
Transcriptomic Responses of Salvia hispanica to the Infestation of Red Spider Mites (Tetranychus neocaledonicus)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Morphological Responses of Leaves to Insect Herbivory
2.2. Summary of RNA-Seq
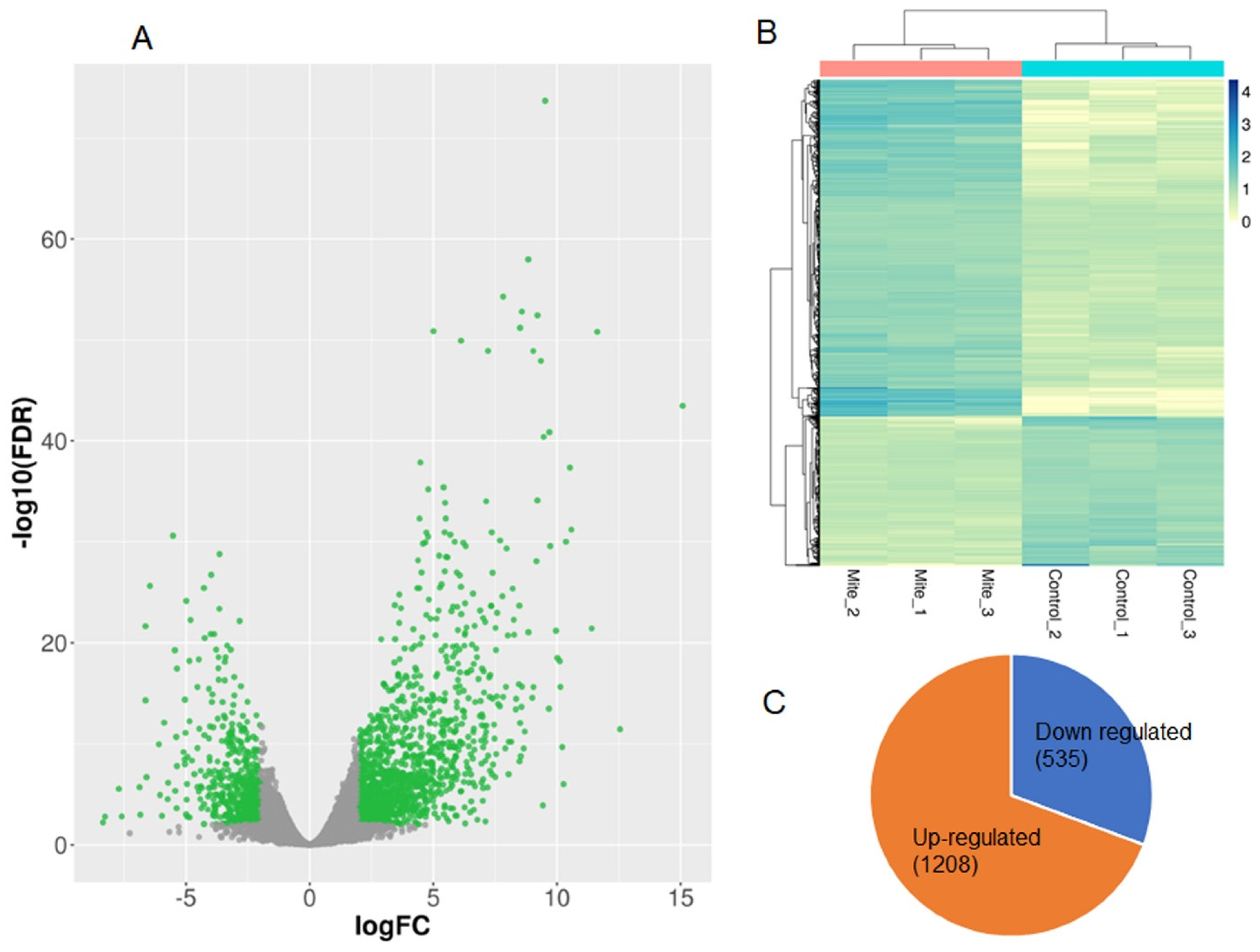
2.3. Identification of Differential Expression Transcripts
2.4. Validation of DEGs Using qRT-PCR
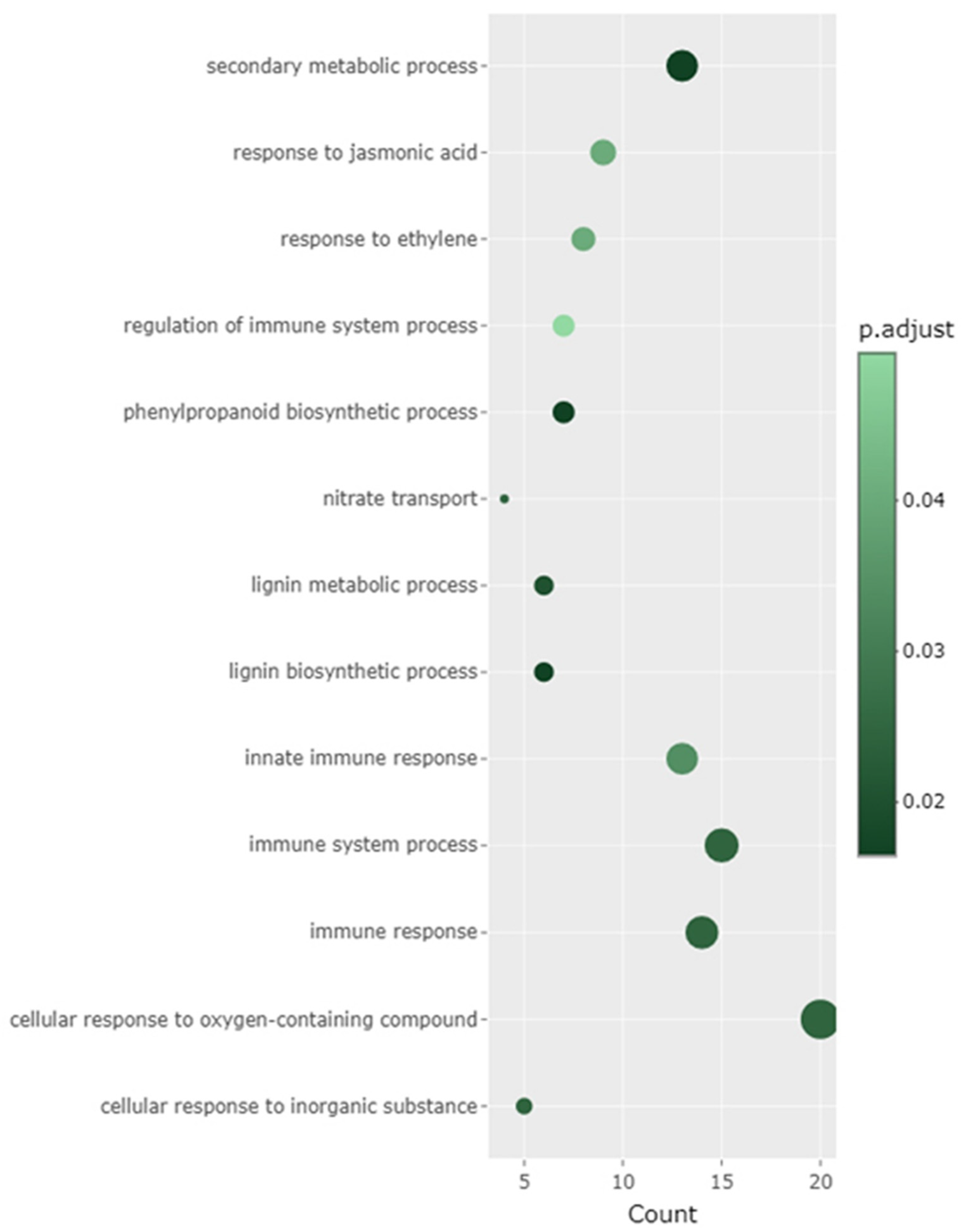
2.5. Overall Enriched Gene Ontology (GO) Based on All DEGs
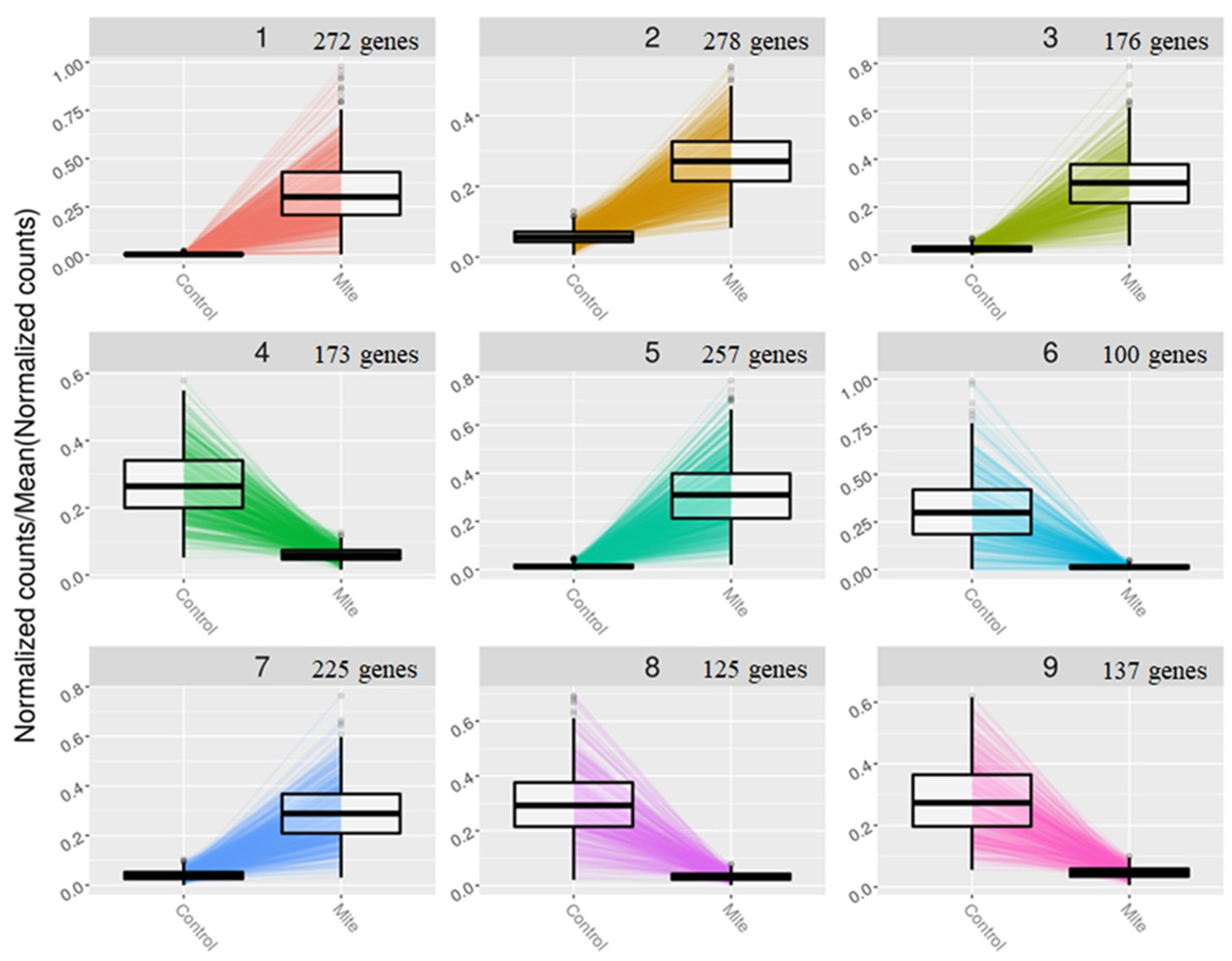
2.6. Construction of Gene Clusters Showing Associated Expression Patterns
2.7. Transcription Factors in Spider Mite Response
3. Discussion
3.1. DEGs in Control and Mite-Infested Samples
3.2. Pathways Involved in the Response to the Infestation of Tetranychus neocaledonicus
3.3. TFs Involved in the Response to the Infestation of Tetranychus neocaledonicus
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction, Library Preparation, and Sequencing
4.3. Bioinformatic Analyses of RNA-Seq Data
4.4. Validation of RNA-Seq Data Using qRT-PCR
4.5. Functional Annotation of DEGs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coates, W.; Ayerza, R. Production potential of chia in northwestern Argentina. Ind. Crops Prod. 1996, 5, 229–233. [Google Scholar] [CrossRef]
- Ayerza, R.; Coates, W. An ω-3 fatty acid enriched chia diet: Influence on egg fatty acid composition, cholesterol and oil content. Can. J. Anim. Sci. 1999, 79, 53–58. [Google Scholar] [CrossRef]
- Wang, L.; Lee, M.; Sun, F.; Song, Z.; Yang, Z.; Yue, G.H. A chromosome-level genome assembly of chia provides insights into high omega-3 content and coat color variation of its seeds. Plant Commun. 2022, 3, 100326. [Google Scholar] [CrossRef] [PubMed]
- Yue, G.H.; Lai, C.C.; Lee, M.; Wang, L.; Song, Z.J. Developing first microsatellites and analysing genetic diversity in six chia (Salvia hispanica L.) cultivars. Genet. Resour. Crop Evol. 2022, 69, 1303–1312. [Google Scholar] [CrossRef]
- Jamshidi, A.; Amato, M.; Ahmadi, A.; Bochicchio, R.; Rossi, R. Chia (Salvia hispanica L.) as a novel forage and feed source: A review. Ital. J. Agron. 2019, 14, 1297. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Bains, K. Chia (Salvia hispanica L.)—A rediscovered ancient grain, from Aztecs to food laboratories. Nutr. Food Sci. 2019, 50, 463–479. [Google Scholar] [CrossRef]
- Peláez, P.; Orona-Tamayo, D.; Montes-Hernández, S.; Valverde, M.E.; Paredes-López, O.; Cibrián-Jaramillo, A. Comparative transcriptome analysis of cultivated and wild seeds of Salvia hispanica (chia). Sci. Rep. 2019, 9, 9761. [Google Scholar] [CrossRef] [Green Version]
- Rachana, R.R.; Manjunatha, M.J.; Devi, S.G.; Naik, M. Seasonal incidence of red spider mite Tetranychus neocaledonicus Andre and its natural enemies. Karnataka J. Agric. Sci. 2009, 22, 213–214. [Google Scholar]
- Oliveira Briozo, M.E.; Silva, J.F.; Barros Ferraz, J.C.; Ramalho Silva, P.R.; Da Silva Melo, J.W.; De França, S.M. Biology and life table of Tetranychus neocaledonicus André (1933) (Acari: Tetranychidae) in different hosts. Syst. Appl. Acarol. 2023, 28, 497–507. [Google Scholar]
- Kant, M.R.; Ament, K.; Sabelis, M.W.; Haring, M.A.; Schuurink, R.C. Differential timing of spider mite-induced direct and indirect defenses in tomato plants. Plant Physiol. 2004, 135, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, A.; Raje, R.; Datta, S.; Sultana, R.; Ontagodi, T. Conventional and molecular approaches towards genetic improvement in pigeonpea for insects resistance. Am. J. Plant Sci. 2013, 4, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Ashra, H.; Nair, S. Review: Trait plasticity during plant-insect interactions: From molecular mechanisms to impact on community dynamics. Plant Sci. 2022, 317, 111188. [Google Scholar] [CrossRef] [PubMed]
- Ayerza, R.; Coates, W. Chia: Rediscovering a Forgotten Crop of the Aztecs; University of Arizona Press: Tucson, AZ, USA, 2005. [Google Scholar]
- Kautz, S.; Trisel, J.A.; Ballhorn, D.J. Jasmonic acid enhances plant cyanogenesis and resistance to herbivory in lima bean. J. Chem. Ecol. 2014, 40, 1186–1196. [Google Scholar] [CrossRef] [PubMed]
- Malka, S.K.; Cheng, Y. Possible interactions between the biosynthetic pathways of indole glucosinolate and auxin. Front. Plant Sci. 2017, 8, 2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Jin, J.; Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids Res. 2019, 48, D1104–D1113. [Google Scholar] [CrossRef]
- Sun, A.; Yu, B.; Zhang, Q.; Peng, Y.; Yang, J.; Sun, Y.; Qin, P.; Jia, T.; Smeekens, S.; Teng, S. MYC2-activated trichome birefringence-like37 acetylates cell walls and enhances herbivore resistance. Plant Physiol. 2020, 184, 1083–1096. [Google Scholar] [CrossRef]
- Yuan, Y.; Teng, Q.; Zhong, R.; Ye, Z.-H. TBL3 and TBL31, two Arabidopsis duf231 domain proteins, are required for 3- o -monoacetylation of xylan. Plant Cell Physiol. 2015, 57, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Zhang, C.-X.; Chen, R.; He, S.Y. Challenging battles of plants with phloem-feeding insects and prokaryotic pathogens. Proc. Natl. Acad. Sci. USA 2019, 116, 23390–23397. [Google Scholar] [CrossRef]
- Molina, A.; Miedes, E.; Bacete, L.; Rodríguez, T.; Mélida, H.; Denancé, N.; Sánchez-Vallet, A.; Rivière, M.-P.; López, G.; Freydier, A.; et al. Arabidopsis cell wall composition determines disease resistance specificity and fitness. Proc. Natl. Acad. Sci. USA 2021, 118, e2010243118. [Google Scholar] [CrossRef]
- Züst, T.; Agrawal, A.A. Trade-Offs Between Plant Growth and Defense Against Insect Herbivory: An Emerging Mechanistic Synthesis. Annu. Rev. Plant Biol. 2017, 68, 513–534. [Google Scholar] [CrossRef] [Green Version]
- Arena, G.D.; Ramos-González, P.L.; Rogerio, L.A.; Ribeiro-Alves, M.; Casteel, C.L.; Freitas-Astúa, J.; Machado, M.A. Making a better home: Modulation of plant defensive response by brevipalpus mites. Front. Plant Sci. 2018, 9, 1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Duan, G.; Li, C.; Liu, L.; Han, G.; Zhang, Y.; Wang, C. The crosstalks between jasmonic acid and other plant hormone signaling highlight the involvement of jasmonic acid as a core component in plant response to biotic and abiotic stresses. Front. Plant Sci. 2019, 10, 1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raza, A.; Charagh, S.; Zahid, Z.; Mubarik, M.S.; Javed, R.; Siddiqui, M.H.; Hasanuzzaman, M. Jasmonic acid: A key frontier in conferring abiotic stress tolerance in plants. Plant Cell Rep. 2021, 40, 1513–1541. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Major, I.T.; Howe, G.A. Resolution of growth–defense conflict: Mechanistic insights from jasmonate signaling. Curr. Opin. Plant Biol. 2018, 44, 72–81. [Google Scholar] [CrossRef]
- Sözen, C.; Schenk, S.T.; Boudsocq, M.; Chardin, C.; Almeida-Trapp, M.; Krapp, A.; Hirt, H.; Mithöfer, A.; Colcombet, J. Wounding and insect feeding trigger two independent mapk pathways with distinct regulation and kinetics. Plant Cell 2020, 32, 1988–2003. [Google Scholar] [CrossRef] [Green Version]
- Mostafa, S.; Wang, Y.; Zeng, W.; Jin, B. Plant responses to herbivory, wounding, and infection. Int. J. Mol. Sci. 2022, 23, 7031. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, F.; Chen, L.; Pan, Y.; Sun, L.; Bao, N.; Zhang, T.; Cui, C.-X.; Qiu, Z.; Zhang, Y.; et al. Jasmonate-mediated wound signalling promotes plant regeneration. Nat. Plants 2019, 5, 491–497. [Google Scholar] [CrossRef]
- Browse, J.; Wager, A. Social network: JAZ protein interactions expand our knowledge of Jasmonate signaling. Front. Plant Sci. 2012, 3, 41. [Google Scholar]
- Ruan, J.; Zhou, Y.; Zhou, M.; Yan, J.; Khurshid, M.; Weng, W.; Cheng, J.; Zhang, K. Jasmonic acid signaling pathway in plants. Int. J. Mol. Sci. 2019, 20, 2479. [Google Scholar] [CrossRef] [Green Version]
- Melotto, M.; Mecey, C.; Niu, Y.; Chung, H.S.; Katsir, L.; Yao, J.; Zeng, W.; Thines, B.; Staswick, P.; Browse, J.; et al. A critical role of two positively charged amino acids in the Jas motif of Arabidopsis JAZ proteins in mediating coronatine- and jasmonoyl isoleucine-dependent interactions with the COI1 F-box protein. Plant J. 2008, 55, 979–988. [Google Scholar] [CrossRef] [Green Version]
- Thines, B.; Katsir, L.; Melotto, M.; Niu, Y.; Mandaokar, A.; Liu, G.; Nomura, K.; He, S.Y.; Howe, G.A.; Browse, J. JAZ repressor proteins are targets of the SCFCOI1 complex during jasmonate signalling. Nature 2007, 448, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Villarroel, C.A.; Jonckheere, W.; Alba, J.M.; Glas, J.J.; Dermauw, W.; Haring, M.A.; Van Leeuwen, T.; Schuurink, R.C.; Kant, M.R. Salivary proteins of spider mites suppress defenses in Nicotiana benthamiana and promote mite reproduction. Plant J. 2016, 86, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, F.; Melotto, M.; Yao, J.; He, S.Y. Jasmonate signaling and manipulation by pathogens and insects. J. Exp. Bot. 2017, 68, 1371–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Wang, D.-D.; Fang, X.; Chen, X.-Y.; Mao, Y.-B. Plant specialized metabolism regulated by Jasmonate signaling. Plant Cell Physiol. 2019, 60, 2638–2647. [Google Scholar] [CrossRef] [PubMed]
- Divekar, P.A.; Narayana, S.; Divekar, B.A.; Kumar, R.; Gadratagi, B.G.; Ray, A.; Singh, A.K.; Rani, V.; Singh, V.; Singh, A.K.; et al. Plant secondary metabolites as defense tools against herbivores for sustainable crop protection. Int. J. Mol. Sci. 2022, 23, 2690. [Google Scholar] [CrossRef]
- Savatin, D.V.; Gramegna, G.; Modesti, V.; Cervone, F. Wounding in the plant tissue: The defense of a dangerous passage. Front. Plant Sci. 2014, 5, 470. [Google Scholar] [CrossRef] [Green Version]
- Zagrobelny, M.; Bak, S.; Rasmussen, A.V.; Jørgensen, B.; Naumann, C.M.; Møller, B.L. Cyanogenic glucosides and plant–insect interactions. Phytochemistry 2004, 65, 293–306. [Google Scholar] [CrossRef]
- Gossner, M.M.; Beenken, L.; Arend, K.; Begerow, D.; Peršoh, D. Insect herbivory facilitates the establishment of an invasive plant pathogen. ISME Commun. 2021, 1, 6. [Google Scholar] [CrossRef]
- Singh, K.B.; Foley, R.C.; Oñate-Sánchez, L. Transcription factors in plant defense and stress responses. Curr. Opin. Plant Biol. 2002, 5, 430–436. [Google Scholar] [CrossRef]
- Agarwal, P.; Reddy, M.P.; Chikara, J. WRKY: Its structure, evolutionary relationship, DNA-binding selectivity, role in stress tolerance and development of plants. Mol. Biol. Rep. 2011, 38, 3883–3896. [Google Scholar] [CrossRef]
- Jiang, J.; Ma, S.; Ye, N.; Jiang, M.; Cao, J.; Zhang, J. WRKY transcription factors in plant responses to stresses. J. Integr. Plant Biol. 2017, 59, 86–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eulgem, T.; Somssich, I.E. Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 2007, 10, 366–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Chen, C.; Fan, B.; Chen, Z. Physical and functional interactions between pathogen-induced Arabidopsis WRKY18, WRKY40, and WRKY60 transcription factors. Plant Cell 2006, 18, 1310–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloth, K.J.; Wiegers, G.L.; Busscher-Lange, J.; van Haarst, J.C.; Kruijer, W.; Bouwmeester, H.J.; Dicke, M.; Jongsma, M.A. AtWRKY22 promotes susceptibility to aphids and modulates salicylic acid and jasmonic acid signalling. J. Exp. Bot. 2016, 67, 3383–3396. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Osbourn, A.; Ma, P. MYB transcription factors as regulators of phenylpropanoid metabolism in plants. Mol. Plant 2015, 8, 689–708. [Google Scholar] [CrossRef] [Green Version]
- Seo, M.-S.; Kim, J.S. Understanding of MYB transcription factors involved in glucosinolate biosynthesis in Brassicaceae. Molecules 2017, 22, 1549. [Google Scholar] [CrossRef] [Green Version]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Nuruzzaman, M.; Sharoni, A.M.; Kikuchi, S. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol. 2013, 4, 248. [Google Scholar] [CrossRef] [Green Version]
- Meraj, T.A.; Fu, J.; Raza, M.A.; Zhu, C.; Shen, Q.; Xu, D.; Wang, Q. Transcriptional factors regulate plant stress responses through mediating secondary metabolism. Genes 2020, 11, 346. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.Y.; Jang, Y.J.; Park, O.K. AP2/ERF Family Transcription Factors ORA59 and RAP2.3 Interact in the Nucleus and Function Together in Ethylene Responses. Front. Plant Sci. 2018, 9, 1675. [Google Scholar] [CrossRef] [Green Version]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2014, 31, 166–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chia, J.M.; Bartfai, R.; Christoffels, A.; Yue, G.H.; Ding, K.; Ho, M.Y.; Hill, J.A.; Stupka, E.; Orban, L. Comparative analysis of the testis and ovary transcriptomes in zebrafish by combining experimental and computational tools. Comp. Funct. Genom. 2004, 5, 403–418. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Lee, M.; Ye, B.; Yue, G.H. Genes, pathways and networks responding to drought stress in oil palm roots. Sci. Rep. 2020, 10, 21303. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.; Tanaseichuk, O.; Benner, C. Chanda, 663 SK Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [Green Version]
- Cassan, O.; Lèbre, S.; Martin, A. Inferring and analyzing gene regulatory networks from multi-factorial expression data: A complete and interactive suite. BMC Genom. 2021, 22, 387. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.; Wang, L.; Yue, G.H. Transcriptomic Responses of Salvia hispanica to the Infestation of Red Spider Mites (Tetranychus neocaledonicus). Int. J. Mol. Sci. 2023, 24, 12261. https://doi.org/10.3390/ijms241512261
Lee M, Wang L, Yue GH. Transcriptomic Responses of Salvia hispanica to the Infestation of Red Spider Mites (Tetranychus neocaledonicus). International Journal of Molecular Sciences. 2023; 24(15):12261. https://doi.org/10.3390/ijms241512261
Chicago/Turabian StyleLee, May, Le Wang, and Gen Hua Yue. 2023. "Transcriptomic Responses of Salvia hispanica to the Infestation of Red Spider Mites (Tetranychus neocaledonicus)" International Journal of Molecular Sciences 24, no. 15: 12261. https://doi.org/10.3390/ijms241512261