
Dysregulated Signalling Pathways Driving Anticancer Drug Resistance
Abstract
: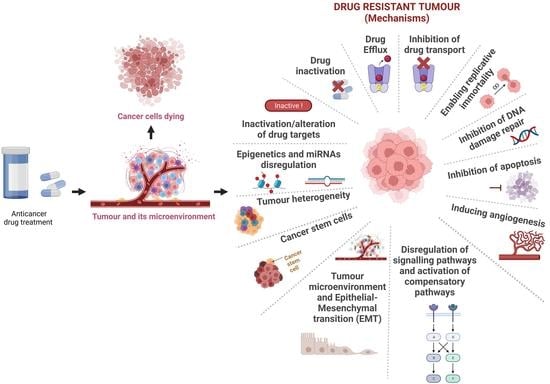
1. Introduction
2. Evolution of Cancer
3. Drug Resistance in Cancer
3.1. Intrinsic Drug Resistance
3.2. Acquired Drug Resistance
3.3. Altered Drug Targets
3.4. Modified Drug Metabolism
3.5. Enhanced Drug Efflux
3.6. Repair of DNA Damage
3.7. Epigenetics Modifications
3.8. Slow Growing Cells
3.9. Undruggable Targets
3.10. Tumour Microenvironment
3.11. Epithelial-Mesenchymal Transition
3.12. Multidrug Resistance
4. Altered Signalling Pathways Involved in Drug Resistance to Cancer
4.1. Wnt/β-Catenin Pathway
4.2. The JAK/STAT Signalling Pathway
4.3. PI3K/Akt/mTOR Pathway
4.4. MAPK Pathway
5. Strategies to Overcome Drug Resistance in Cancer
5.1. Circulating Tumour DNA
5.2. Immunotherapy
5.3. Nanotechnology
5.4. Gene Editing
5.5. Computational Strategies
5.6. miRNAs
5.7. Targeting Signalling Pathways
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| ABCC5 | ATP binding cassette subfamily C member 5 |
| aCD47 | CD47 antibody |
| AhR | Aryl hydrocarbon receptor |
| ALK | Anaplastic lymphoma kinase |
| APEX1 | Apurinic/apyrimidinic endodeoxyribonuclease 1 |
| ASIC1a | Acid sensing ion channel 1a |
| AST | Adeno-to-squamous transdifferentiation |
| ATR | Ataxia telangiectasia and Rad3-related |
| ATP | Adenosine triphosphate |
| BAK1 | BCL2 antagonist/killer1 |
| BBB | Blood-brain barrier |
| BCRP/ABCG2 | Breast cancer resistance protein |
| BLP | Bionic lipoprotein |
| BRCA1/2 | Breast cancer gene1/2 |
| CAFs | Cancer-associated fibroblasts |
| CAR | Chimeric antigen receptor |
| CDKs | Cyclin-dependent kinases |
| cfDNA | Cell-free DNA |
| CHD4 | Chromodomain helicase DNA-binding protein 4 |
| CHK1 | Checkpoint kinase 1 |
| CML | Chronic myeloid leukaemia |
| CML-BP | Blastic phase of chronic myeloid leukaemia |
| CNS | Central nervous system |
| CQ | Chloroquine |
| CRC | Colorectal cancer |
| CSCs | Cancer stem cells |
| ctDNA | Circulating tumour DNA |
| CTLA4 | Cytotoxic T-lymphocyte-associated protein 4 |
| CYP | Cytochrome P450 |
| CYP3A4 | CYP isoforms 3A4 |
| DEHP | Di(2-ethylhexyl) phthalate |
| DNMT1/UHRF1 | DNA methyltransferases1/ Ubiquitin-like containing PHD Ring Finger 1 |
| Dox | Doxorubicin |
| Dox-Mel PL | Doxorubicin-melittin polymersome |
| DTEPs | Drug-tolerant expanded persisters |
| DTPs | Drug-tolerant persisters |
| ECM | Extracellular matrix |
| EGFL7 | EGF such as domain multiple 7 |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| Egr-1 | Early growth response-1 |
| EMT | Epithelial-mesenchymal transition |
| EOC | Epithelial ovarian cancer |
| ER+ | Oestrogen receptor positive |
| 5-FU | 5-fluorouracil |
| FAM46A | Family with sequence similarity 46, member A |
| FGFR4 | Fibroblast growth factor receptor 4 |
| FGFR1 | Fibroblast growth factor receptor 1 |
| FLT3 | FMS-related tyrosine kinase 3 receptor |
| FUT6 | Fucosyltransferase VI |
| GDF-15 | Growth differentiation factor-15 |
| GPER | G-protein coupled oestrogen receptor |
| GR/OR | Gefitinib/osimertinib-resistant |
| GST | Glutathione S-transferase |
| H3K27ac | Histone H3 lysine 27 trimethylation acetylation |
| H3K27me3 | Histone H3 lysine 27 trimethylation |
| HCC | Hepatocellular carcinoma |
| HGF | Hepatocyte growth factor |
| HER2/erbB2 | Human epidermal growth factor receptor 2 |
| HER2+ | Human epidermal growth factor receptor 2 positive |
| HKDC1 | Hexokinase domain containing protein-1 |
| iANG-1 | Intracellular angiopoietin-1 |
| IFN | Interferons |
| IFN-γ | Interferon-gamma |
| IGFR | Insulin-like growth factor receptor |
| IGF-1R | Insulin-like growth factor 1 receptor |
| ISR | Integrated stress response |
| JAK/STAT | Janus kinase/signal transducers and activators of transcription |
| KDM5A | Lysine demethylase 5A |
| lncRNA | Long non-coding RNAs |
| lncRNA UCA1 | LncRNA urothelial carcinoma-associated 1 |
| LSD1/KDM1A | Lysine demethylase 1A |
| m6A | N6-methyladenosine |
| MAPK/ERK | Mitogen-activated protein kinase/extracellular signal-regulated kinase |
| MAPK1 | Mitogen-activated protein kinase 1 |
| MARM2.0 | Second-generation MAPK Adaptive Resistance Model |
| MDR | Multiple drug resistant |
| MDSCs | Myeloid-derived suppressor cells |
| Mel | Melittin |
| miRNAs | MicroRNAs |
| MITF | Melanocyte inducing transcription factor |
| MRD | Minimal residual disease |
| MRP1/ABCC1 | Multidrug resistance-associated protein 1 |
| MSCs | Mesenchymal stromal cells |
| MUC17 | Mucin 17 |
| MVP | Major vault protein |
| ncRNAs | Non-coding RNAs |
| NEAT1 | Nuclear paraspeckle assembly transcript 1 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NGFR | Nerve growth factor receptor |
| NGS | Next-generation sequencing |
| NK | Natural killer |
| NLGN3 | Neuroligin-3 |
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| NSCLC | Non-small-cell lung cancer |
| PCR | Polymerase chain reaction |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 |
| PD-1 | Programmed-cell death protein-1 |
| PDGF | Platelet-derived growth factor |
| PDGFR | Platelet-derived growth factor receptor |
| PD-L1 | Programmed death ligand 1 |
| P-gp/MDR1/ABCB1 | P-glycoprotein |
| pHe | Extracellular pH |
| pHi | Intracellular pH |
| PI3K/Akt/mTOR | Phosphoinositol-3-kinase/protein kinase B/mammalian target of rapamycin |
| PTKs | Protein tyrosine kinases |
| REG4 | Regenerating gene 4 |
| ROS | Reactive oxygen species |
| ROS1 | Ros oncogene 1 |
| RTKs | Receptor tyrosine kinases |
| rTMB | Relative tumour mutation burden |
| SNP | Single nucleotide polymorphism |
| SOD2 | Superoxide dismutase 2 |
| SOX2 | SRY-Box transcription factor 2 |
| TAMs | Tumour-associated macrophages |
| Tβ4 | Thymosin 4 |
| TFF3 | Trefoil factor 3 |
| TGF-β | Transforming growth factor-beta |
| TIS | Therapy-induced senescence |
| TKIs | Tyrosine kinase inhibitors |
| TME | Tumour microenvironment |
| TNBC | Triple negative breast cancer |
| Treg | Regulatory T |
| TYMS | Thymidylate synthase |
| UGT | Uridine diphosphoglucuronosyltransferase |
| Wnt | Wingless-related integration site |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| VESPA | Virtual Enrichment-based Signalling Protein-activity Analysis |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal. Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef]
- Kaemmerer, E.; Loessner, D.; Avery, V.M. Addressing the tumour microenvironment in early drug discovery: A strategy to overcome drug resistance and identify novel targets for cancer therapy. Drug Discov. Today 2021, 26, 663–676. [Google Scholar] [CrossRef]
- Bejarano, L.; Jordao, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y.G.; Brozovic, A.; Goncalves, A.C.; Jurkovicova, D.; Line, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updat. 2019, 46, 100645. [Google Scholar] [CrossRef] [PubMed]
- Rafaeva, M.; Erler, J.T. Framing cancer progression: Influence of the organ- and tumour-specific matrisome. FEBS J. 2020, 287, 1454–1477. [Google Scholar] [CrossRef]
- Siddiqui, I.A.; Sanna, V.; Ahmad, N.; Sechi, M.; Mukhtar, H. Resveratrol nanoformulation for cancer prevention and therapy. Ann. N. Y. Acad. Sci. 2015, 1348, 20–31. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Cho, Y.; Kim, Y.K. Cancer Stem Cells as a Potential Target to Overcome Multidrug Resistance. Front. Oncol. 2020, 10, 764. [Google Scholar] [CrossRef]
- Liu, Y.; Hock, J.M.; Van Beneden, R.J.; Li, X. Aberrant overexpression of FOXM1 transcription factor plays a critical role in lung carcinogenesis induced by low doses of arsenic. Mol. Carcinog. 2014, 53, 380–391. [Google Scholar] [CrossRef]
- Hou, Y.; Zhu, Q.; Li, Z.; Peng, Y.; Yu, X.; Yuan, B.; Liu, Y.; Liu, Y.; Yin, L.; Peng, Y.; et al. The FOXM1-ABCC5 axis contributes to paclitaxel resistance in nasopharyngeal carcinoma cells. Cell Death Dis. 2017, 8, e2659. [Google Scholar] [CrossRef]
- Modi, A.; Purohit, P.; Roy, D.; Vishnoi, J.R.; Pareek, P.; Elhence, P.; Singh, P.; Sharma, S.; Sharma, P.; Misra, S. FOXM1 mediates GDF-15 dependent stemness and intrinsic drug resistance in breast cancer. Mol. Biol. Rep. 2022, 49, 2877–2888. [Google Scholar] [CrossRef]
- Bergamino, M.A.; Lopez-Knowles, E.; Morani, G.; Tovey, H.; Kilburn, L.; Schuster, E.F.; Alataki, A.; Hills, M.; Xiao, H.; Holcombe, C.; et al. HER2-enriched subtype and novel molecular subgroups drive aromatase inhibitor resistance and an increased risk of relapse in early ER+/HER2+ breast cancer. eBioMedicine 2022, 83, 104205. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Esteva, F.J.; Valero, V.; Booser, D.; Guerra, L.T.; Murray, J.L.; Pusztai, L.; Cristofanilli, M.; Arun, B.; Esmaeli, B.; Fritsche, H.A.; et al. Phase II study of weekly docetaxel and trastuzumab for patients with HER-2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Joshi, J.P.; Brown, N.E.; Griner, S.E.; Nahta, R. Growth differentiation factor 15 (GDF15)-mediated HER2 phosphorylation reduces trastuzumab sensitivity of HER2-overexpressing breast cancer cells. Biochem. Pharmacol. 2011, 82, 1090–1099. [Google Scholar] [CrossRef] [PubMed]
- Pernas, S.; Tolaney, S.M. HER2-positive breast cancer: New therapeutic frontiers and overcoming resistance. Ther. Adv. Med. Oncol. 2019, 11, 1758835919833519. [Google Scholar] [CrossRef]
- Huang, D.; Duan, H.; Huang, H.; Tong, X.; Han, Y.; Ru, G.; Qu, L.; Shou, C.; Zhao, Z. Cisplatin resistance in gastric cancer cells is associated with HER2 upregulation-induced epithelial-mesenchymal transition. Sci. Rep. 2016, 6, 20502. [Google Scholar] [CrossRef]
- Witta, S.E.; Gemmill, R.M.; Hirsch, F.R.; Coldren, C.D.; Hedman, K.; Ravdel, L.; Helfrich, B.; Dziadziuszko, R.; Chan, D.C.; Sugita, M.; et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006, 66, 944–950. [Google Scholar] [CrossRef]
- Sayan, A.E.; Griffiths, T.R.; Pal, R.; Browne, G.J.; Ruddick, A.; Yagci, T.; Edwards, R.; Mayer, N.J.; Qazi, H.; Goyal, S.; et al. SIP1 protein protects cells from DNA damage-induced apoptosis and has independent prognostic value in bladder cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 14884–14889. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Milman, N.; Ginini, L.; Gil, Z. Exosomes and their role in tumorigenesis and anticancer drug resistance. Drug Resist. Updat. 2019, 45, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Bernards, R. Feedback and redundancy in receptor tyrosine kinase signaling: Relevance to cancer therapies. Trends Biochem. Sci. 2014, 39, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef]
- Challagundla, K.B.; Wise, P.M.; Neviani, P.; Chava, H.; Murtadha, M.; Xu, T.; Kennedy, R.; Ivan, C.; Zhang, X.; Vannini, I.; et al. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J. Natl. Cancer Inst. 2015, 107, djv135. [Google Scholar] [CrossRef]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Chen, R.; Tian, X. Autocrine EGF and TGF-α promote primary and acquired resistance to ALK/c-Met kinase inhibitors in non-small-cell lung cancer. Pharmacol. Res. Perspect. 2023, 11, e01047. [Google Scholar] [CrossRef]
- Fu, R.; Jiang, S.; Li, J.; Chen, H.; Zhang, X. Activation of the HGF/c-MET axis promotes lenvatinib resistance in hepatocellular carcinoma cells with high c-MET expression. Med. Oncol. 2020, 37, 24. [Google Scholar] [CrossRef]
- Shi, Q.; Xuhong, J.; Luo, T.; Ge, J.; Liu, F.; Lan, Y.; Chen, Q.; Tang, P.; Fan, L.; Chen, L.; et al. PIK3CA mutations are associated with pathologic complete response rate to neoadjuvant pyrotinib and trastuzumab plus chemotherapy for HER2-positive breast cancer. Br. J. Cancer 2023, 128, 121–129. [Google Scholar] [CrossRef]
- Wu, Q.; Ma, J.; Wei, J.; Meng, W.; Wang, Y.; Shi, M. FOXD1-AS1 regulates FOXD1 translation and promotes gastric cancer progression and chemoresistance by activating the PI3K/AKT/mTOR pathway. Mol. Oncol. 2021, 15, 299–316. [Google Scholar] [CrossRef]
- Sun, R.; Meng, Y.; Xu, R.; Li, Y.; Xu, X.; Li, Z.; Zuo, D. Construction of crizotinib resistant models with CD74-ROS1 D2033N and CD74-ROS1 S1986F point mutations to explore resistance mechanism and treatment strategy. Cell Signal 2023, 101, 110497. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Vergara-Gomez, L.; Bizama, C.; Zhong, J.; Buchegger, K.; Suarez, F.; Rosa, L.; Ili, C.; Weber, H.; Obreque, J.; Espinoza, K.; et al. A Novel Gemcitabine-Resistant Gallbladder Cancer Model Provides Insights into Molecular Changes Occurring during Acquired Resistance. Int. J. Mol. Sci. 2023, 24, 7238. [Google Scholar] [CrossRef]
- Topham, J.T.; O’Callaghan, C.J.; Feilotter, H.; Kennecke, H.F.; Lee, Y.S.; Li, W.; Banks, K.C.; Quinn, K.; Renouf, D.J.; Jonker, D.J.; et al. Circulating Tumor DNA Identifies Diverse Landscape of Acquired Resistance to Anti-Epidermal Growth Factor Receptor Therapy in Metastatic Colorectal Cancer. J. Clin. Oncol. 2023, 41, 485–496. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019, 25, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, P.; Gao, C.; Chen, J.; Li, J.; Chen, Z.; Xu, M.; Shao, J.; Zhang, Y.; Xie, J. Down-regulation of LRP1B in colon cancer promoted the growth and migration of cancer cells. Exp. Cell Res. 2017, 357, 1–8. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, P.; Zhao, M.; Cao, H.; Zhao, Y.; Ji, M.; Hou, P.; Chen, M. EGFL7 drives the evolution of resistance to EGFR inhibitors in lung cancer by activating NOTCH signaling. Cell Death Dis. 2022, 13, 910. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Fan, J.; Qin, K.; Zhou, Y.; Yang, J.; Ma, Y.; Shi, M.; Jin, J. LncRNA SNHG7 Regulates Mesenchymal Stem Cell through the Notch1/Jagged1/Hes-1 Signaling Pathway and Influences Folfirinox Resistance in Pancreatic Cancer. Front. Oncol. 2021, 11, 719855. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.H.; Tsai, C.F.; Hsu, C.Y.; Kuo, P.L.; Lee, J.N.; Chai, C.Y.; Hou, M.F.; Chang, C.C.; Long, C.Y.; Ko, Y.C.; et al. Phthalates stimulate the epithelial to mesenchymal transition through an HDAC6-dependent mechanism in human breast epithelial stem cells. Toxicol. Sci. 2012, 128, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.H.; Hsu, C.Y.; Yang, P.J.; Chiu, C.C.; Liang, S.S.; Ou-Yang, F.; Kan, J.Y.; Hou, M.F.; Wang, T.N.; Tsai, E.M. DEHP mediates drug resistance by directly targeting AhR in human breast cancer. Biomed. Pharmacother. 2022, 145, 112400. [Google Scholar] [CrossRef] [PubMed]
- Doane, A.S.; Danso, M.; Lal, P.; Donaton, M.; Zhang, L.; Hudis, C.; Gerald, W.L. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene 2006, 25, 3994–4008. [Google Scholar] [CrossRef]
- Gao, C.; Zhang, L.; Xu, Y.; Ma, X.; Chen, P.; Chen, Z.S.; Wei, L. I13 overrides resistance mediated by the T315I mutation in chronic myeloid leukemia by direct BCR-ABL inhibition. Front. Pharmacol. 2023, 14, 1183052. [Google Scholar] [CrossRef]
- El-Damasy, A.K.; Jin, H.; Park, J.W.; Kim, H.J.; Khojah, H.; Seo, S.H.; Lee, J.H.; Bang, E.K.; Keum, G. Overcoming the imatinib-resistant BCR-ABL mutants with new ureidobenzothiazole chemotypes endowed with potent and broad-spectrum anticancer activity. J. Enzym. Inhib. Med. Chem. 2023, 38, 2189097. [Google Scholar] [CrossRef]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef]
- Soverini, S.; De Benedittis, C.; Papayannidis, C.; Paolini, S.; Venturi, C.; Iacobucci, I.; Luppi, M.; Bresciani, P.; Salvucci, M.; Russo, D.; et al. Drug resistance and BCR-ABL kinase domain mutations in Philadelphia chromosome-positive acute lymphoblastic leukemia from the imatinib to the second-generation tyrosine kinase inhibitor era: The main changes are in the type of mutations, but not in the frequency of mutation involvement. Cancer 2014, 120, 1002–1009. [Google Scholar] [CrossRef]
- Steelman, L.S.; Pohnert, S.C.; Shelton, J.G.; Franklin, R.A.; Bertrand, F.E.; McCubrey, J.A. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 2004, 18, 189–218. [Google Scholar] [CrossRef]
- Warsch, W.; Grundschober, E.; Sexl, V. Adding a new facet to STAT5 in CML: Multitasking for leukemic cells. Cell Cycle 2013, 12, 1813–1814. [Google Scholar] [CrossRef] [PubMed]
- Chien, S.H.; Liu, H.M.; Chen, P.M.; Ko, P.S.; Lin, J.S.; Chen, Y.J.; Lee, L.H.; Hsiao, L.T.; Chiou, T.J.; Gau, J.P.; et al. The landscape of BCR-ABL mutations in patients with Philadelphia chromosome-positive leukaemias in the era of second-generation tyrosine kinase inhibitors. Hematol. Oncol. 2020, 38, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Hao, S.; Liu, X.; Li, Y.; Zheng, X. Feiyiliu Mixture sensitizes EGFR(Del19/T790M/C797S) mutant non-small cell lung cancer to osimertinib by attenuating the PRC1/Wnt/EGFR pathway. Front. Pharmacol. 2023, 14, 1093017. [Google Scholar] [CrossRef]
- Hatlen, M.A.; Schmidt-Kittler, O.; Sherwin, C.A.; Rozsahegyi, E.; Rubin, N.; Sheets, M.P.; Kim, J.L.; Miduturu, C.; Bifulco, N.; Brooijmans, N.; et al. Acquired On-Target Clinical Resistance Validates FGFR4 as a Driver of Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, S.; Wang, Y.; Zhao, Y.; Li, Q. Protein tyrosine kinase inhibitor resistance in malignant tumors: Molecular mechanisms and future perspective. Signal Transduct. Target. Ther. 2022, 7, 329. [Google Scholar] [CrossRef]
- Michael, M.; Doherty, M.M. Tumoral Drug Metabolism: Overview and Its Implications for Cancer Therapy. J. Clin. Oncol. 2005, 23, 205–229. [Google Scholar] [CrossRef]
- Kawahara, B.; Mascharak, P.K. Inhibition of Cytochrome P450 by Carbon Monoxide: Relevance to Drug Resistance in Human Breast Cancer Therapy. Med. Res. Arch. 2023, 11, 1–12. [Google Scholar] [CrossRef]
- Hofman, J.; Vagiannis, D.; Chen, S.; Guo, L. Roles of CYP3A4, CYP3A5 and CYP2C8 drug-metabolizing enzymes in cellular cytostatic resistance. Chem. Biol. Interact. 2021, 340, 109448. [Google Scholar] [CrossRef]
- Li, M.; Sun, S.; Bian, Z.; Yao, S.; Liu, M.; You, X.; Li, M. SNHG15 promotes chemoresistance and glycolysis in colorectal cancer. Pathol. Res. Pract. 2023, 246, 154480. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef]
- Xiang, L.W.; Xue, H.; Ha, M.W.; Yu, D.Y.; Xiao, L.J.; Zheng, H.C. The effects of REG4 expression on chemoresistance of ovarian cancer. J. Obstet. Gynaecol. 2022, 42, 3149–3157. [Google Scholar] [CrossRef] [PubMed]
- Kathawala, R.J.; Gupta, P.; Ashby, C.R., Jr.; Chen, Z.S. The modulation of ABC transporter-mediated multidrug resistance in cancer: A review of the past decade. Drug Resist. Updat. 2015, 18, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wilkens, S. Structure and mechanism of ABC transporters. F1000Prime Rep. 2015, 7, 14. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Pastan, I.H. The Role of Multidrug Resistance Efflux Pumps in Cancer: Revisiting a JNCI Publication Exploring Expression of the MDR1 (P-glycoprotein) Gene. J. Natl. Cancer Inst. 2015, 107, djv222. [Google Scholar] [CrossRef]
- Duan, H.; Liu, Y.; Gao, Z.; Huang, W. Recent advances in drug delivery systems for targeting cancer stem cells. Acta Pharm. Sin. B 2021, 11, 55–70. [Google Scholar] [CrossRef]
- Wang, J.Q.; Wu, Z.X.; Yang, Y.; Teng, Q.X.; Li, Y.D.; Lei, Z.N.; Jani, K.A.; Kaushal, N.; Chen, Z.S. ATP-binding cassette (ABC) transporters in cancer: A review of recent updates. J. Evid. Based Med. 2021, 14, 232–256. [Google Scholar] [CrossRef]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.H.; Chen, Z.S. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updat. 2016, 27, 14–29. [Google Scholar] [CrossRef]
- Xue, X.; Liang, X.J. Overcoming drug efflux-based multidrug resistance in cancer with nanotechnology. Chin. J. Cancer 2012, 31, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Lampada, A.; O’Prey, J.; Szabadkai, G.; Ryan, K.M.; Hochhauser, D.; Salomoni, P. mTORC1-independent autophagy regulates receptor tyrosine kinase phosphorylation in colorectal cancer cells via an mTORC2-mediated mechanism. Cell Death Differ. 2017, 24, 1045–1062. [Google Scholar] [CrossRef]
- Tazzari, P.L.; Cappellini, A.; Ricci, F.; Evangelisti, C.; Papa, V.; Grafone, T.; Martinelli, G.; Conte, R.; Cocco, L.; McCubrey, J.A.; et al. Multidrug resistance-associated protein 1 expression is under the control of the phosphoinositide 3 kinase/Akt signal transduction network in human acute myelogenous leukemia blasts. Leukemia 2007, 21, 427–438. [Google Scholar] [CrossRef]
- Tomiyasu, H.; Watanabe, M.; Sugita, K.; Goto-Koshino, Y.; Fujino, Y.; Ohno, K.; Sugano, S.; Tsujimoto, H. Regulations of ABCB1 and ABCG2 expression through MAPK pathways in acute lymphoblastic leukemia cell lines. Anticancer. Res. 2013, 33, 5317–5323. [Google Scholar] [PubMed]
- Zhu, M.M.; Tong, J.L.; Xu, Q.; Nie, F.; Xu, X.T.; Xiao, S.D.; Ran, Z.H. Increased JNK1 signaling pathway is responsible for ABCG2-mediated multidrug resistance in human colon cancer. PLoS ONE 2012, 7, e41763. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Huang, W.; Wang, X.; Zhang, J.; Tao, T.; Zheng, Y.; Liu, S.; Yang, J.; Chen, Z.-S.; Cai, C.-Y.; et al. Hsa-miR-3178/RhoB/PI3K/Akt, a novel signaling pathway regulates ABC transporters to reverse gemcitabine resistance in pancreatic cancer. Mol. Cancer 2022, 21, 112. [Google Scholar] [CrossRef]
- Zhang, L.; Guo, X.; Zhang, D.; Fan, Y.; Qin, L.; Dong, S.; Zhang, L. Upregulated miR-132 in Lgr5+ gastric cancer stem cell-like cells contributes to cisplatin-resistance via SIRT1/CREB/ABCG2 signaling pathway. Mol. Carcinog. 2017, 56, 2022–2034. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Ajani, J.A.; Song, S. Drug resistance and Cancer stem cells. Cell Commun. Signal 2021, 19, 19. [Google Scholar] [CrossRef]
- Garcia-Mayea, Y.; Mir, C.; Masson, F.; Paciucci, R.; ME, L.L. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin. Cancer Biol. 2020, 60, 166–180. [Google Scholar] [CrossRef]
- Cojoc, M.; Mäbert, K.; Muders, M.H.; Dubrovska, A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin. Cancer Biol. 2015, 31, 16–27. [Google Scholar] [CrossRef]
- Kim, J.K.; Jeon, H.Y.; Kim, H. The molecular mechanisms underlying the therapeutic resistance of cancer stem cells. Arch. Pharm. Res. 2015, 38, 389–401. [Google Scholar] [CrossRef]
- Moitra, K.; Lou, H.; Dean, M. Multidrug efflux pumps and cancer stem cells: Insights into multidrug resistance and therapeutic development. Clin. Pharmacol. Ther. 2011, 89, 491–502. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef]
- Clark-Corrigall, J.; Myssina, S.; Michaelis, M.; Cinatl, J., Jr.; Ahmed, S.; Carr-Wilkinson, J. Elevated Expression of LGR5 and WNT Signaling Factors in Neuroblastoma Cells With Acquired Drug Resistance. Cancer Investig. 2023, 41, 173–182. [Google Scholar] [CrossRef]
- Abdin, S.M.; Tolba, M.F.; Zaher, D.M.; Omar, H.A. Nuclear factor-κB signaling inhibitors revert multidrug-resistance in breast cancer cells. Chem. Biol. Interact. 2021, 340, 109450. [Google Scholar] [CrossRef] [PubMed]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Salehan, M.R.; Morse, H.R. DNA damage repair and tolerance: A role in chemotherapeutic drug resistance. Br. J. Biomed. Sci. 2013, 70, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- De Angelis, P.M.; Svendsrud, D.H.; Kravik, K.L.; Stokke, T. Cellular response to 5-fluorouracil (5-FU) in 5-FU-resistant colon cancer cell lines during treatment and recovery. Mol. Cancer 2006, 5, 20. [Google Scholar] [CrossRef]
- de Angelis, P.M.; Fjell, B.; Kravik, K.L.; Haug, T.; Tunheim, S.H.; Reichelt, W.; Beigi, M.; Clausen, O.P.; Galteland, E.; Stokke, T. Molecular characterizations of derivatives of HCT116 colorectal cancer cells that are resistant to the chemotherapeutic agent 5-fluorouracil. Int. J. Oncol. 2004, 24, 1279–1288. [Google Scholar] [CrossRef]
- Lowery, C.D.; Dowless, M.; Renschler, M.; Blosser, W.; VanWye, A.B.; Stephens, J.R.; Iversen, P.W.; Lin, A.B.; Beckmann, R.P.; Krytska, K.; et al. Broad Spectrum Activity of the Checkpoint Kinase 1 Inhibitor Prexasertib as a Single Agent or Chemopotentiator across a Range of Preclinical Pediatric Tumor Models. Clin. Cancer Res. 2019, 25, 2278–2289. [Google Scholar] [CrossRef]
- Booth, L.; Cruickshanks, N.; Ridder, T.; Dai, Y.; Grant, S.; Dent, P. PARP and CHK inhibitors interact to cause DNA damage and cell death in mammary carcinoma cells. Cancer Biol. Ther. 2013, 14, 458–465. [Google Scholar] [CrossRef]
- Lee, H.J.; Cao, Y.; Pham, V.; Blackwood, E.; Wilson, C.; Evangelista, M.; Klijn, C.; Stokoe, D.; Settleman, J. Ras-MEK Signaling Mediates a Critical Chk1-Dependent DNA Damage Response in Cancer Cells. Mol. Cancer Ther. 2017, 16, 694–704. [Google Scholar] [CrossRef]
- Gonzalez-Conchas, G.A.; Rodriguez-Romo, L.; Hernandez-Barajas, D.; Gonzalez-Guerrero, J.F.; Rodriguez-Fernandez, I.A.; Verdines-Perez, A.; Templeton, A.J.; Ocana, A.; Seruga, B.; Tannock, I.F.; et al. Epidermal growth factor receptor overexpression and outcomes in early breast cancer: A systematic review and a meta-analysis. Cancer Treat. Rev. 2018, 62, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; He, J.; Yuan, Z.; Wang, S.; Peng, R.; Shi, Y.; Teng, X.; Qin, T. EGFR expression correlates with decreased disease-free survival in triple-negative breast cancer: A retrospective analysis based on a tissue microarray. Med. Oncol. 2012, 29, 401–405. [Google Scholar] [CrossRef]
- Rimawi, M.F.; Shetty, P.B.; Weiss, H.L.; Schiff, R.; Osborne, C.K.; Chamness, G.C.; Elledge, R.M. Epidermal growth factor receptor expression in breast cancer association with biologic phenotype and clinical outcomes. Cancer 2010, 116, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Wright, G.; Bryant, H.; Wiggins, L.A.; Schuler, M.; Gassman, N.R. EGFR signaling promotes resistance to CHK1 inhibitor prexasertib in triple negative breast cancer. Cancer Drug Resist. 2020, 3, 980–991. [Google Scholar] [CrossRef]
- Wilting, R.H.; Dannenberg, J.H. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist. Updat. 2012, 15, 21–38. [Google Scholar] [CrossRef]
- Zeller, C.; Brown, R. Therapeutic modulation of epigenetic drivers of drug resistance in ovarian cancer. Ther. Adv. Med. Oncol. 2010, 2, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Wu, J.; Zhou, Z.; Li, J.; Liu, H.; Zhang, H.; Zhang, J.; Huang, W.; He, Y.; Zhu, S.; Huo, M.; et al. CHD4 promotes acquired chemoresistance and tumor progression by activating the MEK/ERK axis. Drug Resist. Updat. 2023, 66, 100913. [Google Scholar] [CrossRef]
- Fan, M.; Yan, P.S.; Hartman-Frey, C.; Chen, L.; Paik, H.; Oyer, S.L.; Salisbury, J.D.; Cheng, A.S.; Li, L.; Abbosh, P.H.; et al. Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer Res. 2006, 66, 11954–11966. [Google Scholar] [CrossRef]
- Ohata, Y.; Shimada, S.; Akiyama, Y.; Mogushi, K.; Nakao, K.; Matsumura, S.; Aihara, A.; Mitsunori, Y.; Ban, D.; Ochiai, T.; et al. Acquired Resistance with Epigenetic Alterations Under Long-Term Antiangiogenic Therapy for Hepatocellular Carcinoma. Mol. Cancer Ther. 2017, 16, 1155–1165. [Google Scholar] [CrossRef]
- Huang, M.; Chen, C.; Geng, J.; Han, D.; Wang, T.; Xie, T.; Wang, L.; Wang, Y.; Wang, C.; Lei, Z.; et al. Targeting KDM1A attenuates Wnt/β-catenin signaling pathway to eliminate sorafenib-resistant stem-like cells in hepatocellular carcinoma. Cancer Lett. 2017, 398, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Wu, J.; Dombkowski, A.; Zhang, K.; Holowatyj, A.; Boerner, J.L.; Yang, Z.Q. Genomic amplification and a role in drug-resistance for the KDM5A histone demethylase in breast cancer. Am. J. Transl. Res. 2012, 4, 247–256. [Google Scholar] [PubMed]
- Lu, Y.; Liu, Y.; Oeck, S.; Glazer, P.M. Hypoxia Promotes Resistance to EGFR Inhibition in NSCLC Cells via the Histone Demethylases, LSD1 and PLU-1. Mol. Cancer Res. 2018, 16, 1458–1469. [Google Scholar] [CrossRef]
- Tuncel, G.; Kalkan, R. Importance of m N6-methyladenosine (m6A) RNA modification in cancer. Med. Oncol. 2019, 36, 36. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Li, H.; Zhang, D.; Xu, L.; Liu, H.; Hao, X.; Yan, X.; Liao, H.; Chen, X.; Xie, K.; et al. KIAA1429 contributes to liver cancer progression through N6-methyladenosine-dependent post-transcriptional modification of GATA3. Mol. Cancer 2019, 18, 186. [Google Scholar] [CrossRef]
- Yue, Y.; Liu, J.; Cui, X.; Cao, J.; Luo, G.; Zhang, Z.; Cheng, T.; Gao, M.; Shu, X.; Ma, H.; et al. VIRMA mediates preferential m6A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018, 4, 10. [Google Scholar] [CrossRef]
- Lin, X.; Ye, R.; Li, Z.; Zhang, B.; Huang, Y.; Du, J.; Wang, B.; Meng, H.; Xian, H.; Yang, X.; et al. KIAA1429 promotes tumorigenesis and gefitinib resistance in lung adenocarcinoma by activating the JNK/ MAPK pathway in an m6A-dependent manner. Drug Resist. Updat. 2023, 66, 100908. [Google Scholar] [CrossRef]
- Lin, S.; Ruan, H.; Qin, L.; Zhao, C.; Gu, M.; Wang, Z.; Liu, B.; Wang, H.; Wang, J. Acquired resistance to EGFR-TKIs in NSCLC mediates epigenetic downregulation of MUC17 by facilitating NF-κB activity via UHRF1/DNMT1 complex. Int. J. Biol. Sci. 2023, 19, 832–851. [Google Scholar] [CrossRef]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e819. [Google Scholar] [CrossRef] [PubMed]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.e237. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.R.; Nelson, P.S. Cellular senescence and cancer chemotherapy resistance. Drug Resist. Updat. 2012, 15, 123–131. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002, 109, 335–346. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.Y.; Wu, D.Y. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005, 65, 2795–2803. [Google Scholar] [CrossRef]
- Sabisz, M.; Skladanowski, A. Cancer stem cells and escape from drug-induced premature senescence in human lung tumor cells: Implications for drug resistance and in vitro drug screening models. Cell Cycle 2009, 8, 3208–3217. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv. Cancer Res. 2019, 141, 43–84. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, C.; Ling, S.; Wei, R.; Wang, J.; Xu, X. The metabolic flexibility of quiescent CSC: Implications for chemotherapy resistance. Cell Death Dis. 2021, 12, 835. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Takeishi, S.; Nakayama, K.I. To wake up cancer stem cells, or to let them sleep, that is the question. Cancer Sci. 2016, 107, 875–881. [Google Scholar] [CrossRef]
- Xu, Z.Y.; Tang, J.N.; Xie, H.X.; Du, Y.A.; Huang, L.; Yu, P.F.; Cheng, X.D. 5-Fluorouracil chemotherapy of gastric cancer generates residual cells with properties of cancer stem cells. Int. J. Biol. Sci. 2015, 11, 284–294. [Google Scholar] [CrossRef]
- Kobayashi, A.; Okuda, H.; Xing, F.; Pandey, P.R.; Watabe, M.; Hirota, S.; Pai, S.K.; Liu, W.; Fukuda, K.; Chambers, C.; et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 2011, 208, 2641–2655. [Google Scholar] [CrossRef]
- Yang, A.; Qin, S.; Schulte, B.A.; Ethier, S.P.; Tew, K.D.; Wang, G.Y. MYC Inhibition Depletes Cancer Stem-like Cells in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 6641–6650. [Google Scholar] [CrossRef]
- Civenni, G.; Malek, A.; Albino, D.; Garcia-Escudero, R.; Napoli, S.; Di Marco, S.; Pinton, S.; Sarti, M.; Carbone, G.M.; Catapano, C.V. RNAi-mediated silencing of Myc transcription inhibits stem-like cell maintenance and tumorigenicity in prostate cancer. Cancer Res. 2013, 73, 6816–6827. [Google Scholar] [CrossRef]
- Wu, C.H.; van Riggelen, J.; Yetil, A.; Fan, A.C.; Bachireddy, P.; Felsher, D.W. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc. Natl. Acad. Sci. USA 2007, 104, 13028–13033. [Google Scholar] [CrossRef]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, A.; Beer, S.; Mandl, S.; Bachmann, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Gray-Schopfer, V.; Wellbrock, C.; Marais, R. Melanoma biology and new targeted therapy. Nature 2007, 445, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Henriques, V.; Martins, T.; Link, W.; Ferreira, B.I. The Emerging Therapeutic Landscape of Advanced Melanoma. Curr. Pharm. Des. 2018, 24, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 313–322. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef]
- Limberg, J.; Egan, C.E.; Gray, K.D.; Singh, M.; Loewenstein, Z.; Yang, Y.; Riascos, M.C.; Al Asadi, H.; Safe, P.; El Eshaky, S.; et al. Activation of the JAK/STAT Pathway Leads to BRAF Inhibitor Resistance in BRAFV600E Positive Thyroid Carcinoma. Mol. Cancer Res. 2023, 21, 397–410. [Google Scholar] [CrossRef]
- Corrales, E.; Levit-Zerdoun, E.; Metzger, P.; Mertes, R.; Lehmann, A.; Münch, J.; Lemke, S.; Kowar, S.; Boerries, M. PI3K/AKT signaling allows for MAPK/ERK pathway independency mediating dedifferentiation-driven treatment resistance in melanoma. Cell Commun. Signal 2022, 20, 187. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, Y.; Aguilar, A.; Bernard, D.; Yang, C.Y. Targeting the MDM2-p53 Protein-Protein Interaction for New Cancer Therapy: Progress and Challenges. Cold Spring Harb. Perspect. Med. 2017, 7, a026245. [Google Scholar] [CrossRef]
- Khoury, K.; Dömling, A. P53 mdm2 inhibitors. Curr. Pharm. Des. 2012, 18, 4668–4678. [Google Scholar] [CrossRef]
- Liu, D.P.; Song, H.; Xu, Y. A common gain of function of p53 cancer mutants in inducing genetic instability. Oncogene 2010, 29, 949–956. [Google Scholar] [CrossRef]
- Tung, M.C.; Lin, P.L.; Wang, Y.C.; He, T.Y.; Lee, M.C.; Yeh, S.D.; Chen, C.Y.; Lee, H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6, 41692–41705. [Google Scholar] [CrossRef]
- Sauer, L.; Gitenay, D.; Vo, C.; Baron, V.T. Mutant p53 initiates a feedback loop that involves Egr-1/EGF receptor/ERK in prostate cancer cells. Oncogene 2010, 29, 2628–2637. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.H.; Harrison, T.A.; Phipps, A.I.; Steinfelder, R.; Trinh, Q.M.; Qu, C.; Banbury, B.L.; Georgeson, P.; Grasso, C.S.; Giannakis, M.; et al. Landscape of somatic single nucleotide variants and indels in colorectal cancer and impact on survival. Nat. Commun. 2020, 11, 3644. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e123. [Google Scholar] [CrossRef] [PubMed]
- Vita, M.; Henriksson, M. The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol. 2006, 16, 318–330. [Google Scholar] [CrossRef]
- Ben-David, E.; Bester, A.C.; Shifman, S.; Kerem, B. Transcriptional dynamics in colorectal carcinogenesis: New insights into the role of c-Myc and miR17 in benign to cancer transformation. Cancer Res. 2014, 74, 5532–5540. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, J.; Yin, J.; Gan, Y.; Xu, S.; Gu, Y.; Huang, W. Alternative approaches to target Myc for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 117. [Google Scholar] [CrossRef]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.N.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Yap, T.A.; Gerlinger, M.; Futreal, P.A.; Pusztai, L.; Swanton, C. Intratumor heterogeneity: Seeing the wood for the trees. Sci. Transl. Med. 2012, 4, 127ps110. [Google Scholar] [CrossRef]
- McGranahan, N.; Favero, F.; de Bruin, E.C.; Birkbak, N.J.; Szallasi, Z.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015, 7, 283ra254. [Google Scholar] [CrossRef] [PubMed]
- Schiavon, G.; Hrebien, S.; Garcia-Murillas, I.; Cutts, R.J.; Pearson, A.; Tarazona, N.; Fenwick, K.; Kozarewa, I.; Lopez-Knowles, E.; Ribas, R.; et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci. Transl. Med. 2015, 7, 313ra182. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Laughney, A.M.; Hu, J.; Campbell, N.R.; Bakhoum, S.F.; Setty, M.; Lavallée, V.P.; Xie, Y.; Masilionis, I.; Carr, A.J.; Kottapalli, S.; et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat. Med. 2020, 26, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017, 169, 750–765.e717. [Google Scholar] [CrossRef] [PubMed]
- Costa, N.L.; Valadares, M.C.; Souza, P.P.; Mendonça, E.F.; Oliveira, J.C.; Silva, T.A.; Batista, A.C. Tumor-associated macrophages and the profile of inflammatory cytokines in oral squamous cell carcinoma. Oral. Oncol. 2013, 49, 216–223. [Google Scholar] [CrossRef]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z.G. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013, 23, 898–914. [Google Scholar] [CrossRef]
- Dituri, F.; Mazzocca, A.; Giannelli, G.; Antonaci, S. PI3K functions in cancer progression, anticancer immunity and immune evasion by tumors. Clin. Dev. Immunol. 2011, 2011, 947858. [Google Scholar] [CrossRef]
- Lu, H.; Clauser, K.R.; Tam, W.L.; Fröse, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A.; et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 2014, 16, 1105–1117. [Google Scholar] [CrossRef]
- Stahl, M.; Schupp, J.; Jäger, B.; Schmid, M.; Zissel, G.; Müller-Quernheim, J.; Prasse, A. Lung collagens perpetuate pulmonary fibrosis via CD204 and M2 macrophage activation. PLoS ONE 2013, 8, e81382. [Google Scholar] [CrossRef]
- Bollyky, P.L.; Wu, R.P.; Falk, B.A.; Lord, J.D.; Long, S.A.; Preisinger, A.; Teng, B.; Holt, G.E.; Standifer, N.E.; Braun, K.R.; et al. ECM components guide IL-10 producing regulatory T-cell (TR1) induction from effector memory T-cell precursors. Proc. Natl. Acad. Sci. USA 2011, 108, 7938–7943. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Zhang, C.J.; Zhu, N.; Du, K.; Yin, Y.F.; Tan, X.; Liao, D.F.; Qin, L. Lipid metabolism and carcinogenesis, cancer development. Am. J. Cancer Res. 2018, 8, 778–791. [Google Scholar] [PubMed]
- Ma, L.; Hernandez, M.O.; Zhao, Y.; Mehta, M.; Tran, B.; Kelly, M.; Rae, Z.; Hernandez, J.M.; Davis, J.L.; Martin, S.P.; et al. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell 2019, 36, 418–430.e416. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Rodriguez-Pascual, J.; Ayuso-Sacido, A.; Belda-Iniesta, C. Drug resistance in cancer immunotherapy: New strategies to improve checkpoint inhibitor therapies. Cancer Drug Resist. 2019, 2, 980–993. [Google Scholar] [CrossRef]
- Mandal, R.; Şenbabaoğlu, Y.; Desrichard, A.; Havel, J.J.; Dalin, M.G.; Riaz, N.; Lee, K.W.; Ganly, I.; Hakimi, A.A.; Chan, T.A.; et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight 2016, 1, e89829. [Google Scholar] [CrossRef]
- Liu, C.; Wang, M.; Zhang, H.; Li, C.; Zhang, T.; Liu, H.; Zhu, S.; Chen, J. Tumor microenvironment and immunotherapy of oral cancer. Eur. J. Med. Res. 2022, 27, 198. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, H.; Deng, Y.; Tai, Y.; Zeng, K.; Zhang, Y.; Liu, W.; Zhang, Q.; Yang, Y. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 422. [Google Scholar] [CrossRef]
- Skolekova, S.; Matuskova, M.; Bohac, M.; Toro, L.; Durinikova, E.; Tyciakova, S.; Demkova, L.; Gursky, J.; Kucerova, L. Cisplatin-induced mesenchymal stromal cells-mediated mechanism contributing to decreased antitumor effect in breast cancer cells. Cell Commun. Signal 2016, 14, 4. [Google Scholar] [CrossRef]
- Eun, J.W.; Yoon, J.H.; Ahn, H.R.; Kim, S.; Kim, Y.B.; Lim, S.B.; Park, W.; Kang, T.W.; Baek, G.O.; Yoon, M.G.; et al. Cancer-associated fibroblast-derived secreted phosphoprotein 1 contributes to resistance of hepatocellular carcinoma to sorafenib and lenvatinib. Cancer Commun 2023, 43, 455–479. [Google Scholar] [CrossRef]
- Amin, T.; Viol, F.; Krause, J.; Fahl, M.; Eggers, C.; Awwad, F.; Schmidt, B.; Benten, D.; Ungefroren, H.; Fraune, C.; et al. Cancer-Associated Fibroblasts Induce Proliferation and Therapeutic Resistance to Everolimus in Neuroendocrine Tumors through STAT3 Activation. Neuroendocrinology 2023, 113, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.; Wang, J.; Li, Y.; Lu, Z.; Huang, J.; Sun, S.; Mao, S.; Lei, Y.; Zang, R.; Sun, N.; et al. Cisplatin-activated PAI-1 secretion in the cancer-associated fibroblasts with paracrine effects promoting esophageal squamous cell carcinoma progression and causing chemoresistance. Cell Death Dis. 2018, 9, 759. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, L.; Jiang, H.; Li, Q.; Wang-Gillam, A.; Yu, J.; Head, R.; Liu, J.; Ruzinova, M.B.; Lim, K.H. Tumor-Stroma IL1β-IRAK4 Feedforward Circuitry Drives Tumor Fibrosis, Chemoresistance, and Poor Prognosis in Pancreatic Cancer. Cancer Res. 2018, 78, 1700–1712. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e816. [Google Scholar] [CrossRef]
- Han, M.E.; Kim, H.J.; Shin, D.H.; Hwang, S.H.; Kang, C.D.; Oh, S.O. Overexpression of NRG1 promotes progression of gastric cancer by regulating the self-renewal of cancer stem cells. J. Gastroenterol. 2015, 50, 645–656. [Google Scholar] [CrossRef]
- Bai, S.; Zhao, Y.; Chen, W.; Peng, W.; Wang, Y.; Xiong, S.; Aruna; Li, Y.; Yang, Y.; Chen, S.; et al. The stromal-tumor amplifying STC1-Notch1 feedforward signal promotes the stemness of hepatocellular carcinoma. J. Transl. Med. 2023, 21, 236. [Google Scholar] [CrossRef]
- Lawal, B.; Wu, A.T.; Chen, C.H.; George, T.A.; Wu, S.Y. Identification of INFG/STAT1/NOTCH3 as γ-Mangostin’s potential targets for overcoming doxorubicin resistance and reducing cancer-associated fibroblasts in triple-negative breast cancer. Biomed. Pharmacother. 2023, 163, 114800. [Google Scholar] [CrossRef]
- Correia, A.L.; Bissell, M.J. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist. Updat. 2012, 15, 39–49. [Google Scholar] [CrossRef]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef]
- Sermeus, A.; Cosse, J.P.; Crespin, M.; Mainfroid, V.; de Longueville, F.; Ninane, N.; Raes, M.; Remacle, J.; Michiels, C. Hypoxia induces protection against etoposide-induced apoptosis: Molecular profiling of changes in gene expression and transcription factor activity. Mol. Cancer 2008, 7, 27. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Magnussen, A.L.; Mills, I.G. Vascular normalisation as the stepping stone into tumour microenvironment transformation. Br. J. Cancer 2021, 125, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Bakshi, A.K.; Mittapelly, N.; Gautam, S.; Marwaha, D.; Rai, N.; Singh, N.; Tiwari, P.; Agarwal, N.; Kumar, A.; et al. Recent updates on innovative approaches to overcome drug resistance for better outcomes in cancer. J. Control Release 2022, 346, 43–70. [Google Scholar] [CrossRef]
- Qiu, G.Z.; Jin, M.Z.; Dai, J.X.; Sun, W.; Feng, J.H.; Jin, W.L. Reprogramming of the Tumor in the Hypoxic Niche: The Emerging Concept and Associated Therapeutic Strategies. Trends Pharmacol. Sci. 2017, 38, 669–686. [Google Scholar] [CrossRef]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Veliça, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1α/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell 2017, 32, 669–683.e665. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, S.; Gao, L.; Zhi, K.; Ren, W. The Molecular Basis and Therapeutic Aspects of Cisplatin Resistance in Oral Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 761379. [Google Scholar] [CrossRef]
- Bhandari, V.; Li, C.H.; Bristow, R.G.; Boutros, P.C.; Aaltonen, L.A.; Abascal, F.; Abeshouse, A.; Aburatani, H.; Adams, D.J.; Agrawal, N.; et al. Divergent mutational processes distinguish hypoxic and normoxic tumours. Nat. Commun. 2020, 11, 737. [Google Scholar] [CrossRef]
- Deben, C.; Deschoolmeester, V.; De Waele, J.; Jacobs, J.; Van den Bossche, J.; Wouters, A.; Peeters, M.; Rolfo, C.; Smits, E.; Lardon, F.; et al. Hypoxia-Induced Cisplatin Resistance in Non-Small Cell Lung Cancer Cells Is Mediated by HIF-1α and Mutant p53 and Can Be Overcome by Induction of Oxidative Stress. Cancers 2018, 10, 126. [Google Scholar] [CrossRef]
- Parmakhtiar, B.; Burger, R.A.; Kim, J.H.; Fruehauf, J.P. HIF Inactivation of p53 in Ovarian Cancer Can Be Reversed by Topotecan, Restoring Cisplatin and Paclitaxel Sensitivity. Mol. Cancer Res. 2019, 17, 1675–1686. [Google Scholar] [CrossRef]
- Mao, X.Y.; Jin, M.Z.; Chen, J.F.; Zhou, H.H.; Jin, W.L. Live or let die: Neuroprotective and anti-cancer effects of nutraceutical antioxidants. Pharmacol. Ther. 2018, 183, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Peck, B.; Schulze, A. Lipid Metabolism at the Nexus of Diet and Tumor Microenvironment. Trends Cancer 2019, 5, 693–703. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Lu, Y.; Wu, X.; Pan, T.; Yu, Z.; Hou, J.; Wu, A.; Li, J.; Yang, Z.; Li, C.; et al. The cross-talk between tumor cells and activated fibroblasts mediated by lactate/BDNF/TrkB signaling promotes acquired resistance to anlotinib in human gastric cancer. Redox Biol. 2021, 46, 102076. [Google Scholar] [CrossRef]
- Sharma, M.; Astekar, M.; Soi, S.; Manjunatha, B.S.; Shetty, D.C.; Radhakrishnan, R. pH Gradient Reversal: An Emerging Hallmark of Cancers. Recent. Pat. Anticancer. Drug Discov. 2015, 10, 244–258. [Google Scholar] [CrossRef]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A. The chemistry, physiology and pathology of pH in cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130099. [Google Scholar] [CrossRef]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Wojtkowiak, J.W.; Verduzco, D.; Schramm, K.J.; Gillies, R.J. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol. Pharm. 2011, 8, 2032–2038. [Google Scholar] [CrossRef]
- Taylor, S.; Spugnini, E.P.; Assaraf, Y.G.; Azzarito, T.; Rauch, C.; Fais, S. Microenvironment acidity as a major determinant of tumor chemoresistance: Proton pump inhibitors (PPIs) as a novel therapeutic approach. Drug Resist. Updat. 2015, 23, 69–78. [Google Scholar] [CrossRef]
- de Bem Prunes, B.; Nunes, J.S.; da Silva, V.P.; Laureano, N.K.; Gonçalves, D.R.; Machado, I.S.; Barbosa, S.; Lamers, M.L.; Rados, P.V.; Kurth, I.; et al. The role of tumor acidification in aggressiveness, cell dissemination and treatment resistance of oral squamous cell carcinoma. Life Sci. 2022, 288, 120163. [Google Scholar] [CrossRef] [PubMed]
- Peppicelli, S.; Toti, A.; Giannoni, E.; Bianchini, F.; Margheri, F.; Del Rosso, M.; Calorini, L. Metformin is also effective on lactic acidosis-exposed melanoma cells switched to oxidative phosphorylation. Cell Cycle 2016, 15, 1908–1918. [Google Scholar] [CrossRef]
- LaMonte, G.; Tang, X.; Chen, J.L.-Y.; Wu, J.; Ding, C.-K.C.; Keenan, M.M.; Sangokoya, C.; Kung, H.-N.; Ilkayeva, O.; Boros, L.G.; et al. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab. 2013, 1, 23. [Google Scholar] [CrossRef]
- Mazzio, E.A.; Boukli, N.; Rivera, N.; Soliman, K.F. Pericellular pH homeostasis is a primary function of the Warburg effect: Inversion of metabolic systems to control lactate steady state in tumor cells. Cancer Sci. 2012, 103, 422–432. [Google Scholar] [CrossRef]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef]
- Sauvant, C.; Nowak, M.; Wirth, C.; Schneider, B.; Riemann, A.; Gekle, M.; Thews, O. Acidosis induces multi-drug resistance in rat prostate cancer cells (AT1) in vitro and in vivo by increasing the activity of the p-glycoprotein via activation of p38. Int. J. Cancer 2008, 123, 2532–2542. [Google Scholar] [CrossRef]
- Williams, A.C.; Collard, T.J.; Paraskeva, C. An acidic environment leads to p53 dependent induction of apoptosis in human adenoma and carcinoma cell lines: Implications for clonal selection during colorectal carcinogenesis. Oncogene 1999, 18, 3199–3204. [Google Scholar] [CrossRef] [PubMed]
- Federici, C.; Petrucci, F.; Caimi, S.; Cesolini, A.; Logozzi, M.; Borghi, M.; D’Ilio, S.; Lugini, L.; Violante, N.; Azzarito, T.; et al. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PLoS ONE 2014, 9, e88193. [Google Scholar] [CrossRef] [PubMed]
- Hanoun, M.; Maryanovich, M.; Arnal-Estapé, A.; Frenette, P.S. Neural regulation of hematopoiesis, inflammation, and cancer. Neuron 2015, 86, 360–373. [Google Scholar] [CrossRef]
- Amit, M.; Na’ara, S.; Gil, Z. Mechanisms of cancer dissemination along nerves. Nat. Rev. Cancer 2016, 16, 399–408. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef]
- Mao, X.Y.; Li, Q.Q.; Gao, Y.F.; Zhou, H.H.; Liu, Z.Q.; Jin, W.L. Gap junction as an intercellular glue: Emerging roles in cancer EMT and metastasis. Cancer Lett. 2016, 381, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.S.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, A.; Bussink, J.; Rowan, A.E.; Span, P.N. The mechanical microenvironment in cancer: How physics affects tumours. Semin. Cancer Biol. 2015, 35, 62–70. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Yang, X.H.; Flores, L.M.; Li, Q.; Zhou, P.; Xu, F.; Krop, I.E.; Hemler, M.E. Disruption of laminin-integrin-CD151-focal adhesion kinase axis sensitizes breast cancer cells to ErbB2 antagonists. Cancer Res. 2010, 70, 2256–2263. [Google Scholar] [CrossRef]
- Wang, J.P.; Hielscher, A. Fibronectin: How Its Aberrant Expression in Tumors May Improve Therapeutic Targeting. J. Cancer 2017, 8, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Park, C.C.; Hilsenbeck, S.G.; Ward, R.; Rimawi, M.F.; Wang, Y.C.; Shou, J.; Bissell, M.J.; Osborne, C.K.; Schiff, R. β1 integrin mediates an alternative survival pathway in breast cancer cells resistant to lapatinib. Breast Cancer Res. 2011, 13, R84. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: Key players in cancer and potential therapeutic strategy. Signal Transduct. Target. Ther. 2020, 5, 145. [Google Scholar] [CrossRef]
- Vaidya, F.U.; Sufiyan Chhipa, A.; Mishra, V.; Gupta, V.K.; Rawat, S.G.; Kumar, A.; Pathak, C. Molecular and cellular paradigms of multidrug resistance in cancer. Cancer Rep. 2022, 5, e1291. [Google Scholar] [CrossRef]
- Zhang, Y.; Pan, Q.; Shao, Z. Extracellular vesicles derived from cancer-associated fibroblasts carry tumor-promotive microRNA-1228-3p to enhance the resistance of hepatocellular carcinoma cells to sorafenib. Hum. Cell 2023, 36, 296–311. [Google Scholar] [CrossRef]
- Qin, W.; Wang, L.; Tian, H.; Wu, X.; Xiao, C.; Pan, Y.; Fan, M.; Tai, Y.; Liu, W.; Zhang, Q.; et al. CAF-derived exosomes transmitted Gremlin-1 promotes cancer progression and decreases the sensitivity of hepatoma cells to sorafenib. Mol. Carcinog. 2022, 61, 764–775. [Google Scholar] [CrossRef]
- Shi, L.; Zhu, W.; Huang, Y.; Zhuo, L.; Wang, S.; Chen, S.; Zhang, B.; Ke, B. Cancer-associated fibroblast-derived exosomal microRNA-20a suppresses the PTEN/PI3K-AKT pathway to promote the progression and chemoresistance of non-small cell lung cancer. Clin. Transl. Med. 2022, 12, e989. [Google Scholar] [CrossRef]
- Zhao, Q.; Huang, L.; Qin, G.; Qiao, Y.; Ren, F.; Shen, C.; Wang, S.; Liu, S.; Lian, J.; Wang, D.; et al. Cancer-associated fibroblasts induce monocytic myeloid-derived suppressor cell generation via IL-6/exosomal miR-21-activated STAT3 signaling to promote cisplatin resistance in esophageal squamous cell carcinoma. Cancer Lett. 2021, 518, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Wang, Z.; Chai, Q.; Li, Z.; Zhang, M.; Zhang, Y.; Zhang, L.; Tang, Q.; Zhu, H.; Sui, H. Exosomes derived from MDR cells induce cetuximab resistance in CRC via PI3K/AKT signaling-mediated Sox2 and PD-L1 expression. Exp. Ther. Med. 2023, 25, 86. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Xie, S.L.; Fan, S.; Zhang, S.Y.; Chen, W.X.; Li, Q.X.; Pan, G.K.; Zhang, H.Q.; Wang, W.W.; Weng, B.; Zhang, Z.; et al. SOX8 regulates cancer stem-like properties and cisplatin-induced EMT in tongue squamous cell carcinoma by acting on the Wnt/β-catenin pathway. Int. J. Cancer 2018, 142, 1252–1265. [Google Scholar] [CrossRef]
- Usman, S.; Jamal, A.; Teh, M.T.; Waseem, A. Major Molecular Signaling Pathways in Oral Cancer Associated with Therapeutic Resistance. Front. Oral. Health 2020, 1, 603160. [Google Scholar] [CrossRef]
- Huang, L.; Wu, R.L.; Xu, A.M. Epithelial-mesenchymal transition in gastric cancer. Am. J. Transl. Res. 2015, 7, 2141–2158. [Google Scholar] [PubMed]
- Kirave, P.; Gondaliya, P.; Kulkarni, B.; Rawal, R.; Garg, R.; Jain, A.; Kalia, K. Exosome mediated miR-155 delivery confers cisplatin chemoresistance in oral cancer cells via epithelial-mesenchymal transition. Oncotarget 2020, 11, 1157–1171. [Google Scholar] [CrossRef] [PubMed]
- Mousset, A.; Lecorgne, E.; Bourget, I.; Lopez, P.; Jenovai, K.; Cherfils-Vicini, J.; Dominici, C.; Rios, G.; Girard-Riboulleau, C.; Liu, B.; et al. Neutrophil extracellular traps formed during chemotherapy confer treatment resistance via TGF-β activation. Cancer Cell 2023, 41, 757–775.e710. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Liu, Y.; He, J.; Gao, T.; Li, L.; He, S. Family with sequence similarity 46 member a confers chemo-resistance to ovarian carcinoma via TGF-β/Smad2 signaling. Bioengineered 2022, 13, 10629–10639. [Google Scholar] [CrossRef]
- Wo, L.; Zhang, B.; You, X.; Hu, Y.; Gu, Z.; Zhang, M.; Wang, Q.; Lv, Z.; Zhao, H. Up-regulation of LncRNA UCA1 by TGF-β promotes doxorubicin resistance in breast cancer cells. Immunopharmacol. Immunotoxicol. 2022, 44, 492–499. [Google Scholar] [CrossRef]
- Tang, Y.A.; Chen, Y.F.; Bao, Y.; Mahara, S.; Yatim, S.; Oguz, G.; Lee, P.L.; Feng, M.; Cai, Y.; Tan, E.Y.; et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E5990–E5999. [Google Scholar] [CrossRef]
- Wang, M.Q.; Chen, Y.R.; Xu, H.W.; Zhan, J.R.; Suo, D.Q.; Wang, J.J.; Ma, Y.Z.; Guan, X.Y.; Li, Y.; Zhu, S.L. HKDC1 upregulation promotes glycolysis and disease progression, and confers chemoresistance onto gastric cancer. Cancer Sci. 2023, 114, 1365–1377. [Google Scholar] [CrossRef]
- Momeny, M.; Yousefi, H.; Eyvani, H.; Moghaddaskho, F.; Salehi, A.; Esmaeili, F.; Alishahi, Z.; Barghi, F.; Vaezijoze, S.; Shamsaiegahkani, S.; et al. Blockade of nuclear factor-κB (NF-κB) pathway inhibits growth and induces apoptosis in chemoresistant ovarian carcinoma cells. Int. J. Biochem. Cell Biol. 2018, 99, 1–9. [Google Scholar] [CrossRef]
- Boustan, A.; Jahangiri, R.; Ghalehno, A.D.; Khorsandi, M.; Mosaffa, F.; Jamialahmadi, K. Expression analysis elucidates the roles of Nicastrin, Notch4, and Hes1 in prognosis and endocrine-therapy resistance in ER-positive breast cancer patients. Res. Pharm. Sci. 2023, 18, 78–88. [Google Scholar] [CrossRef]
- Deng, J.; Bai, X.; Feng, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Inhibition of PI3K/Akt/mTOR signaling pathway alleviates ovarian cancer chemoresistance through reversing epithelial-mesenchymal transition and decreasing cancer stem cell marker expression. BMC Cancer 2019, 19, 618. [Google Scholar] [CrossRef]
- Scripture, C.D.; Figg, W.D. Drug interactions in cancer therapy. Nat. Rev. Cancer 2006, 6, 546–558. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Zheng, Y.; Ma, L.; Tian, L.; Sun, Q. Clinically-Relevant ABC Transporter for Anti-Cancer Drug Resistance. Front. Pharmacol. 2021, 12, 648407. [Google Scholar] [CrossRef]
- Li, Y.; Liu, R.; Han, X.; Xu, W.; Liu, Y. PLAGL2 increases adriamycin resistance and EMT in breast cancer cells by activating the Wnt pathway. Genes. Genom. 2023, 45, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Quintanal-Villalonga, A.; Taniguchi, H.; Zhan, Y.A.; Hasan, M.M.; Chavan, S.S.; Meng, F.; Uddin, F.; Allaj, V.; Manoj, P.; Shah, N.S.; et al. Comprehensive molecular characterization of lung tumors implicates AKT and MYC signaling in adenocarcinoma to squamous cell transdifferentiation. J. Hematol. Oncol. 2021, 14, 170. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Han, X.; Chen, Y.; Tong, X.; Xue, Y.; Yao, S.; Tang, S.; Pan, Y.; Sun, Y.; Wang, X.; et al. Oxidative stress-triggered Wnt signaling perturbation characterizes the tipping point of lung adeno-to-squamous transdifferentiation. Signal Transduct. Target. Ther. 2023, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Lepore Signorile, M.; Grossi, V.; Di Franco, S.; Forte, G.; Disciglio, V.; Fasano, C.; Sanese, P.; De Marco, K.; Susca, F.C.; Mangiapane, L.R.; et al. Pharmacological targeting of the novel β-catenin chromatin-associated kinase p38α in colorectal cancer stem cell tumorspheres and organoids. Cell Death Dis. 2021, 12, 316. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, C.; Wang, G.; Chen, J.; Ju, S.; Huang, J.; Wang, X. SNORD1C maintains stemness and 5-FU resistance by activation of Wnt signaling pathway in colorectal cancer. Cell Death Discov. 2022, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Min, H.Y.; Yong, Y.S.; Ann, J.; Nguyen, C.T.; La, M.T.; Hyun, S.Y.; Le, H.T.; Kim, H.; Kwon, H.; et al. A novel C-terminal heat shock protein 90 inhibitor that overcomes STAT3-Wnt-β-catenin signaling-mediated drug resistance and adverse effects. Theranostics 2022, 12, 105–125. [Google Scholar] [CrossRef]
- Guo, W.; Li, W.; Yuan, L.; Mei, X.; Hu, W. MicroRNA-106a-3p Induces Apatinib Resistance and Activates Janus-Activated Kinase 2 (JAK2)/Signal Transducer and Activator of Transcription 3 (STAT3) by Targeting the SOCS System in Gastric Cancer. Med. Sci. Monit. 2019, 25, 10122–10128. [Google Scholar] [CrossRef]
- He, H.; Song, F.; Gao, Q.; Lu, Z.; Yuan, Y.; Li, X.; Chen, L.; Jia, C.; Yang, R.; Yang, J.; et al. The APEX1/miRNA-27a-5p axis plays key roles in progression, metastasis and targeted chemotherapy of gastric cancer. Int. J. Pharm. 2021, 599, 120446. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Liu, P.; Hu, C.; Zhou, Z.; Wang, P.; Wang, Y.; Zhang, Y.; Ran, Y.; Li, P.; Zhao, J.; et al. Intracellular angiopoietin-1 promotes TKI-resistance via activation of JAK/STAT5 pathway in chronic myeloid leukemia. Oncogene 2023, 42, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, H.; Meng, J.; Guo, F.; Ren, D.; Wu, H.; Jin, X. S-palmitoylation of PCSK9 induces sorafenib resistance in liver cancer by activating the PI3K/AKT pathway. Cell Rep. 2022, 40, 111194. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xia, X. Long noncoding RNA NEAT1 suppresses sorafenib sensitivity of hepatocellular carcinoma cells via regulating miR-335-c-Met. J. Cell Physiol. 2019, 234, 14999–15009. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Shinonaga, R.; Sakaguchi, H.; Kitagawa, Y.; Yoshida, K. NEAT1-SOD2 Axis Confers Sorafenib and Lenvatinib Resistance by Activating AKT in Liver Cancer Cell Lines. Curr. Issues Mol. Biol. 2023, 45, 1073–1085. [Google Scholar] [CrossRef]
- Enomoto, K.; Hirayama, S.; Kumashiro, N.; Jing, X.; Kimura, T.; Tamagawa, S.; Matsuzaki, I.; Murata, S.I.; Hotomi, M. Synergistic Effects of Lenvatinib (E7080) and MEK Inhibitors against Anaplastic Thyroid Cancer in Preclinical Models. Cancers 2021, 13, 862. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, T.; Wu, C.; Xia, Q.; Xu, D. ASIC1a mediates the drug resistance of human hepatocellular carcinoma via the Ca2+/PI3-kinase/AKT signaling pathway. Lab. Invest. 2017, 97, 53–69. [Google Scholar] [CrossRef]
- Vijayakumar, G.; Swetha, U.S.; Sudhagar, S. Tamoxifen modulates mitochondrial dynamics through AMPK and MAPK during nutrition deprivation. Cell Biol. Int. 2022, 46, 1661–1671. [Google Scholar] [CrossRef]
- Rekha, P.; Gupta, A.; Goud, K.S.; Biswas, B.; Bhattar, S.; Vijayakumar, G.; Selvaraju, S. GPER induces mitochondrial fission through p44/42 MAPK—Drp1 pathway in breast cancer cells. Biochem. Biophys. Res. Commun. 2023, 643, 16–23. [Google Scholar] [CrossRef]
- Troiani, T.; Napolitano, S.; Vitagliano, D.; Morgillo, F.; Capasso, A.; Sforza, V.; Nappi, A.; Ciardiello, D.; Ciardiello, F.; Martinelli, E. Primary and acquired resistance of colorectal cancer cells to anti-EGFR antibodies converge on MEK/ERK pathway activation and can be overcome by combined MEK/EGFR inhibition. Clin. Cancer Res. 2014, 20, 3775–3786. [Google Scholar] [CrossRef]
- Wang, Q.; Liao, C.; Tan, Z.; Li, X.; Guan, X.; Li, H.; Tian, Z.; Liu, J.; An, J. FUT6 inhibits the proliferation, migration, invasion, and EGF-induced EMT of head and neck squamous cell carcinoma (HNSCC) by regulating EGFR/ERK/STAT signaling pathway. Cancer Gene Ther. 2023, 30, 182–191. [Google Scholar] [CrossRef]
- Pan, Z.; Wang, K.; Wang, X.; Jia, Z.; Yang, Y.; Duan, Y.; Huang, L.; Wu, Z.X.; Zhang, J.Y.; Ding, X. Cholesterol promotes EGFR-TKIs resistance in NSCLC by inducing EGFR/Src/Erk/SP1 signaling-mediated ERRα re-expression. Mol. Cancer 2022, 21, 77. [Google Scholar] [CrossRef]
- Udagawa, S.; Ooki, A.; Shinozaki, E.; Fukuda, K.; Yamaguchi, K.; Osumi, H. Circulating Tumor DNA: The Dawn of a New Era in the Optimization of Chemotherapeutic Strategies for Metastatic Colo-Rectal Cancer Focusing on RAS Mutation. Cancers 2023, 15, 1473. [Google Scholar] [CrossRef] [PubMed]
- Draškovič, T.; Zidar, N.; Hauptman, N. Circulating Tumor DNA Methylation Biomarkers for Characterization and Determination of the Cancer Origin in Malignant Liver Tumors. Cancers 2023, 15, 859. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef]
- Lin, F.; Xiong, M.; Hao, W.; Song, Y.; Liu, R.; Yang, Y.; Yuan, X.; Fan, D.; Zhang, Y.; Hao, M.; et al. A Novel Blockade CD47 Antibody with Therapeutic Potential for Cancer. Front. Oncol. 2020, 10, 615534. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chan, S.L.; Kudo, M.; Lau, G.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Dao, T.V.; Toni, E.N.D.; et al. Phase 3 randomized, open-label, multicenter study of tremelimumab (T) and durvalumab (D) as first-line therapy in patients (pts) with unresectable hepatocellular carcinoma (uHCC): HIMALAYA. J. Clin. Oncol. 2022, 40, 379. [Google Scholar] [CrossRef]
- Nilsson, M.B.; Yang, Y.; Heeke, S.; Patel, S.A.; Poteete, A.; Udagawa, H.; Elamin, Y.Y.; Moran, C.A.; Kashima, Y.; Arumugam, T.; et al. CD70 is a therapeutic target upregulated in EMT-associated EGFR tyrosine kinase inhibitor resistance. Cancer Cell 2023, 41, 340–355.e346. [Google Scholar] [CrossRef]
- Marks, D.K.; Gartrell, R.D.; El Asmar, M.; Boboila, S.; Hart, T.; Lu, Y.; Pan, Q.; Yu, J.; Hibshoosh, H.; Guo, H.; et al. Akt Inhibition Is Associated with Favorable Immune Profile Changes within the Tumor Microenvironment of Hormone Receptor Positive, HER2 Negative Breast Cancer. Front. Oncol. 2020, 10, 968. [Google Scholar] [CrossRef]
- Owusu-Brackett, N.; Zhao, M.; Akcakanat, A.; Evans, K.W.; Yuca, E.; Dumbrava, E.I.; Janku, F.; Meric-Bernstam, F. Targeting PI3Kβ alone and in combination with chemotherapy or immunotherapy in tumors with PTEN loss. Oncotarget 2020, 11, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Ju, S.; Kim, J.; Jung, Y. Chloroquine prevents hypoxic accumulation of HIF-1α by inhibiting ATR kinase: Implication in chloroquine-mediated chemosensitization of colon carcinoma cells under hypoxia. Pharmacol. Rep. 2023, 75, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Cocco, S.; Leone, A.; Roca, M.S.; Lombardi, R.; Piezzo, M.; Caputo, R.; Ciardiello, C.; Costantini, S.; Bruzzese, F.; Sisalli, M.J.; et al. Inhibition of autophagy by chloroquine prevents resistance to PI3K/AKT inhibitors and potentiates their antitumor effect in combination with paclitaxel in triple negative breast cancer models. J. Transl. Med. 2022, 20, 290. [Google Scholar] [CrossRef]
- Dong, Q.; Han, D.; Li, B.; Yang, Y.; Ren, L.; Xiao, T.; Zhang, J.; Li, Z.; Yang, H.; Liu, H. Bionic lipoprotein loaded with chloroquine-mediated blocking immune escape improves antitumor immunotherapy. Int. J. Biol. Macromol. 2023, 240, 124342. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Yetisgin, A.A.; Cetinel, S.; Zuvin, M.; Kosar, A.; Kutlu, O. Therapeutic Nanoparticles and Their Targeted Delivery Applications. Molecules 2020, 25, 2193. [Google Scholar] [CrossRef]
- Majidinia, M.; Mirza-Aghazadeh-Attari, M.; Rahimi, M.; Mihanfar, A.; Karimian, A.; Safa, A.; Yousefi, B. Overcoming multidrug resistance in cancer: Recent progress in nanotechnology and new horizons. IUBMB Life 2020, 72, 855–871. [Google Scholar] [CrossRef] [PubMed]
- Han, E.; Kim, D.; Cho, Y.; Lee, S.; Kim, J.; Kim, H. Development of Polymersomes Co-Delivering Doxorubicin and Melittin to Overcome Multidrug Resistance. Molecules 2023, 28, 1087. [Google Scholar] [CrossRef]
- Zhou, S.; Ma, Y.; Xu, R.; Tang, X. Nanoparticles Loaded with GSK1059615 Combined with Sorafenib Inhibited Programmed Cell Death 1 Ligand 1 Expression by Negatively Regulating the PI3K/Akt/NF-κB Pathway, Thereby Reversing the Drug Resistance of Hepatocellular Carcinoma to Sorafenib. J. Biomed. Nanotechnol. 2022, 18, 693–704. [Google Scholar] [CrossRef]
- Lv, W.; Wu, H.; Zhang, Y.; Li, H.; Shu, H.; Su, C.; Zhu, Y.; Wang, T.; Nie, F. cRGD-targeted gold-based nanoparticles overcome EGFR-TKI resistance of NSCLC via low-temperature photothermal therapy combined with sonodynamic therapy. Biomater. Sci. 2023, 11, 1677–1691. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Buhimschi, A.D.; Armstrong, H.A.; Toure, M.; Jaime-Figueroa, S.; Chen, T.L.; Lehman, A.M.; Woyach, J.A.; Johnson, A.J.; Byrd, J.C.; Crews, C.M. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 2018, 57, 3564–3575. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhao, X.; Ding, N.; Gao, H.; Wu, Y.; Yang, Y.; Zhao, M.; Hwang, J.; Song, Y.; Liu, W.; et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018, 28, 779–781. [Google Scholar] [CrossRef]
- Desai, A.; Lovly, C.M. Strategies to overcome resistance to ALK inhibitors in non-small cell lung cancer: A narrative review. Transl. Lung Cancer Res. 2023, 12, 615–628. [Google Scholar] [CrossRef]
- Yang, T.; Curtis, S.; Bai, A.; Young, A.; Derosier, D.; Ripley, S.; Bai, S. CRISPR/Cas9 targeting liposomes knocked down multidrug resistance proteins in brain endothelial cells as a model to predict potential pharmacoresistance. Colloids Surf. B Biointerfaces 2023, 222, 113103. [Google Scholar] [CrossRef]
- Huang, S.; Ma, Z.; Zhou, Q.; Wang, A.; Gong, Y.; Li, Z.; Wang, S.; Yan, Q.; Wang, D.; Hou, B.; et al. Genome-Wide CRISPR/Cas9 Library Screening Identified that DUSP4 Deficiency Induces Lenvatinib Resistance in Hepatocellular Carcinoma. Int. J. Biol. Sci. 2022, 18, 4357–4371. [Google Scholar] [CrossRef]
- Roy, S.K.; Srivastava, S.; Hancock, A.; Shrivastava, A.; Morvant, J.; Shankar, S.; Srivastava, R.K. Inhibition of ribosome assembly factor PNO1 by CRISPR/Cas9 technique suppresses lung adenocarcinoma and Notch pathway: Clinical application. J. Cell Mol. Med. 2023, 27, 365–378. [Google Scholar] [CrossRef]
- Qureshi, R.; Zou, B.; Alam, T.; Wu, J.; Lee, V.H.F.; Yan, H. Computational Methods for the Analysis and Prediction of EGFR-Mutated Lung Cancer Drug Resistance: Recent Advances in Drug Design, Challenges and Future Prospects. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 20, 238–255. [Google Scholar] [CrossRef]
- Huang, Y.Q.; Sun, P.; Chen, Y.; Liu, H.X.; Hao, G.F.; Song, B.A. Bioinformatics toolbox for exploring target mutation-induced drug resistance. Brief. Bioinform. 2023, 24, bbad033. [Google Scholar] [CrossRef]
- Fröhlich, F.; Gerosa, L.; Muhlich, J.; Sorger, P.K. Mechanistic model of MAPK signaling reveals how allostery and rewiring contribute to drug resistance. Mol. Syst. Biol. 2023, 19, e10988. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, G.; Li, W.; Turunen, M.; He, J.; Subramaniam, P.S.; Pampou, S.; Griffin, A.T.; Karan, C.; Kerwin, P.; Murray, D.; et al. Network-based elucidation of colon cancer drug resistance by phosphoproteomic time-series analysis. bioRxiv 2023. preprint. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, Y.; Li, H.; Zhou, Y.; Shi, S.; Chen, Z.; He, X.; Zhang, H.; Li, F.; Yin, J.; et al. DRESIS: The first comprehensive landscape of drug resistance information. Nucleic Acids Res. 2023, 51, D1263–D1275. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, Y.; Guo, L.; Liu, C.; Wang, P.; Ren, W. Exosomal microRNA-107 reverses chemotherapeutic drug resistance of gastric cancer cells through HMGA2/mTOR/P-gp pathway. BMC Cancer 2021, 21, 1290. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Chen, L.; Chen, R.; Xu, F.; Huang, Z.; Huang, R.; Wang, W.; Xu, Q. miR-4486 enhances cisplatin sensitivity of gastric cancer cells by restraining the JAK3/STAT3 signalling pathway. J. Chemother. 2022, 34, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Fang, Y.; Wang, X.; Han, Y.; Du, F.; Li, C.; Hu, H.; Liu, H.; Liu, Q.; Wang, J.; et al. miR-302a Inhibits Metastasis and Cetuximab Resistance in Colorectal Cancer by Targeting NFIB and CD44. Theranostics 2019, 9, 8409–8425. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Khan, F.; Upadhyay, T.K.; Seungjoon, M.; Park, M.N.; Kim, B. New insights about the PDGF/PDGFR signaling pathway as a promising target to develop cancer therapeutic strategies. Biomed. Pharmacother. 2023, 161, 114491. [Google Scholar] [CrossRef]
- Yang, Y.; Deng, Y.; Chen, X.; Zhang, J.; Chen, Y.; Li, H.; Wu, Q.; Yang, Z.; Zhang, L.; Liu, B. Inhibition of PDGFR by CP-673451 induces apoptosis and increases cisplatin cytotoxicity in NSCLC cells via inhibiting the Nrf2-mediated defense mechanism. Toxicol. Lett. 2018, 295, 88–98. [Google Scholar] [CrossRef]
- Yin, L.; He, J.; Xue, J.; Na, F.; Tong, R.; Wang, J.; Gao, H.; Tang, F.; Mo, X.; Deng, L.; et al. PDGFR-β inhibitor slows tumor growth but increases metastasis in combined radiotherapy and Endostar therapy. Biomed. Pharmacother. 2018, 99, 615–621. [Google Scholar] [CrossRef]
- Wong, J.P.; Todd, J.R.; Finetti, M.A.; McCarthy, F.; Broncel, M.; Vyse, S.; Luczynski, M.T.; Crosier, S.; Ryall, K.A.; Holmes, K.; et al. Dual Targeting of PDGFRα and FGFR1 Displays Synergistic Efficacy in Malignant Rhabdoid Tumors. Cell Rep. 2016, 17, 1265–1275. [Google Scholar] [CrossRef]
- Kun, E.; Tsang, Y.T.M.; Ng, C.W.; Gershenson, D.M.; Wong, K.K. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat. Rev. 2021, 92, 102137. [Google Scholar] [CrossRef] [PubMed]
- Dudka, W.; Hoser, G.; Mondal, S.S.; Turos-Korgul, L.; Swatler, J.; Kusio-Kobialka, M.; Wołczyk, M.; Klejman, A.; Brewinska-Olchowik, M.; Kominek, A.; et al. Targeting integrated stress response with ISRIB combined with imatinib treatment attenuates RAS/RAF/MAPK and STAT5 signaling and eradicates chronic myeloid leukemia cells. BMC Cancer 2022, 22, 1254. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Carlomagno, F.; Anaganti, S.; Guida, T.; Salvatore, G.; Troncone, G.; Wilhelm, S.M.; Santoro, M. BAY 43-9006 inhibition of oncogenic RET mutants. J. Natl. Cancer Inst. 2006, 98, 326–334. [Google Scholar] [CrossRef]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef]
- Strumberg, D.; Clark, J.W.; Awada, A.; Moore, M.J.; Richly, H.; Hendlisz, A.; Hirte, H.W.; Eder, J.P.; Lenz, H.J.; Schwartz, B. Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: A review of four phase I trials in patients with advanced refractory solid tumors. Oncologist 2007, 12, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Takayama, T.; Mazzaferro, V.; Chau, G.Y.; Yang, J.; Kudo, M.; Cai, J.; Poon, R.T.; Han, K.H.; Tak, W.Y.; et al. Adjuvant sorafenib for hepatocellular carcinoma after resection or ablation (STORM): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2015, 16, 1344–1354. [Google Scholar] [CrossRef]
- Tovar, V.; Cornella, H.; Moeini, A.; Vidal, S.; Hoshida, Y.; Sia, D.; Peix, J.; Cabellos, L.; Alsinet, C.; Torrecilla, S.; et al. Tumour initiating cells and IGF/FGF signalling contribute to sorafenib resistance in hepatocellular carcinoma. Gut 2017, 66, 530–540. [Google Scholar] [CrossRef]
- Wang, F.; Bank, T.; Malnassy, G.; Arteaga, M.; Shang, N.; Dalheim, A.; Ding, X.; Cotler, S.J.; Denning, M.F.; Nishimura, M.I.; et al. Inhibition of insulin-like growth factor 1 receptor enhances the efficacy of sorafenib in inhibiting hepatocellular carcinoma cell growth and survival. Hepatol. Commun. 2018, 2, 732–746. [Google Scholar] [CrossRef]
- Peng, S.W.; Ngo, M.T.; Kuo, Y.C.; Teng, M.H.; Guo, C.L.; Lai, H.C.; Chang, T.S.; Huang, Y.H. Niclosamide Revitalizes Sorafenib through Insulin-like Growth Factor 1 Receptor (IGF-1R)/Stemness and Metabolic Changes in Hepatocellular Carcinoma. Cancers 2023, 15, 931. [Google Scholar] [CrossRef]
- Chen, W.; Mook, R.A., Jr.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell Signal 2018, 41, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhou, X.; Xu, H.; Shi, X.; Zhao, J.; Yang, M.; Zhang, L.; Jin, X.; Hu, Y.; Li, X.; et al. Niclosamide Inhibits Cell Growth and Enhances Drug Sensitivity of Hepatocellular Carcinoma Cells via STAT3 Signaling Pathway. J. Cancer 2018, 9, 4150–4155. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lou, W.; Armstrong, C.; Zhu, Y.; Evans, C.P.; Gao, A.C. Niclosamide suppresses cell migration and invasion in enzalutamide resistant prostate cancer cells via Stat3-AR axis inhibition. Prostate 2015, 75, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.; Chen, Y.K.; Hsu, Y.J.; Lin, B.R. Niclosamide inhibits the cell proliferation and enhances the responsiveness of esophageal cancer cells to chemotherapeutic agents. Oncol. Rep. 2020, 43, 549–561. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interventions | NCT Number | Type of Cancer | Study Title |
|---|---|---|---|
| Targeting multiple RTKs | |||
| Famitinib (a RTKI against multiple targets, e.g., VEGFR 2/3, PDGFR and stem cell factor receptor (c-kit)). Sunitinib (a RTKI against multiple targets, e.g., VEGFR and PDGFR) | NCT04409223 | Gastrointestinal Stromal Tumours | Efficacy and Safety of Famitinib Versus Sunitinib in the Treatment of Advanced Gastrointestinal Stromal Tumour Patients After Failure of Imatinib |
| AL3818 (a RTKI against multiple targets, e.g., VEGFR, FGFR, PDGFR and c-kit). Paclitaxel, Pegylated Liposomal Doxorubicin (PLD), Topotecan, Carboplatin | NCT02584478 | Endometrial Carcinoma, Ovarian Carcinoma, Fallopian Tube Carcinoma, Primary Peritoneal Carcinoma, Cervical Carcinoma | Phase 1/2a/3 Evaluation of Adding AL3818 to Standard Platinum-Based Chemotherapy in Subjects With Recurrent or Metastatic Endometrial, Ovarian, Fallopian, Primary Peritoneal or Cervical Carcinoma (AL3818-US-002) |
| Regorafenib (a RTKI against multiple targets, e.g., VEGF1/2/3, PDGFR, FGFR, KIT, RET, RAF-1, BRAF) Nivolumab, Docetaxel, Paclitaxel, Irinotecan, Trifluridine/Tipracil | NCT04879368 | Gastro-Oesophageal Cancer | RegoNivo vs Standard of Care Chemotherapy in AGOC |
| Targeting EGFR | |||
| ASK120067 (Third generation EGFR TKI) Gefitinib (First generation EGFR TKI) ASK120067, Placebo Gefitinib 250 mg, Gefitinib, Placebo ASK120067 | NCT04143607 | Locally Advanced or Metastatic NSCLC | ASK120067 Versus Gefitinib as First-line Treatment for EGFRm Locally Advanced or Metastatic NSCLC |
| Aumolertinib (Third generation EGFR TKI) Osimertinib (Second generation EGFR TKI) Pemetrexed, Cisplatin, Carboplatin. Paclitaxel, Nab paclitaxel, Gemcitabine | NCT05493501 | NSCLC | Aumolertinib With Chemotherapy or Alone Compared With Osimertinib in Patients With Epidermal Growth Factor Receptor-Mutant Non-Small Cell Lung Cancer |
| Gefitinib (First generation EGFR TKI) Afatinib (Second generation ErbB family inhibitor Erlotinib (First generation EGFR TKI) Metformin Hydrochloride, Placebo | NCT05445791 | NSCLC | Metformin Plus Tyrosine Kinase Inhibitors for Treatment of Patients With Non-small Cell Lung Cancer With EGFR Mutations |
| Targeting EGFR and HER-2 | |||
| Pyrotinib (EGFR and HER2 inhibitor) Trastuzumab (HER2 inhibitor) | NCT05346861 | HER2-positive Breast Cancer, Metastatic Breast Cancer | Pyrotinib Rechallenge in Her2-positive Metastatic Breast Cancer Pretreated With Pyrotinib and Trastuzumab |
| Targeting EGFR and VEGFR | |||
| Gefitinib (First generation EGFR TKI) Apatinib (VEGFR-2 TKIs) Placebo | NCT02824458 | Non-Squamous NSCLC | A Study of Gefitinib With or Without Apatinib in Patients With Advanced Non-squamous Non-Small-Cell Lung Cancer Harboring EGFR Mutations |
| Osimertinib (Second generation EGFR TKI) Bevacizumab (anti-VEGF monoclonal antibody) | NCT04181060 | Advanced Lung Non-Squamous Non-Small Cell Carcinoma, Metastatic Lung Non-Squamous Non-Small Cell Carcinoma, Recurrent Lung Non-Squamous Non-Small Cell Carcinoma, Stage IIIB Lung Cancer AJCC v8, Stage IV Lung Cancer AJCC v8 | Osimertinib With or Without Bevacizumab as Initial Treatment for Patients With EGFR-Mutant Lung Cancer |
| Targeting BCR-ABL and JAK | |||
| Dasatinib (small-molecule, BCR-ABL inhibitor) Ruxolitinib (JAK inhibitor) Prednisone, Vincristine, Daunorubicin. Pegaspargase. Erwinase®, Cyclophosphamide, Cytarabine, Mercaptopurine, Methotrexate, Blinatumomab, Bortezomib, Dexamethasone, Doxorubicin, Etoposide, Clofarabine, Vorinostat. Idarubicin. Nelarabine, Thioguanine, Asparaginase Erwinia chrysanthemi (recombinant)-rywn, Calaspargase Pegol | NCT03117751 | Acute Lymphoblastic Leukemia, Acute Lymphoblastic Lymphoma | Total Therapy XVII for Newly Diagnosed Patients With Acute Lymphoblastic Leukemia and Lymphoma |
| Targeting VEGF pathway | |||
| IBI305 (anti-VEGF monoclonal antibody) Sintilimab, Pemetrexed, Cisplatin, Placebo1, Placebo2 | NCT03802240 | Non-Squamous NSCLC | Sintilimab ± IBI305 Plus Chemotherapy (Pemetrexed + Cisplatin) for EGFRm + Locally Advanced or Metastasis Non-Squamous NSCLC Patients After EGFR-TKI Treatment Failure |
| BD0801 (anti-VEGF monoclonal antibody) Paclitaxel, Placebo, Topotecan, doxorubicin liposome | NCT04908787 | Ovarian Cancer | A Phase III Study of BD0801 Combined With Chemotherapy in Recurrent, Platinum-resistant Epithelial Ovarian Cancer |
| Targeting mTOR | |||
| Everolimus (mTOR inhibitor) Letrozole, Bicalutamide, Itraconazole | NCT03458221 | Recurrent Ovarian Cancer, Signal Transduction Pathway Deregulation, Therapy-Associated Cancer | Signal TrAnsduction Pathway Activity Analysis in OVarian cancER |
| Targeting HER3 | |||
| Patritumab Deruxtecan (HER3-DXd antibody attached to topoisomerase I inhibitor) Platinum-based chemotherapy | NCT05338970 | Non-Squamous NSCLC, EGFR L858R, EGFR Exon 19 Deletion | HERTHENA-Lung02: A Study of Patritumab Deruxtecan Versus Platinum-based Chemotherapy in Metastatic or Locally Advanced EGFRm NSCLC After Failure of EGFR TKI Therapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bou Antoun, N.; Chioni, A.-M. Dysregulated Signalling Pathways Driving Anticancer Drug Resistance. Int. J. Mol. Sci. 2023, 24, 12222. https://doi.org/10.3390/ijms241512222
Bou Antoun N, Chioni A-M. Dysregulated Signalling Pathways Driving Anticancer Drug Resistance. International Journal of Molecular Sciences. 2023; 24(15):12222. https://doi.org/10.3390/ijms241512222
Chicago/Turabian StyleBou Antoun, Nauf, and Athina-Myrto Chioni. 2023. "Dysregulated Signalling Pathways Driving Anticancer Drug Resistance" International Journal of Molecular Sciences 24, no. 15: 12222. https://doi.org/10.3390/ijms241512222