576 mice. Neurosci. Lett. 2010, 471, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Izzo, A.; Nitti, M.; Mollo, N.; Paladino, S.; Procaccini, C.; Faicchia, D.; Calì, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; et al. Metformin restores the mitochondrial network and reverses mitochondrial dysfunction in Down syndrome cells. Hum. Mol. Genet. 2017, 26, 1056–1069. [Google Scholar] [CrossRef] [Green Version]
- Mollo, N.; Nitti, M.; Zerillo, L.; Faicchia, D.; Micillo, T.; Accarino, R.; Secondo, A.; Petrozziello, T.; Calì, G.; Cicatiello, R.; et al. Pioglitazone Improves Mitochondrial Organization and Bioenergetics in Down Syndrome Cells. Front. Genet. 2019, 10, 606. [Google Scholar] [CrossRef] [Green Version]
- Butterick, T.A.; Hocum Stone, L.; Duffy, C.; Holley, C.; Cabrera, J.A.; Crampton, M.; Ward, H.B.; Kelly, R.F.; McFalls, E.O. Pioglitazone increases PGC1-α signaling within chronically ischemic myocardium. Basic Res. Cardiol. 2016, 111, 37. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Weaver, I.C.G.; Gauthier-Fisher, A.; Wang, H.; He, L.; Yeomans, J.; Wondisford, F.; Kaplan, D.R.; Miller, F.D. CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and Rubinstein-Taybi syndrome brain. Dev. Cell 2010, 18, 114–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhowail, A.; Alsikhan, R.; Alsaud, M.; Aldubayan, M.; Rabbani, S.I. Protective Effects of Pioglitazone on Cognitive Impairment and the Underlying Mechanisms: A Review of Literature. Drug Des. Devel. Ther. 2022, 16, 2919–2931. [Google Scholar] [CrossRef]
- Zaki, M.E.; El-Bassyouni, H.T.; Tosson, A.M.S.; Youness, E.; Hussein, J. Coenzyme Q10 and pro-inflammatory markers in children with Down syndrome: Clinical and biochemical aspects. J. Pediatr. 2017, 93, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Patterson, D. Folate metabolism and the risk of Down syndrome. Downs Syndr. Res. Pract. 2008, 12, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Tiano, L.; Padella, L.; Santoro, L.; Carnevali, P.; Principi, F.; Bruge, F.; Gabrielli, O.; Littarru, G.P. Prolonged coenzyme Q10 treatment in Down syndrome patients: Effect on DNA oxidation. Neurobiol. Aging 2012, 33, e1–e8. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, L.; Xin, H.; Guo, Y.; Zhu, B.; Su, L.; Wang, S.; Zeng, J.; Chen, Q.; Deng, R.; et al. Mitochondria-targeting folic acid-modified nanoplatform based on mesoporous carbon and a bioactive peptide for improved colorectal cancer treatment. Acta Biomater. 2022, 152, 453–472. [Google Scholar] [CrossRef]
- Blehaut, H.; Mircher, C.; Ravel, A.; Conte, M.; de Portzamparc, V.; Poret, G.; Huon de Kermadec, F.; Rethore, M.-O.; Sturtz, F.G. Effect of leucovorin (Folinic acid) on the developmental quotient of children with down’s syndrome (Trisomy 21) and influence of thyroid status. PLoS ONE 2010, 5, e8394. [Google Scholar] [CrossRef] [Green Version]
- Ellis, J.M.; Tan, H.K.; Gilbert, R.E.; Muller, D.P.R.; Henley, W.; Moy, R.; Pumphrey, R.; Ani, C.; Davies, S.; Edwards, V.; et al. Supplementation with antioxidants and folinic acid for children with Down’s syndrome: Randomized controlled trial. BMJ 2008, 336, 594–597. [Google Scholar] [CrossRef] [Green Version]
- Larsen, E.L.; Padella, L.; Bergholdt, H.K.M.; Henriksen, T.; Santoro, L.; Gabrielli, O.; Poulsen, H.E.; Littarru, G.P.; Orlando, P.; Tiano, L. The effect of long-term treatment with coenzyme Q10 on nucleic acid modifications by oxidation in children with Down syndrome. Neurobiol. Aging 2018, 67, 159–161. [Google Scholar] [CrossRef]
- Armstrong, D.D. Review of Rett syndrome. J. Neuropathol. Exp. Neurol. 1997, 56, 843–849. [Google Scholar] [CrossRef]
- Feldman, D.; Banerjee, A.; Sur, M. Developmental dynamics of Rett syndrome. Neural Plast. 2016, 2016, 6154080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, H.; Cobb, S.; Downs, J. Clinical and biological progress over 50 years in Rett syndrome. Nat. Rev. Neurol. 2017, 13, 37–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, W.E.; Tierney, E.; Rohde, C.A.; Suarez-Pedraza, M.C.; Clarke, M.A.; Salorio, C.F.; Bibat, G.; Bukelis, I.; Naram, D.; Lanham, D.C.; et al. Social impairments in Rett syndrome: Characteristics and relationship with clinical severity. J. Intellect. Disabil. Res. 2012, 56, 233–247. [Google Scholar] [CrossRef]
- Sanmann, J.N.; Schaefer, G.B.; Buehler, B.A.; Sanger, W.G. Algorithmic approach for methyl-CpG binding protein 2 (MECP2) gene testing in patients with neurodevelopmental disabilities. J. Child Neurol. 2012, 27, 346–354. [Google Scholar] [CrossRef]
- Young, J.I.; Hong, E.P.; Castle, J.C.; Crespo-Barreto, J.; Bowman, A.B.; Rose, M.F.; Kang, D.; Richman, R.; Johnson, J.M.; Berget, S.; et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. USA 2005, 102, 17551–17558. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Rikhye, R.V.; Breton-Provencher, V.; Tang, X.; Li, C.; Li, K. Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, E7287–E7296. [Google Scholar] [CrossRef] [Green Version]
- Marina, N.; Turovsky, E.; Christie, I.N.; Hosford, P.S.; Hadjihambi, A.; Korsak, A.; Ang, R.; Mastitskaya, S.; Sheikhbahaei, S.; Theparambil, S.M.; et al. Brain metabolic sensing and metabolic signaling at the level of an astrocyte. Glia 2018, 66, 1185–1199. [Google Scholar] [CrossRef] [Green Version]
- Dotti, M.T.; Manneschi, L.; Malandrini, A.; De Stefano, N.; Caznerale, F.; Federico, A. Mitochondrial dysfunction in Rett syndrome. An ultrastructural and biochemical study. Brain Dev. 1993, 15, 103–106. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, B.; Valenti, D.; Chiodi, V.; Ferrante, A.; de Bari, L.; Fiorentini, C.; Domenici, M.R.; Ricceri, L.; Vacca, R.A.; Fabbri, A.; et al. Modulation of Rho GTPases rescues brain mitochondrial dysfunction, cognitive deficits and aberrant synaptic plasticity in female mice modeling Rett syndrome. Eur. Neuropsychopharmacol. 2015, 25, 889–901. [Google Scholar] [CrossRef]
- Dave, A.; Pillai, P.P. Docosahexaenoic acid increased MeCP2 mediated mitochondrial respiratory complexes II and III enzyme activities in cortical astrocytes. J. Biochem. Mol. Toxicol. 2022, 36, e23002. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; de Bari, L.; Vigli, D.; Lacivita, E.; Leopoldo, M.; Laviola, G. Stimulation of the brain serotonin receptor 7 rescues mitochondrial dysfunction in female mice from two models of Rett syndrome. Neuropharmacology 2017, 121, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Felice, C.; Ciccoli, L.; Leoncini, S.; Signorini, C.; Rossi, M.; Vannuccini, L. Systemic oxidative stress in classic Rett syndrome. Free Radic. Biol. Med. 2009, 47, 440–448. [Google Scholar] [CrossRef]
- Signorini, C.; Leoncini, S.; De Felice, C.; Pecorelli, A.; Meloni, I.; Ariani, F.; Mari, F.; Amabile, S.; Paccagnini, E.; Gentile, M.; et al. Redox imbalance and morphological changes in skin fibroblasts in typical Rett syndrome. Oxid. Med. Cell Longev. 2014, 2014, 195935. [Google Scholar] [CrossRef] [Green Version]
- Festerling, K.; Can, K.; Kügler, S.; Müller, M. Overshooting Subcellular Redox-Responses in Rett-Mouse Hippocampus during Neurotransmitter Stimulation. Cells 2020, 9, 2539. [Google Scholar] [CrossRef]
- Bebensee, D.F.; Can, K.; Müller, M. Increased Mitochondrial Mass and Cytosolic Redox Imbalance in Hippocampal Astrocytes of a Mouse Model of Rett Syndrome: Subcellular Changes Revealed by Ratiometric Imaging of JC-1 and roGFP1 Fluorescence. Oxid. Med. Cell Longev. 2017, 2017, 3064016. [Google Scholar] [CrossRef] [Green Version]
- Belichenko, P.V.; Wright, E.E.; Belichenko, N.P.; Masliah, E.; Li, H.H.; Mobley, W.C.; Francke, U. Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: Evidence for disruption of neuronal networks. J. Comp. Neurol. 2009, 514, 240–258. [Google Scholar] [CrossRef]
- Crivellari, I.; Pecorelli, A.; Cordone, V.; Marchi, S.; Pinton, P.; Hayek, J.; Cervellati, C.; Valacchi, G. Impaired mitochondrial quality control in Rett Syndrome. Arch. Biochem. Biophys. 2021, 700, 108790. [Google Scholar] [CrossRef]
- Sandweiss, A.J.; Brandt, V.L.; Zoghbi, H.Y. Advances in understanding of Rett syndrome and MECP2 duplication syndrome: Prospects for future therapies. Lancet Neurol. 2020, 19, 689–698. [Google Scholar] [CrossRef]
- Shulyakova, N.; Andreazza, A.C.; Mills, L.R.; Eubanks, J.H. Mitochondrial dysfunction in the pathogenesis of Rett syndrome: Implications for mitochondria-targeted therapies. Front. Cell Neurosci. 2017, 11, 58. [Google Scholar] [CrossRef] [Green Version]
- Zuliani, I.; Urbinati, C.; Valenti, D.; Quattrini, M.C.; Medici, V.; Cosentino, L.; Pietraforte, D.; Di Domenico, F.; Perluigi, M.; Vacca, R.A.; et al. The Anti-Diabetic Drug Metformin Rescues Aberrant Mitochondrial Activity and Restrains Oxidative Stress in a Female Mouse Model of Rett Syndrome. Int. J. Mol. Sci. 2020, 9, 1669. [Google Scholar] [CrossRef] [PubMed]
- Urbinati, C.; Lanzillotta, C.; Cosentino, L.; Valenti, D.; Quattrini, M.C.; Di Crescenzo, L.; Prestia, F.; Pietraforte, D.; Perluigi, M.; Di Domenico, F.; et al. Chronic treatment with the anti-diabetic drug metformin rescues impaired brain mitochondrial activity and selectively ameliorates defective cognitive flexibility in a female mouse model of Rett syndrome. Neuropharmacology 2023, 224, 109350. [Google Scholar] [CrossRef]
- Urbinati, C.; Cosentino, L.; Germinario, E.A.P.; Valenti, D.; Vigli, D.; Ricceri, L.; Laviola, G.; Fiorentini, C.; Vacca, R.A.; Fabbri, A.; et al. Treatment with the Bacterial Toxin CNF1 Selectively Rescues Cognitive and Brain Mitochondrial Deficits in a Female Mouse Model of Rett Syndrome Carrying a MeCP2-Null Mutation. Int. J. Mol. Sci. 2021, 22, 6739. [Google Scholar] [CrossRef] [PubMed]
- Garber, K.B.; Visootsak, J.; Warren, S.T. Fragile X syndrome. Eur. J. Hum. Genet. 2008, 16, 666–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penagarikano, O.; Mulle, J.G.; Warren, S.T. The pathophysiology of Fragile X syndrome. Annu. Rev. Genomics Hum. Genet. 2007, 8, 109–129. [Google Scholar] [CrossRef] [Green Version]
- Maurin, T.; Zongaro, S.; Bardoni, B. Fragile X Syndrome: From molecular pathology to therapy. Neurosci. Biobehav. Rev. 2014, 46, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Dahlhaus, R. Of men and mice: Modeling the fragile X syndrome. Front. Mol. Neurosci. 2018, 11, 41. [Google Scholar] [CrossRef]
- Raspa, M.; Wheeler, A.C.; Riley, C. Public Health Literature Review of Fragile X Syndrome. Pediatrics 2017, 139 (Suppl. S3), S153–S171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, J.D.; Zhao, X. The molecular biology of FMRP: New insights into Fragile X syndrome. Nat. Rev. Neurosci. 2021, 22, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Till, S.M. The developmental roles of FMRP. Biochem. Soc. Trans. 2010, 38, 507–510. [Google Scholar] [CrossRef]
- Geng, J.; Khaket, T.P.; Pan, J.; Li, W.; Zhang, Y.; Ping, Y.; Cobos Sillero, M.I.; Lu, B. Dev Deregulation of ER-mitochondria contact formation and mitochondrial calcium homeostasis mediated by VDAC in fragile X syndrome. Cell 2023, 58, 597–615.e10. [Google Scholar] [CrossRef]
- Bassell, G.J.; Warren, S.T. Fragile X syndrome: Loss of local mRNA regulation alters synaptic development and function. Neuron 2008, 60, 201–214. [Google Scholar] [CrossRef] [Green Version]
- Bulow, P.; Murphy, T.J.; Bassell, G.J.; Wenner, P. Homeostatic Intrinsic Plasticity Is Functionally Altered in Fmr1 KO Cortical Neurons. Cell Rep. 2019, 26, 1378–1388.e13. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Wang, F.; Li, M.; Sah, N.; Stockton, M.; Tidei, J.J.; Gao, Y.; Korabelnikov, T.; Kannan, S.; Vevea, J.D.; et al. Reduced mitochondrial fusion and Huntingtin levels contribute to impaired dendritic maturation and behavioural deficits in Fmr1-mutant mice. Nat. Neurosci. 2019, 22, 386–400. [Google Scholar] [CrossRef]
- Ascano, M., Jr.; Mukherjee, N.; Bandaru, P.; Miller, J.B.; Nusbaum, J.D.; Corcoran, D.L.; Langlois, C.; Munschauer, M.; Dewell, S.; Hafner, M.; et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 2012, 492, 382–386. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, K.K.; Wang, A.; Wang, L.; Tracey, M.; Kleiner, G.; Quinzii, C.M.; Sun, L.; Yang, G.; Perez-Zoghbi, J.F.; Licznerski, P.; et al. Inefficient thermogenic mitochondrial respiration due to futile proton leak in a mouse model of Fragile X syndrome. FASEB J. 2020, 34, 7404–7426. [Google Scholar] [CrossRef]
- Licznerski, P.; Park, H.A.; Rolyan, H.; Chen, R.; Mnatsakanyan, N.; Miranda, P.; Graham, M.; Wu, J.; Cruz-Reyes, N.; Mehta, N.; et al. ATP Synthase c-Subunit Leak Causes Aberrant Cellular Metabolism in Fragile X Syndrome. Cell 2020, 182, 1170–1185.e9. [Google Scholar] [CrossRef]
- Ha, B.G.; Heo, J.-Y.; Jang, Y.-J.; Park, T.-S.; Choi, J.-Y.; Jang, W.Y.; Jeong, S.-J. Depletion of Mitochondrial Components from Extracellular Vesicles Secreted from Astrocytes in a Mouse Model of Fragile X Syndrome. Int. J. Mol. Sci. 2021, 22, 410. [Google Scholar] [CrossRef]
- Bülow, P.; Wenner, P.A.; Faundez, V.; Bassell, G.J. Mitochondrial Structure and Polarity in Dendrites and the Axon Initial Segment Are Regulated by Homeostatic Plasticity and Dysregulated in Fragile X Syndrome. Front. Cell Dev. Biol. 2021, 9, 702020. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valenti, D.; Vacca, R.A. Brain Mitochondrial Bioenergetics in Genetic Neurodevelopmental Disorders: Focus on Down, Rett and Fragile X Syndromes. Int. J. Mol. Sci. 2023, 24, 12488. https://doi.org/10.3390/ijms241512488
Valenti D, Vacca RA. Brain Mitochondrial Bioenergetics in Genetic Neurodevelopmental Disorders: Focus on Down, Rett and Fragile X Syndromes. International Journal of Molecular Sciences. 2023; 24(15):12488. https://doi.org/10.3390/ijms241512488
Chicago/Turabian StyleValenti, Daniela, and Rosa Anna Vacca. 2023. "Brain Mitochondrial Bioenergetics in Genetic Neurodevelopmental Disorders: Focus on Down, Rett and Fragile X Syndromes" International Journal of Molecular Sciences 24, no. 15: 12488. https://doi.org/10.3390/ijms241512488