
Circadian Regulation of Macrophages and Osteoclasts in Rheumatoid Arthritis

Abstract
:1. Introduction
2. Overview of Rheumatoid Arthritis
3. Roles of Macrophages in the Pathogenesis of RA
4. Outline of Mammalian Circadian Clocks
5. Circadian Regulation of Inflammatory Macrophages
6. Circadian Regulation of Macrophages by the CLOCK/BMAL1 Complex
7. Circadian Regulation of Macrophages by PER, CRY, and Rev-erb
8. Osteoclastogenesis and Inflammation
9. Circadian Regulation of Osteoclastogenesis

{kind=link}
{kind=link}
{kind=link}
| Depletion Methods | Bone Mass | Osteoclasto-Genesis | References |
|---|---|---|---|
| Ctsk promoter-driven Bmal1 KO | Increased | Decreased | [57] |
| Bmal1 knockdown in RAW264.7 cells | Not applicable | Increased | [60] |
| Ctsk promoter-driven Bmal1 KO | Normal | Normal | [61] |
| Bmal1-/- (germline) | Decreased | Increased | [63] |
| Bmal1-/- (germline) | Decreased | Increased | [65] |
| Osx promoter-driven Bmal1 KO | Decreased | Increased | [65] |
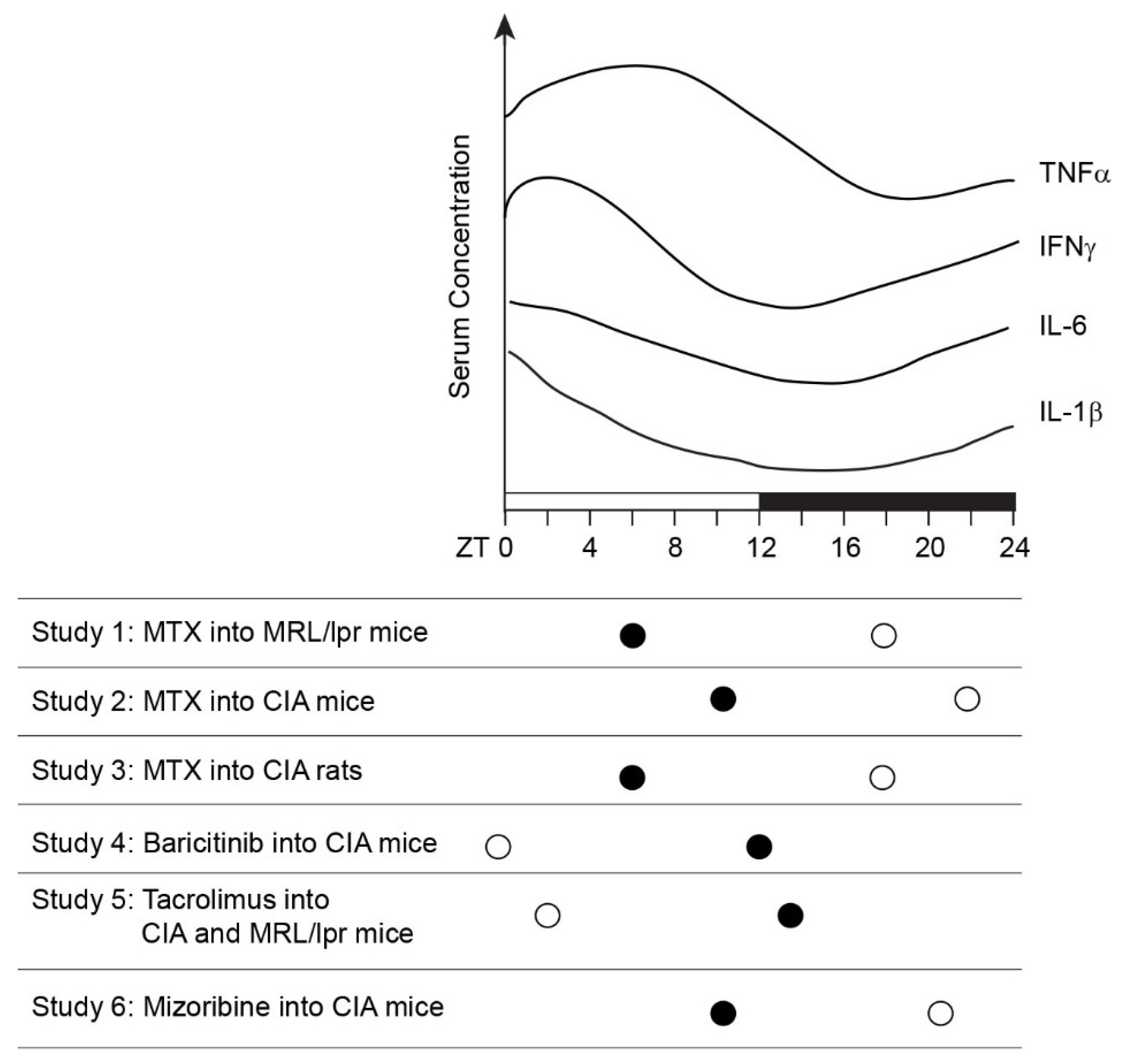
10. Chronotherapy

11. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTH | adrenocorticotropic hormone |
| ACPA | anti-citrullinated protein antibody |
| AP-1 | activator protein 1 |
| BMDM | bone marrow-derived monocyte/macrophage |
| CAPRA | Circadian Administration of Prednisone in RA |
| CIA | collagen-induced arthritis |
| Ctsk | cathepsin K |
| CCL2 | C-C- motif ligand 2 |
| CRP | C-reactive protein |
| CXCL1 | C-X-C motif ligand 1 |
| DMARD | disease-modifying anti-rheumatic drug |
| GM-CSF | granulocyte macrophage-colony stimulating factor |
| HALO | hours after the light was turned on |
| IFN | interferon |
| IFR3 | interferon regulatory factor 3 |
| IL-1 | interleukin-1 |
| FLS | fibroblast-like synoviocyte |
| H3K27ac | acetylation of lysine 27 in histone H3 |
| H3K27me3 | trimethylation of lysine 27 in histone H3 |
| HMGB1 | high mobility group box 1 |
| JAK | Janus kinase |
| KO | knockout |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| M-CSF | macrophage-colony stimulating factor |
| MLS | macrophage-like synoviocyte |
| MMP | matrix metalloproteinase |
| MTX | methotrexate |
| Nfatc1 | nuclear factor of activated T cells 1 |
| NF-kB | nuclear factor kappa B subunit 1 |
| NK cell | natural killer cell |
| NKT cell | natural killer T cell |
| Opg | osteoprotegerin |
| PAS | Per-Armt-Sim domain |
| PKA | protein kinase A |
| PRR | pattern recognition receptor |
| RA | rheumatoid arthritis |
| RANKL | receptor activator of nuclear factor-κB ligand |
| Rev-erbα | reverse orientation c-erbAα |
| RORα | retinoic acid receptor-related orphan receptor α |
| RRE | Rev-erb/ROR response element |
| SAA | serum amyloid A |
| TGFβ | transforming growth factor β |
| TLR2 | toll-like receptor 2 |
| WT | wild type |
| ZT | Zeitgeber Time |
References
- England, B.R.; Mikuls, T.R. Clinical features of rheumatoid arthritis. In Firestein & Kelley’s Textbook of Rheumatology, 11th ed.; Firestein, G.S., Budd, R.C., Gabriel, S.E., McInnes, I.B., O’Dell, J.R., Koretzky, G., Eds.; Elsevier: Philadelphia, PA, USA, 2020; pp. 1236–1257. [Google Scholar]
- Straub, R.H.; Cutolo, M. Circadian rhythms in rheumatoid arthritis: Implications for pathophysiology and therapeutic management. Arthritis Rheum. 2007, 56, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Petrovsky, N.; McNair, P.; Harrison, L.C. Diurnal rhythms of pro-inflammatory cytokines: Regulation by plasma cortisol and therapeutic implications. Cytokine 1998, 10, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.T.; Pierre, K.K.; Schlesinger, N.; Androulakis, I.P. The Potential of Circadian Realignment in Rheumatoid Arthritis. Crit. Rev. Biomed. Eng. 2016, 44, 177–191. [Google Scholar] [CrossRef] [Green Version]
- Arvidson, N.G.; Gudbjornsson, B.; Elfman, L.; Ryden, A.C.; Totterman, T.H.; Hallgren, R. Circadian rhythm of serum interleukin-6 in rheumatoid arthritis. Ann. Rheum. Dis. 1994, 53, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Madhok, R.; Crilly, A.; Watson, J.; Capell, H.A. Serum interleukin 6 levels in rheumatoid arthritis: Correlations with clinical and laboratory indices of disease activity. Ann. Rheum. Dis. 1993, 52, 232–234. [Google Scholar] [CrossRef]
- Cutolo, M. Glucocorticoids and chronotherapy in rheumatoid arthritis. RMD Open 2016, 2, e000203. [Google Scholar] [CrossRef] [Green Version]
- Ursini, F.; De Giorgi, A.; D’Onghia, M.; De Giorgio, R.; Fabbian, F.; Manfredini, R. Chronobiology and Chronotherapy in Inflammatory Joint Diseases. Pharmaceutics 2021, 13, 1832. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef]
- Sparks, J.A. Rheumatoid Arthritis. Ann. Intern. Med. 2019, 170, ITC1–ITC16. [Google Scholar] [CrossRef]
- Gravallese, E.M.; Firestein, G.S. Rheumatoid Arthritis—Common Origins, Divergent Mechanisms. N. Engl. J. Med. 2023, 388, 529–542. [Google Scholar] [CrossRef]
- Firestein, G.S. Etiology of rheumatoid arthritis. In Firestein & Kelley’s Textbook of Rheumatology, 11th ed.; Firestein, G.S., Budd, R.C., Gabriel, S.E., McInnes, I.B., O’Dell, J.R., Koretzky, G., Eds.; Elsevier: Philadelphia, PA, USA, 2020; pp. 1181–1199. [Google Scholar]
- Firestein, G.S. Pathogenesis of rheumatoid arthritis. In Firestein & Kelley’s Textbook of Rheumatology, 11th ed.; Firestein, G.S., Budd, R.C., Gabriel, S.E., McInnes, I.B., O’Dell, J.R., Koretzky, G., Eds.; Elsevier: Philadelphia, PA, USA, 2020; pp. 1200–1235. [Google Scholar]
- Winter, D.R.; Montgomery, A.; Perlman, H. Monnuclear phagocytes. In Firestein & Kelley’s Textbook of Rheumatology, 11th ed.; Firestein, G.S., Budd, R.C., Gabriel, S.E., McInnes, I.B., O’Dell, J.R., Koretzky, G., Eds.; Elsevier: Philadelphia, PA, USA, 2020; pp. 150–160. [Google Scholar]
- Tu, J.; Hong, W.; Zhang, P.; Wang, X.; Korner, H.; Wei, W. Ontology and Function of Fibroblast-Like and Macrophage-Like Synoviocytes: How Do They Talk to Each Other and Can They Be Targeted for Rheumatoid Arthritis Therapy? Front. Immunol. 2018, 9, 1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, J.E.; Ray, D.W. The role of the circadian clock in rheumatoid arthritis. Arthritis Res. Ther. 2013, 15, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Hashimoto, T.; Sakai, Y.; Hashiramoto, A. Involvement of the circadian rhythm and inflammatory cytokines in the pathogenesis of rheumatoid arthritis. J. Immunol. Res. 2014, 2014, 282495. [Google Scholar] [CrossRef] [PubMed]
- Ingpen, M.L. The quantitative measurement of joint changes in rheumatoid arthritis. Ann. Phys. Med. 1968, 9, 322–327. [Google Scholar] [CrossRef]
- Cajochen, C.; Krauchi, K.; Wirz-Justice, A. Role of melatonin in the regulation of human circadian rhythms and sleep. J. Neuroendocr. 2003, 15, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Zisapel, N. New perspectives on the role of melatonin in human sleep, circadian rhythms and their regulation. Br. J. Pharmacol. 2018, 175, 3190–3199. [Google Scholar] [CrossRef] [Green Version]
- Lakatos, P.; Blumsohn, A.; Eastell, R.; Tarjan, G.; Shinoda, H.; Stern, P.H. Circadian rhythm of in vitro bone-resorbing activity in human serum. J. Clin. Endocrinol. Metab. 1995, 80, 3185–3190. [Google Scholar] [CrossRef]
- Perry, M.G.; Kirwan, J.R.; Jessop, D.S.; Hunt, L.P. Overnight variations in cortisol, interleukin 6, tumour necrosis factor alpha and other cytokines in people with rheumatoid arthritis. Ann. Rheum. Dis. 2009, 68, 63–68. [Google Scholar] [CrossRef]
- Siouti, E.; Andreakos, E. The many facets of macrophages in rheumatoid arthritis. Biochem. Pharmacol. 2019, 165, 152–169. [Google Scholar] [CrossRef]
- Ji, R.R.; Chamessian, A.; Zhang, Y.Q. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354, 572–577. [Google Scholar] [CrossRef] [Green Version]
- Patke, A.; Young, M.W.; Axelrod, S. Molecular mechanisms and physiological importance of circadian rhythms. Nat. Rev. Mol. Cell Biol. 2020, 21, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Koronowski, K.B.; Sassone-Corsi, P. Communicating clocks shape circadian homeostasis. Science 2021, 371, eabd0951. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.D.; Fentress, S.J.; Qiu, Y.; Yun, K.; Cox, J.S.; Chawla, A. Circadian gene Bmal1 regulates diurnal oscillations of Ly6C(hi) inflammatory monocytes. Science 2013, 341, 1483–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oishi, Y.; Hayashi, S.; Isagawa, T.; Oshima, M.; Iwama, A.; Shimba, S.; Okamura, H.; Manabe, I. Bmal1 regulates inflammatory responses in macrophages by modulating enhancer RNA transcription. Sci. Rep. 2017, 7, 7086. [Google Scholar] [CrossRef] [PubMed]
- Timmons, G.A.; Carroll, R.G.; O’Siorain, J.R.; Cervantes-Silva, M.P.; Fagan, L.E.; Cox, S.L.; Palsson-McDermott, E.; Finlay, D.K.; Vincent, E.E.; Jones, N.; et al. The Circadian Clock Protein BMAL1 Acts as a Metabolic Sensor In Macrophages to Control the Production of Pro IL-1beta. Front. Immunol. 2021, 12, 700431. [Google Scholar] [CrossRef]
- Alexander, R.K.; Liou, Y.H.; Knudsen, N.H.; Starost, K.A.; Xu, C.; Hyde, A.L.; Liu, S.; Jacobi, D.; Liao, N.S.; Lee, C.H. Bmal1 integrates mitochondrial metabolism and macrophage activation. eLife 2020, 9, e54090. [Google Scholar] [CrossRef]
- Deng, W.; Zhu, S.; Zeng, L.; Liu, J.; Kang, R.; Yang, M.; Cao, L.; Wang, H.; Billiar, T.R.; Jiang, J.; et al. The Circadian Clock Controls Immune Checkpoint Pathway in Sepsis. Cell Rep. 2018, 24, 366–378. [Google Scholar] [CrossRef] [Green Version]
- Kitchen, G.B.; Cunningham, P.S.; Poolman, T.M.; Iqbal, M.; Maidstone, R.; Baxter, M.; Bagnall, J.; Begley, N.; Saer, B.; Hussell, T.; et al. The clock gene Bmal1 inhibits macrophage motility, phagocytosis, and impairs defense against pneumonia. Proc. Natl. Acad. Sci. USA 2020, 117, 1543–1551. [Google Scholar] [CrossRef] [Green Version]
- Bellet, M.M.; Deriu, E.; Liu, J.Z.; Grimaldi, B.; Blaschitz, C.; Zeller, M.; Edwards, R.A.; Sahar, S.; Dandekar, S.; Baldi, P.; et al. Circadian clock regulates the host response to Salmonella. Proc. Natl. Acad. Sci. USA 2013, 110, 9897–9902. [Google Scholar] [CrossRef]
- Xu, H.; Li, H.; Woo, S.L.; Kim, S.M.; Shende, V.R.; Neuendorff, N.; Guo, X.; Guo, T.; Qi, T.; Pei, Y.; et al. Myeloid cell-specific disruption of Period1 and Period2 exacerbates diet-induced inflammation and insulin resistance. J. Biol. Chem. 2014, 289, 16374–16388. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Malkani, G.; Shi, X.; Meyer, M.; Cunningham-Runddles, S.; Ma, X.; Sun, Z.S. The circadian clock Period 2 gene regulates gamma interferon production of NK cells in host response to lipopolysaccharide-induced endotoxic shock. Infect. Immun. 2006, 74, 4750–4756. [Google Scholar] [CrossRef] [Green Version]
- Silver, A.C.; Arjona, A.; Walker, W.E.; Fikrig, E. The circadian clock controls toll-like receptor 9-mediated innate and adaptive immunity. Immunity 2012, 36, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Narasimamurthy, R.; Hatori, M.; Nayak, S.K.; Liu, F.; Panda, S.; Verma, I.M. Circadian clock protein cryptochrome regulates the expression of proinflammatory cytokines. Proc. Natl. Acad. Sci. USA 2012, 109, 12662–12667. [Google Scholar] [CrossRef]
- Gibbs, J.E.; Blaikley, J.; Beesley, S.; Matthews, L.; Simpson, K.D.; Boyce, S.H.; Farrow, S.N.; Else, K.J.; Singh, D.; Ray, D.W.; et al. The nuclear receptor REV-ERBalpha mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc. Natl. Acad. Sci. USA 2012, 109, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Sakurai, T.; Ogasawara, J.; Shirato, K.; Ishibashi, Y.; Oh-ishi, S.; Imaizumi, K.; Haga, S.; Hitomi, Y.; Izawa, T.; et al. Direct and indirect suppression of interleukin-6 gene expression in murine macrophages by nuclear orphan receptor REV-ERBalpha. Sci. World J. 2014, 2014, 685854. [Google Scholar] [CrossRef]
- Sato, S.; Sakurai, T.; Ogasawara, J.; Takahashi, M.; Izawa, T.; Imaizumi, K.; Taniguchi, N.; Ohno, H.; Kizaki, T. A circadian clock gene, Rev-erbalpha, modulates the inflammatory function of macrophages through the negative regulation of Ccl2 expression. J. Immunol. 2014, 192, 407–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirato, K.; Sato, S. Macrophage Meets the Circadian Clock: Implication of the Circadian Clock in the Role of Macrophages in Acute Lower Respiratory Tract Infection. Front. Cell Infect. Microbiol. 2022, 12, 826738. [Google Scholar] [CrossRef] [PubMed]
- Timmons, G.A.; O’Siorain, J.R.; Kennedy, O.D.; Curtis, A.M.; Early, J.O. Innate Rhythms: Clocks at the Center of Monocyte and Macrophage Function. Front. Immunol. 2020, 11, 1743. [Google Scholar] [CrossRef]
- Jerigova, V.; Zeman, M.; Okuliarova, M. Circadian Disruption and Consequences on Innate Immunity and Inflammatory Response. Int. J. Mol. Sci. 2022, 23, 13722. [Google Scholar] [CrossRef]
- Keller, M.; Mazuch, J.; Abraham, U.; Eom, G.D.; Herzog, E.D.; Volk, H.D.; Kramer, A.; Maier, B. A circadian clock in macrophages controls inflammatory immune responses. Proc. Natl. Acad. Sci. USA 2009, 106, 21407–21412. [Google Scholar] [CrossRef]
- Collins, E.J.; Cervantes-Silva, M.P.; Timmons, G.A.; O’Siorain, J.R.; Curtis, A.M.; Hurley, J.M. Post-transcriptional circadian regulation in macrophages organizes temporally distinct immunometabolic states. Genome Res. 2021, 31, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Natoli, G. Molecular control of activation and priming in macrophages. Nat. Immunol. 2016, 17, 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, O.; Ang, C.K.E.; Guan, X.L. Macrophage-Bacteria Interactions-A Lipid-Centric Relationship. Front. Immunol. 2017, 8, 1836. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Halberg, F.; Johnson, E.A.; Brown, B.W.; Bittner, J.J. Susceptibility rhythm to E. coli endotoxin and bioassay. Proc. Soc. Exp. Biol. Med. 1960, 103, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Jacome-Galarza, C.E.; Percin, G.I.; Muller, J.T.; Mass, E.; Lazarov, T.; Eitler, J.; Rauner, M.; Yadav, V.K.; Crozet, L.; Bohm, M.; et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature 2019, 568, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, J.; Xie, X.; Gu, F.; Sui, Z.; Zhang, K.; Yu, T. Recent Advances in Osteoclast Biological Behavior. Front. Cell Dev. Biol. 2021, 9, 788680. [Google Scholar] [CrossRef]
- Blangy, A.; Bompard, G.; Guerit, D.; Marie, P.; Maurin, J.; Morel, A.; Vives, V. The osteoclast cytoskeleton—Current understanding and therapeutic perspectives for osteoporosis. J. Cell Sci. 2020, 133, jcs244798. [Google Scholar] [CrossRef]
- Aureal, M.; Machuca-Gayet, I.; Coury, F. Rheumatoid Arthritis in the View of Osteoimmunology. Biomolecules 2020, 11, 48. [Google Scholar] [CrossRef]
- Maeda, K.; Yoshida, K.; Nishizawa, T.; Otani, K.; Yamashita, Y.; Okabe, H.; Hadano, Y.; Kayama, T.; Kurosaka, D.; Saito, M. Inflammation and Bone Metabolism in Rheumatoid Arthritis: Molecular Mechanisms of Joint Destruction and Pharmacological Treatments. Int. J. Mol. Sci. 2022, 23, 2871. [Google Scholar] [CrossRef]
- Jung, S.M.; Kim, K.W.; Yang, C.W.; Park, S.H.; Ju, J.H. Cytokine-mediated bone destruction in rheumatoid arthritis. J. Immunol. Res. 2014, 2014, 263625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danks, L.; Komatsu, N.; Guerrini, M.M.; Sawa, S.; Armaka, M.; Kollias, G.; Nakashima, T.; Takayanagi, H. RANKL expressed on synovial fibroblasts is primarily responsible for bone erosions during joint inflammation. Ann. Rheum. Dis. 2016, 75, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Meednu, N.; Zhang, H.; Owen, T.; Sun, W.; Wang, V.; Cistrone, C.; Rangel-Moreno, J.; Xing, L.; Anolik, J.H. Production of RANKL by Memory B Cells: A Link Between B Cells and Bone Erosion in Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 805–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.Y.; Wang, J.; Tong, X.; Chen, J.; Wang, B.; Miao, Z.N.; Li, X.; Ye, J.X.; Yuan, F.L. Emerging role of circadian rhythm in bone remodeling. J. Mol. Med. 2019, 97, 19–24. [Google Scholar] [CrossRef]
- Chen, G.; Tang, Q.; Yu, S.; Xie, Y.; Sun, J.; Li, S.; Chen, L. The biological function of BMAL1 in skeleton development and disorders. Life Sci. 2020, 253, 117636. [Google Scholar] [CrossRef]
- Qin, Y.; Chen, Z.H.; Wu, J.J.; Zhang, Z.Y.; Yuan, Z.D.; Guo, D.Y.; Chen, M.N.; Li, X.; Yuan, F.L. Circadian clock genes as promising therapeutic targets for bone loss. Biomed. Pharmacother. 2023, 157, 114019. [Google Scholar] [CrossRef]
- Xu, C.; Ochi, H.; Fukuda, T.; Sato, S.; Sunamura, S.; Takarada, T.; Hinoi, E.; Okawa, A.; Takeda, S. Circadian Clock Regulates Bone Resorption in Mice. J. Bone Miner. Res. 2016, 31, 1344–1355. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Fujihara, Y.; Kondo, H.; Noguchi, T.; Togari, A. Glucocorticoids mediate circadian timing in peripheral osteoclasts resulting in the circadian expression rhythm of osteoclast-related genes. Bone 2014, 61, 1–9. [Google Scholar] [CrossRef]
- Zhao, J.; Zhou, X.; Tang, Q.; Yu, R.; Yu, S.; Long, Y.; Cao, C.; Han, J.; Shi, A.; Mao, J.J.; et al. BMAL1 Deficiency Contributes to Mandibular Dysplasia by Upregulating MMP3. Stem Cell Rep. 2018, 10, 180–195. [Google Scholar] [CrossRef] [Green Version]
- Tsang, K.; Liu, H.; Yang, Y.; Charles, J.F.; Ermann, J. Defective circadian control in mesenchymal cells reduces adult bone mass in mice by promoting osteoclast function. Bone 2019, 121, 172–180. [Google Scholar] [CrossRef]
- Samsa, W.E.; Vasanji, A.; Midura, R.J.; Kondratov, R.V. Deficiency of circadian clock protein BMAL1 in mice results in a low bone mass phenotype. Bone 2016, 84, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Yu, R.; Long, Y.; Zhao, J.; Yu, S.; Tang, Q.; Chen, L. BMAL1 deficiency promotes skeletal mandibular hypoplasia via OPG downregulation. Cell Prolif. 2018, 51, e12470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, G.; Hua, B.; Yang, Y.; Xu, L.; Cai, T.; Sun, N.; Yan, Z.; Lu, C.; Qian, R. The Circadian Gene Clock Regulates Bone Formation Via PDIA3. J. Bone Miner. Res. 2017, 32, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Takarada, T.; Xu, C.; Ochi, H.; Nakazato, R.; Yamada, D.; Nakamura, S.; Kodama, A.; Shimba, S.; Mieda, M.; Fukasawa, K.; et al. Bone Resorption Is Regulated by Circadian Clock in Osteoblasts. J. Bone Miner. Res. 2017, 32, 872–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukasaki, M.; Takayanagi, H. Osteoclast biology in the single-cell era. Inflamm. Regen. 2022, 42, 27. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Patel, M.S.; Bradley, A.; Wagner, E.F.; Karsenty, G. The molecular clock mediates leptin-regulated bone formation. Cell 2005, 122, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Maronde, E.; Schilling, A.F.; Seitz, S.; Schinke, T.; Schmutz, I.; van der Horst, G.; Amling, M.; Albrecht, U. The clock genes Period 2 and Cryptochrome 2 differentially balance bone formation. PLoS ONE 2010, 5, e11527. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Larkin, D.W.; Albrecht, U.; Sun, Z.S.; Sage, M.; Eichele, G.; Lee, C.C.; Bradley, A. The mPer2 gene encodes a functional component of the mammalian circadian clock. Nature 1999, 400, 169–173. [Google Scholar] [CrossRef]
- O’Dell, J.R. Treatment of rheumatoid arthritis. In Firestein & Kelley’s Textbook of Rheumatology, 11th ed.; Firestein, G.S., Budd, R.C., Gabriel, S.E., McInnes, I.B., O’Dell, J.R., Koretzky, G., Eds.; Elsevier: Philadelphia, PA, USA, 2020; pp. 1258–1283. [Google Scholar]
- Van der Goes, M.C.; Jacobs, J.W.G. Glucocorticoid therapy. In Firestein & Kelley’s Textbook of Rheumatology, 11th ed.; Firestein, G.S., Budd, R.C., Gabriel, S.E., McInnes, I.B., O’Dell, J.R., Koretzky, G., Eds.; Elsevier: Philadelphia, PA, USA, 2020; pp. 985–1006. [Google Scholar]
- Jang, D.I.; Lee, A.H.; Shin, H.Y.; Song, H.R.; Park, J.H.; Kang, T.B.; Lee, S.R.; Yang, S.H. The Role of Tumor Necrosis Factor Alpha (TNF-alpha) in Autoimmune Disease and Current TNF-alpha Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef]
- Upchurch, K.S.; Kay, J. Evolution of treatment for rheumatoid arthritis. Rheumatology 2012, 51 (Suppl. S6), vi28–vi36. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, K.; Kavanaugh, A.; Ritchlin, C.T. Anti-cytokine therapies. In Firestein & Kelley’s Textbook of Rheumatology; Firestein, G.S., Budd, R.C., Gabriel, S.E., McInnes, I.B., O’Dell, J.R., Koretzky, G., Eds.; Elsevier: Philadelphia, PA, USA, 2020; pp. 1046–1067. [Google Scholar]
- Reddy, V.; Cohen, S. Intra-cellular targeting agents in rheumatic disease. In Firestein & Kelley’s Textbook of Rheumatology, 11th ed.; Firestein, G.S., Budd, R.C., Gabriel, S.E., McInnes, I.B., O’Dell, J.R., Koretzky, G., Eds.; Elsevier: Philadelphia, PA, USA, 2020; pp. 1091–1110. [Google Scholar]
- Deandrade, J.R.; McCormick, J.N.; Hill, A.G. Small Doses of Prednisolone in the Management of Rheumatoid Arthritis. Ann. Rheum. Dis. 1964, 23, 158–162. [Google Scholar] [CrossRef]
- Cutolo, M. Circadian rhythms and rheumatoid arthritis. Jt. Bone Spine 2019, 86, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Derendorf, H.; Ruebsamen, K.; Clarke, L.; Schaeffler, A.; Kirwan, J.R. Pharmacokinetics of modified-release prednisone tablets in healthy subjects and patients with rheumatoid arthritis. J. Clin. Pharmacol. 2013, 53, 326–333. [Google Scholar] [CrossRef] [Green Version]
- Buttgereit, F.; Doering, G.; Schaeffler, A.; Witte, S.; Sierakowski, S.; Gromnica-Ihle, E.; Jeka, S.; Krueger, K.; Szechinski, J.; Alten, R. Efficacy of modified-release versus standard prednisone to reduce duration of morning stiffness of the joints in rheumatoid arthritis (CAPRA-1): A double-blind, randomised controlled trial. Lancet 2008, 371, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, F.; Mehta, D.; Kirwan, J.; Szechinski, J.; Boers, M.; Alten, R.E.; Supronik, J.; Szombati, I.; Romer, U.; Witte, S.; et al. Low-dose prednisone chronotherapy for rheumatoid arthritis: A randomised clinical trial (CAPRA-2). Ann. Rheum. Dis. 2013, 72, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Iaccarino, L.; Doria, A.; Govoni, M.; Sulli, A.; Marcassa, C. Efficacy of the switch to modified-release prednisone in rheumatoid arthritis patients treated with standard glucocorticoids. Clin. Exp. Rheumatol. 2013, 31, 498–505. [Google Scholar]
- Pfeiffer, B.M.; Krenzer, S.; Dockhorn, R.; Schwenke, R.; Schwenke, H.; Waehrisch, J.; Kraus, E. Impact of modified-release prednisone on functional ability in patients with rheumatoid arthritis. Rheumatol. Int. 2013, 33, 1447–1454. [Google Scholar] [CrossRef]
- Boers, M.; Buttgereit, F. A simple model that suggests possible cost savings when modified-release prednisone 5 mg/day is added to current treatment in patients with active rheumatoid arthritis. Rheumatology 2013, 52, 1435–1437. [Google Scholar] [CrossRef] [Green Version]
- Cannella, A.C.; O’Dell, J.R. Traditional DMARDs: Methotrexate, leflunomide, sulfasalazine, hydroxychloroquine, and combination therapies. In Firestein & Kelley’s Textbook of Rheumatology, 11th ed.; Firestein, G.S., Budd, R.C., Gabriel, S.E., McInnes, I.B., O’Dell, J.R., Koretzky, G., Eds.; Elsevier: Philadelphia, PA, USA, 2020; pp. 1007–1030. [Google Scholar]
- To, H.; Yoshimatsu, H.; Tomonari, M.; Ida, H.; Tsurumoto, T.; Tsuji, Y.; Sonemoto, E.; Shimasaki, N.; Koyanagi, S.; Sasaki, H.; et al. Methotrexate chronotherapy is effective against rheumatoid arthritis. Chronobiol. Int. 2011, 28, 267–274. [Google Scholar] [CrossRef]
- Kanasaki, Y.; Tomonari, M.; Sasaki, H.; To, H. Chronopharmacology of mizoribine in collagen-induced arthritis rats. J. Pharmacol. Sci. 2012, 120, 112–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obayashi, K.; Tomonari, M.; Yoshimatsu, H.; Fukuyama, R.; Ieiri, I.; Higuchi, S.; To, H. Dosing time-dependency of the arthritis-inhibiting effect of tacrolimus in mice. J. Pharmacol. Sci. 2011, 116, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Yaekura, A.; Yoshida, K.; Morii, K.; Oketani, Y.; Okumura, I.; Kaneshiro, K.; Shibanuma, N.; Sakai, Y.; Hashiramoto, A. Chronotherapy targeting cytokine secretion attenuates collagen-induced arthritis in mice. Int. Immunopharmacol. 2020, 84, 106549. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yan, X.; Wang, F.; Ge, F.; Li, Z. Role of methotrexate chronotherapy in collagen-induced rheumatoid arthritis in rats. Z. Rheumatol. 2018, 77, 249–255. [Google Scholar] [CrossRef] [Green Version]
- To, H.; Irie, S.; Tomonari, M.; Watanabe, Y.; Kitahara, T.; Sasaki, H. Therapeutic index of methotrexate depends on circadian cycling of tumour necrosis factor-alpha in collagen-induced arthritic rats and mice. J. Pharm. Pharmacol. 2009, 61, 1333–1338. [Google Scholar] [CrossRef]
- Parnell, A.A.; De Nobrega, A.K.; Lyons, L.C. Translating around the clock: Multi-level regulation of post-transcriptional processes by the circadian clock. Cell Signal 2021, 80, 109904. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kikyo, N. Circadian Regulation of Macrophages and Osteoclasts in Rheumatoid Arthritis. Int. J. Mol. Sci. 2023, 24, 12307. https://doi.org/10.3390/ijms241512307
Kikyo N. Circadian Regulation of Macrophages and Osteoclasts in Rheumatoid Arthritis. International Journal of Molecular Sciences. 2023; 24(15):12307. https://doi.org/10.3390/ijms241512307
Chicago/Turabian StyleKikyo, Nobuaki. 2023. "Circadian Regulation of Macrophages and Osteoclasts in Rheumatoid Arthritis" International Journal of Molecular Sciences 24, no. 15: 12307. https://doi.org/10.3390/ijms241512307