
Whole Transcriptome Analysis of Substantia Nigra in Mice with MPTP-Induced Parkinsonism Bearing Defective Glucocerebrosidase Activity
 ,
, {kind=link}
{kind=link}
{kind=link}
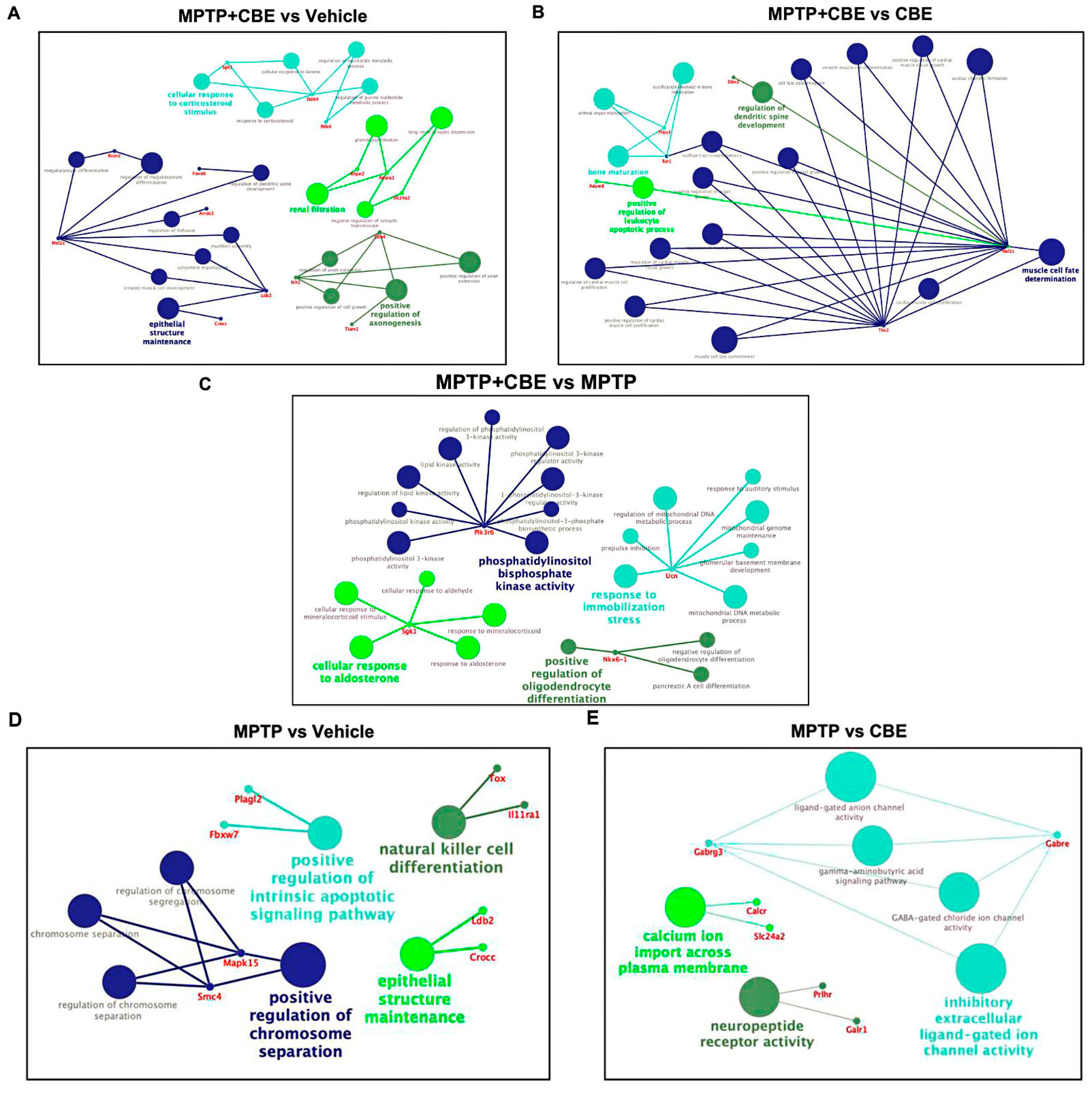{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Changes in the Transcriptome Attributed to the Dysfunction of GCase
2.2. Gene Expression Outliers Highlight Targeted Pathways in Cojoined Influence of MPTP and CBE
2.3. Overlapping Analysis of Enriched Pathways in MPTP-CBE Mice Model and Data Set of RNA-Seq Peripheral Blood Monocyte-Derived Macrophages from L444P/N GBA-PD Patients
3. Discussion
4. Materials and Methods
4.1. Brain Samples from Mice
4.2. RNA Isolation and RNA Sequencing (RNA-Seq)
4.3. Quality Control
4.4. Reads Mapping to Reference Genome
4.5. Quantification of Gene Expression Level
4.6. Analysis of Gene Differential Expression
4.7. GO Enrichment Analysis of Differentially Expressed Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, V.M.-Y.; Trojanowski, J.Q. Mechanisms of Parkinson’s Disease Linked to Pathological α-Synuclein: New Targets for Drug Discovery. Neuron 2006, 52, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Pu, J. Alpha-Synuclein in Parkinson’s Disease: From Pathogenetic Dysfunction to Potential Clinical Application. Park. Dis. 2016, 2016, 1720621. [Google Scholar] [CrossRef] [Green Version]
- Gan-Or, Z.; Giladi, N.; Rozovski, U.; Shifrin, C.; Rosner, M.S.; Gurevich, T.; Bar-Shira, A.; Orr-Urtreger, A. Genotype-Phenotype Correlations between GBA Mutations and Parkinson Disease Risk and Onset. Neurology 2008, 70, 2277–2283. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Anheim, M.; Condroyer, C.; Pollak, P.; Durif, F.; Dupuits, C.; Viallet, F.; Lohmann, E.; Corvol, J.-C.; Honoré, A.; et al. Large-Scale Screening of the Gaucher’s Disease-Related Glucocerebrosidase Gene in Europeans with Parkinson’s Disease. Hum. Mol. Genet. 2011, 20, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sekine, T.; Funayama, M.; Li, L.; Yoshino, H.; Nishioka, K.; Tomiyama, H.; Hattori, N. Clinicogenetic Study of GBA Mutations in Patients with Familial Parkinson’s Disease. Neurobiol. Aging 2014, 35, 935.e3–935.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emelyanov, A.K.; Usenko, T.S.; Tesson, C.; Senkevich, K.A.; Nikolaev, M.A.; Miliukhina, I.V.; Kopytova, A.E.; Timofeeva, A.A.; Yakimovsky, A.F.; Lesage, S.; et al. Mutation Analysis of Parkinson’s Disease Genes in a Russian Data Set. Neurobiol. Aging 2018, 71, 267.e7–267.e10. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, E.; Stubblefield, B.K.; Maniwang, E.; Lopez, G.; Moaven, N.; Goldin, E.; Marugan, J.; Patnaik, S.; Dutra, A.; Southall, N.; et al. Macrophage Models of Gaucher Disease for Evaluating Disease Pathogenesis and Candidate Drugs. Sci. Transl. Med. 2014, 6, 240ra73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pant, D.C.; Aguilera-Albesa, S.; Pujol, A. Ceramide Signalling in Inherited and Multifactorial Brain Metabolic Diseases. Neurobiol. Dis. 2020, 143, 105014. [Google Scholar] [CrossRef]
- Lai, M.; La Rocca, V.; Amato, R.; Freer, G.; Pistello, M. Sphingolipid/Ceramide Pathways and Autophagy in the Onset and Progression of Melanoma: Novel Therapeutic Targets and Opportunities. Int. J. Mol. Sci. 2019, 20, 3436. [Google Scholar] [CrossRef] [Green Version]
- Sabrin, A.; Stiban, J. Roles of Ceramides and Other Sphingolipids in Immune Cell Function and Inflammation. In The Role of Bioactive Lipids in Cancer, Inflammation and Related Diseases; Honn, K.V., Zeldin, D.C., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 169–191. ISBN 978-3-030-21735-8. [Google Scholar]
- Usenko, T.; Bezrukova, A.; Basharova, K.; Panteleeva, A.; Nikolaev, M.; Kopytova, A.; Miliukhina, I.; Emelyanov, A.; Zakharova, E.; Pchelina, S. Comparative Transcriptome Analysis in Monocyte-Derived Macrophages of Asymptomatic GBA Mutation Carriers and Patients with GBA-Associated Parkinson’s Disease. Genes 2021, 12, 1545. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Levy, O.A.; Waters, C.C.; Fahn, S.; Ford, B.; Kuo, S.H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase Activity in Parkinson’s Disease with and without GBA Mutations. Brain 2015, 138, 2648–2658. [Google Scholar] [CrossRef] [Green Version]
- Guedes, L.C.; Chan, R.B.; Gomes, M.A.; Conceição, V.A.; Machado, R.B.; Soares, T.; Xu, Y.; Gaspar, P.; Carriço, J.A.; Alcalay, R.N.; et al. Serum Lipid Alterations in GBA-Associated Parkinson’s Disease. Park. Relat. Disord. 2017, 44, 58–65. [Google Scholar] [CrossRef]
- Pchelina, S.; Emelyanov, A.; Baydakova, G.; Andoskin, P.; Senkevich, K.; Nikolaev, M.; Miliukhina, I.; Yakimovskii, A.; Timofeeva, A.; Fedotova, E.; et al. Oligomeric α-Synuclein and Glucocerebrosidase Activity Levels in GBA-Associated Parkinson’s Disease. Neurosci. Lett. 2017, 636, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Pchelina, S.; Baydakova, G.; Nikolaev, M.; Senkevich, K.; Emelyanov, A.; Kopytova, A.; Miliukhina, I.; Yakimovskii, A.; Timofeeva, A.; Berkovich, O.; et al. Blood Lysosphingolipids Accumulation in Patients with Parkinson’s Disease with Glucocerebrosidase 1 Mutations. Mov. Disord. 2018, 33, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Kopytova, A.E.; Usenko, T.S.; Baydakova, G.V.; Nikolaev, M.A.; Senkevich, K.A.; Izyumchenko, A.D.; Tyurin, A.A.; Miliukhina, I.V.; Emelyanov, A.K.; Zakharova, E.Y.; et al. Could Blood Hexosylsphingosine Be a Marker for Parkinson’s Disease Linked with GBA1 Mutations? Mov. Disord. 2022, 37, 1779–1781. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in Humans Due to a Product of Meperidine-Analog Synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Manchado, A.B.; Villadiego, J.; Romo-Madero, S.; Suárez-Luna, N.; Bermejo-Navas, A.; Rodríguez-Gómez, J.A.; Garrido-Gil, P.; Labandeira-García, J.L.; Echevarría, M.; López-Barneo, J.; et al. Chronic and Progressive Parkinson’s Disease MPTP Model in Adult and Aged Mice. J. Neurochem. 2016, 136, 373–387. [Google Scholar] [CrossRef] [Green Version]
- Mustapha, M.; Mat Taib, C.N. MPTP-Induced Mouse Model of Parkinson’s Disease: A Promising Direction of Therapeutic Strategies. Biomol. Biomed. 2021, 21, 422–433. [Google Scholar] [CrossRef]
- Meredith, G.; Rademacher, D. MPTP Mouse Models of Parkinson’s Disease: An Update. J. Park. Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef]
- Kanfer, J.N.; Legler, G.; Sullivan, J.; Raghavan, S.S.; Mumford, R.A. The Gaucher Mouse. Biochem. Biophys. Res. Commun. 1975, 67, 85–90. [Google Scholar] [CrossRef]
- Mus, L.; Siani, F.; Giuliano, C.; Ghezzi, C.; Cerri, S.; Blandini, F. Development and Biochemical Characterization of a Mouse Model of Parkinson’s Disease Bearing Defective Glucocerebrosidase Activity. Neurobiol. Dis. 2019, 124, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Ugrumov, M.V.; Khaindrava, V.G.; Kozina, E.A.; Kucheryanu, V.G.; Bocharov, E.V.; Kryzhanovsky, G.N.; Kudrin, V.S.; Narkevich, V.B.; Klodt, P.M.; Rayevsky, K.S.; et al. Modeling of Presymptomatic and Symptomatic Stages of Parkinsonism in Mice. Neuroscience 2011, 181, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Pchelina, S.N.; Bezrukova, A.I.; Rudenok, M.M.; Zhuravlev, A.S.; Rybolovlev, I.N.; Baydakova, G.V.; Nikolaev, M.A.; Nesterov, M.S.; Abaimov, D.A.; Usenko, T.S.; et al. Biochemical characterization of double toxic MPTP/CBE presymptomatic Parkison like phenotype in mice. NeuroToxicology 2023. submitted. [Google Scholar]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alieva, A.K.; Filatova, E.V.; Kolacheva, A.A.; Rudenok, M.M.; Slominsky, P.A.; Ugrumov, M.V.; Shadrina, M.I. Transcriptome Profile Changes in Mice with MPTP-Induced Early Stages of Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 6775–6784. [Google Scholar] [CrossRef]
- Alieva, A.K.; Zyrin, V.S.; Rudenok, M.M.; Kolacheva, A.A.; Shulskaya, M.V.; Ugryumov, M.V.; Slominsky, P.A.; Shadrina, M.I. Whole-Transcriptome Analysis of Mouse Models with MPTP-Induced Early Stages of Parkinson’s Disease Reveals Stage-Specific Response of Transcriptome and a Possible Role of Myelin-Linked Genes in Neurodegeneration. Mol. Neurobiol. 2018, 55, 7229–7241. [Google Scholar] [CrossRef]
- Vardi, A.; Zigdon, H.; Meshcheriakova, A.; Klein, A.D.; Yaacobi, C.; Eilam, R.; Kenwood, B.M.; Rahim, A.A.; Massaro, G.; Merrill, A.H., Jr.; et al. Delineating Pathological Pathways in a Chemically Induced Mouse Model of Gaucher Disease. J. Pathol. 2016, 239, 496–509. [Google Scholar] [CrossRef]
- Vardi, A.; Ben-Dor, S.; Cho, S.M.; Kalinke, U.; Spanier, J.; Futerman, A.H. Mice Defective in Interferon Signaling Help Distinguish between Primary and Secondary Pathological Pathways in a Mouse Model of Neuronal Forms of Gaucher Disease. J. Neuroinflammation 2020, 17, 265. [Google Scholar] [CrossRef]
- Xu, Y.-H.; Jia, L.; Quinn, B.; Zamzow, M.; Stringer, K.; Aronow, B.; Sun, Y.; Zhang, W.; Setchell, K.D.R.; Grabowski, G.A. Global Gene Expression Profile Progression in Gaucher Disease Mouse Models. BMC Genom. 2011, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Boddupalli, C.S.; Nair, S.; Belinsky, G.; Gans, J.; Teeple, E.; Nguyen, T.-H.; Mehta, S.; Guo, L.; Kramer, M.L.; Ruan, J.; et al. Neuroinflammation in Neuronopathic Gaucher Disease: Role of Microglia and NK Cells, Biomarkers, and Response to Substrate Reduction Therapy. Elife 2022, 11, e79830. [Google Scholar] [CrossRef] [PubMed]
- Millington, G.W.M. The Role of Proopiomelanocortin (POMC) Neurones in Feeding Behaviour. Nutr. Metab. 2007, 4, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riboldi, G.M.; Vialle, R.A.; Navarro, E.; Udine, E.; de Paiva Lopes, K.; Humphrey, J.; Allan, A.; Parks, M.; Henderson, B.; Astudillo, K.; et al. Transcriptome Deregulation of Peripheral Monocytes and Whole Blood in GBA-Related Parkinson’s Disease. Mol. Neurodegener. 2022, 17, 52. [Google Scholar] [CrossRef] [PubMed]
- Daniel, N.H.; Aravind, A.; Thakur, P. Are Ion Channels Potential Therapeutic Targets for Parkinson’s Disease? Neurotoxicology 2021, 87, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.P.; Bano, S.; Sen, S.; Suchal, K.; Kumar, S.; Nikolajeff, F.; Dey, S.K.; Sharma, V. Altered Neural Cell Junctions and Ion-Channels Leading to Disrupted Neuron Communication in Parkinson’s Disease. NPJ Park. Dis. 2022, 8, 66. [Google Scholar] [CrossRef]
- Miliukhina, I.V.; Usenko, T.S.; Senkevich, K.A.; Nikolaev, M.A.; Timofeeva, A.A.; Agapova, E.A.; Semenov, A.V.; Lubimova, N.E.; Totolyan, A.A.; Pchelina, S.N. Plasma Cytokines Profile in Patients with Parkinson’s Disease Associated with Mutations in GBA Gene. Bull. Exp. Biol. Med. 2020, 168, 423–426. [Google Scholar] [CrossRef]
- Pandey, M.K.; Burrow, T.A.; Rani, R.; Martin, L.J.; Witte, D.; Setchell, K.D.; Mckay, M.A.; Magnusen, A.F.; Zhang, W.; Liou, B.; et al. Complement Drives Glucosylceramide Accumulation and Tissue Inflammation in Gaucher Disease. Nature 2017, 543, 108–112. [Google Scholar] [CrossRef]
- Nagata, M.; Izumi, Y.; Ishikawa, E.; Kiyotake, R.; Doi, R.; Iwai, S.; Omahdi, Z.; Yamaji, T.; Miyamoto, T.; Bamba, T.; et al. Intracellular Metabolite β-Glucosylceramide Is an Endogenous Mincle Ligand Possessing Immunostimulatory Activity. Proc. Natl. Acad. Sci. USA 2017, 114, E3285–E3294. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. ER Stress and the Unfolded Protein Response. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- Omura, T.; Kaneko, M.; Okuma, Y.; Matsubara, K.; Nomura, Y. Endoplasmic Reticulum Stress and Parkinson’s Disease: The Role of HRD1 in Averting Apoptosis in Neurodegenerative Disease. Oxid. Med. Cell. Longev. 2013, 2013, 239854. [Google Scholar] [CrossRef] [Green Version]
- da Costa, C.A.; El Manaa, W.; Duplan, E.; Checler, F. The Endoplasmic Reticulum Stress/Unfolded Protein Response and Their Contributions to Parkinson’s Disease Physiopathology. Cells 2020, 9, 2495. [Google Scholar] [CrossRef]
- Smith, L.; Schapira, A.H. GBA Variants and Parkinson Disease: Mechanisms and Treatments. Cells 2022, 11, 1261. [Google Scholar] [CrossRef]
- Maor, G.; Rencus-Lazar, S.; Filocamo, M.; Steller, H.; Segal, D.; Horowitz, M. Unfolded Protein Response in Gaucher Disease: From Human to Drosophila. Orphanet J. Rare Dis. 2013, 8, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurzawa-Akanbi, M.; Hanson, P.; Blain, P.; Lett, D.; Mckeith, I.; Chinnery, P.; Morris, C. Glucocerebrosidase Mutations Alter the Endoplasmic Reticulum and Lysosomes in Lewy Body Disease. J. Neurochem. 2012, 123, 298–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöndorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. IPSC-Derived Neurons from GBA1-Associated Parkinson’s Disease Patients Show Autophagic Defects and Impaired Calcium Homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martinez, A.; Beavan, M.; Gegg, M.E.; Chau, K.-Y.; Whitworth, A.J.; Schapira, A.H.V. Parkinson Disease-Linked GBA Mutation Effects Reversed by Molecular Chaperones in Human Cell and Fly Models. Sci. Rep. 2016, 6, 31380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, H.J.R.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s IPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, S.-H.; Tasset, I.; Cheng, M.M.; Diaz, A.; Pan, M.-K.; Lieberman, O.J.; Hutten, S.J.; Alcalay, R.N.; Kim, S.; Ximénez-Embún, P.; et al. Mutant Glucocerebrosidase Impairs α-Synuclein Degradation by Blockade of Chaperone-Mediated Autophagy. Sci. Adv. 2022, 8, eabm6393. [Google Scholar] [CrossRef] [PubMed]
- Usenko, T.S.; Bezrukova, A.I.; Bogdanova, D.A.; Nikolaev, M.A.; Miliukhina, I.V.; Gracheva, E.V.; Senkevich, K.A.; Zakharova, E.Y.; Emelyanov, A.K.; Pchelina, S.N. Gene Expression of Lysosomal Membrane Proteins in Parkinson Disease, Associated with Mutations in the Glucocerebrosidase Gene (Gba). Ann. Clin. Exp. Neurol. 2020, 14, 43–49. [Google Scholar] [CrossRef]
- Heras-Sandoval, D.; Pérez-Rojas, J.M.; Hernández-Damián, J.; Pedraza-Chaverri, J. The Role of PI3K/AKT/MTOR Pathway in the Modulation of Autophagy and the Clearance of Protein Aggregates in Neurodegeneration. Cell Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef]
- Xu, F.; Na, L.; Li, Y.; Chen, L. RETRACTED ARTICLE: Roles of the PI3K/AKT/MTOR Signalling Pathways in Neurodegenerative Diseases and Tumours. Cell Biosci. 2020, 10, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, S.; Lim, S. Acupuncture Inhibits the Increase in Alpha-Synuclein by Modulating SGK1 in an MPTP Induced Parkinsonism Mouse Model. Am. J. Chin. Med. 2019, 47, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Gao, Y.; Yan, H.; Liu, G.; Zhou, Y.; Tao, T.; Yue, T.; Pang, C.; Chen, X.; Gao, S.; et al. PDK4 Decrease Neuronal Apoptosis via Inhibiting ROS-ASK1/P38 Pathway in Early Brain Injury after Subarachnoid Hemorrhage. Antioxid. Redox Signal. 2021, 36, 505–524. [Google Scholar] [CrossRef]
- Gao, X.; Gao, Y.-Y.; Wu, L.-Y.; Peng, Z.; Liu, X.-Z.; Chen, X.-X.; Gao, S.; Zhang, H.-S.; Lu, Y.; Hang, C.-H.; et al. High Expression of PDK4 Could Play a Potentially Protective Role by Attenuating Oxidative Stress after Subarachnoid Hemorrhage. J. Clin. Med. 2022, 11, 3974. [Google Scholar] [CrossRef]
- Gong, J.; Zhang, L.; Zhang, Q.; Li, X.; Xia, X.-J.; Liu, Y.-Y.; Yang, Q.-S. Lentiviral Vector-Mediated SHC3 Silencing Exacerbates Oxidative Stress Injury in Nigral Dopamine Neurons by Regulating the PI3K-AKT-FoxO Signaling Pathway in Rats with Parkinson’s Disease. Cell Physiol. Biochem. 2018, 49, 971–984. [Google Scholar] [CrossRef]
- Castel, P.; Ellis, H.; Bago, R.; Toska, E.; Razavi, P.; Carmona, F.J.; Kannan, S.; Verma, C.S.; Dickler, M.; Chandarlapaty, S.; et al. PDK1-SGK1 Signaling Sustains AKT-Independent MTORC1 Activation and Confers Resistance to PI3Kα Inhibition. Cancer Cell 2016, 30, 229–242. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Chen, X.; Wang, Y.; Peng, H.; Wang, Y.; Jing, Y.; Zhang, H. PDK4 Promotes Tumorigenesis Through Activation of CREB-RHEB-MTORC1 Signaling Cascade. J. Biol. Chem. 2014, 289, 29739–29749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Sisqués, L.; Sancho-Balsells, A.; Solana-Balaguer, J.; Campoy-Campos, G.; Vives-Isern, M.; Soler-Palazón, F.; Anglada-Huguet, M.; López-Toledano, M.-Á.; Mandelkow, E.-M.; Alberch, J.; et al. RTP801/REDD1 Contributes to Neuroinflammation Severity and Memory Impairments in Alzheimer’s Disease. Cell Death Dis. 2021, 12, 616. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Hu, Y.; Wang, L.; Zhang, D.; Wang, H.; Dai, Y.; Cui, X.; Zheng, G. DDIT4 Mediates the Proliferation-Promotive Effect of IL-34 in Human Monocytic Leukemia Cells. Blood Sci. 2021, 3, 48–56. [Google Scholar] [CrossRef]
- Malagelada, C.; Ryu, E.; Biswas, S.; Jackson-Lewis, V.; Greene, L. RTP801 Is Elevated in Parkinson Brain Substantia Nigral Neurons and Mediates Death in Cellular Models of Parkinson’s Disease by a Mechanism Involving Mammalian Target of Rapamycin Inactivation. J. Neurosci. 2006, 26, 9996–10005. [Google Scholar] [CrossRef] [Green Version]
- Verma, A.; Kommaddi, R.P.; Gnanabharathi, B.; Hirsch, E.C.; Ravindranath, V. Genes Critical for Development and Differentiation of Dopaminergic Neurons Are Downregulated in Parkinson’s Disease. J. Neural. Transm. 2023, 130, 495–512. [Google Scholar] [CrossRef] [PubMed]
- Li, C.C.; Wu, T.S.; Huang, C.F.; Jang, L.T.; Liu, Y.T.; You, S.T.; Liou, G.G.; Lee, F.J.S. GTP-Binding-Defective ARL4D Alters Mitochondrial Morphology and Membrane Potential. PLoS ONE 2012, 7, e43552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Weng, H.-Y.; Tang, X.-P.; Lin, Y.; Yuan, Y.; Li, Q.; Tang, Z.; Wu, H.-B.; Yang, S.; Li, Y.; et al. ARL4C Stabilized by AKT/MTOR Pathway Promotes the Invasion of PTEN-Deficient Primary Human Glioblastoma. J. Pathol. 2019, 247, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Li, X.; Lai, K.O. Activity and Function of the Prmt8 Protein Arginine Methyltransferase in Neurons. Life 2021, 11, 1132. [Google Scholar] [CrossRef]
- Bogetofte, H.; Ryan, B.J.; Jensen, P.; Schmidt, S.I.; Vergoossen, D.L.E.; Barnkob, M.B.; Kiani, L.N.; Chughtai, U.; Heon-Roberts, R.; Caiazza, M.C.; et al. Post-Translational Proteomics Platform Identifies Neurite Outgrowth Impairments in Parkinson’s Disease GBA-N370S Dopamine Neurons. Cell Rep. 2023, 42, 112180. [Google Scholar] [CrossRef] [PubMed]
- Grigor’eva, E.V.; Kopytova, A.E.; Yarkova, E.S.; Pavlova, S.V.; Sorogina, D.A.; Malakhova, A.A.; Malankhanova, T.B.; Baydakova, G.V.; Zakharova, E.Y.; Medvedev, S.P.; et al. Biochemical Characteristics of IPSC-Derived Dopaminergic Neurons from N370S GBA Variant Carriers with and without Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 4437. [Google Scholar] [CrossRef]
- Mubariz, F.; Saadin, A.; Lingenfelter, N.; Sarkar, C.; Banerjee, A.; Lipinski, M.M.; Awad, O. Deregulation of MTORC1-TFEB Axis in Human IPSC Model of GBA1-Associated Parkinson’s Disease. Front. Neurosci. 2023, 17, 1152503. [Google Scholar] [CrossRef]
- Balestrino, R.; Tunesi, S.; Tesei, S.; Lopiano, L.; Zecchinelli, A.L.; Goldwurm, S. Penetrance of Glucocerebrosidase (GBA) Mutations in Parkinson’s Disease: A Kin Cohort Study. Mov. Disord. 2020, 35, 2111–2114. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Dion, P.; Rouleau, G. Genetic Perspective on the Role of the Autophagy-Lysosome Pathway in Parkinson Disease. Autophagy 2015, 11, 1443–1457. [Google Scholar] [CrossRef] [Green Version]
- Kinghorn, K.; Asghari, A.; Castillo-Quan, J. The Emerging Role of Autophagic-Lysosomal Dysfunction in Gaucher Disease and Parkinson’s Disease. Neural Regen. Res. 2017, 12, 380–384. [Google Scholar] [CrossRef]
- Zhang, Z.-N.; Hui, Z.; Chen, C.; Liang, Y.; Tang, L.-L.; Wang, S.-L.; Xu, C.-C.; Yang, H.; Zhao, Y.; Zhang, J.-S. Mechanism of Autophagy Regulation in MPTP-Induced PD Mice via the MTOR Signaling Pathway by Echinacoside. Neuropsychiatr. Dis. Treat. 2021, 17, 1397–1411. [Google Scholar] [CrossRef]
- Wang, X.-W.; Yuan, L.-J.; Yang, Y.; Zhang, M.; Chen, W.-F. IGF-1 Inhibits MPTP/MPP +-Induced Autophagy on Dopaminergic Neurons through the IGF-1R/PI3K-Akt-MTOR Pathway and GPER. Am. J. Physiol.-Endocrinol. Metab. 2020, 319, E734–E743. [Google Scholar] [CrossRef]
- Deng, H.; Ma, Z. Protective Effects of Berberine against MPTP-Induced Dopaminergic Neuron Injury through Promoting Autophagy in Mice. Food Funct. 2021, 12, 8366–8375. [Google Scholar] [CrossRef]
- Rudenok, M.M.; Alieva, A.K.; Nikolaev, M.A.; Kolacheva, A.A.; Ugryumov, M.V.; Pchelina, S.N.; Slominsky, P.A.; Shadrina, M.I. Possible Involvement of Genes Related to Lysosomal Storage Disorders in the Pathogenesis of Parkinson’s Disease. Mol. Biol. 2019, 53, 24–31. [Google Scholar] [CrossRef]
- Lenihan, J.A.; Saha, O.; Heimer-McGinn, V.; Cryan, J.F.; Feng, G.; Young, P.W. Decreased Anxiety-Related Behaviour but Apparently Unperturbed NUMB Function in Ligand of NUMB Protein-X (LNX) 1/2 Double Knockout Mice. Mol. Neurobiol. 2017, 54, 8090–8109. [Google Scholar] [CrossRef] [PubMed]
- Kovalev, G.I.; Vasileva, E.V.; Salimov, R.M. Comparison of Mouse Behavior in the Open Field, Closed and Elevated Cross-Maze Tests by the Use of Factor Analysi. Zhurnal Vyss. Nervn. Deyatelnosti Im. I.P. Pavlov. 2019, 69, 123–130. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality Control of RNA-Seq Experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 5 June 2023).
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python Framework to Work with High-Throughput Sequencing Data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape Plug-in to Decipher Functionally Grouped Gene Ontology and Pathway Annotation Networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape Plugin: Pathway Insights Using Integrated Experimental and in Silico Data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef] [Green Version]
- Chi, J.-H.; Panner, A.; Cachola, K.; Crane, C.-A.; Murray, J.; Pieper, R.-O.; James, C.-D.; Parsa, A.-T. Increased expression of the glioma-associated antigen ARF4L after loss of the tumor suppressor PTEN. Laboratory investigation. J. Neurosurg. 2008, 108, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Wang, J.; Jones, K.-T.; Ives, H.-E.; Feldman, M.-E.; Yao, L.J.; Shokat, K.-M.; Ashrafi, K.; Pearce, D. mTOR complex-2 activates ENaC by phosphorylating SGK1. J. Am. Soc. Nephrol. 2010, 21, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, L.; Hou, X.; Zhang, Z.; Zhou, X.; Gao, W. Role of Autophagy Mediated by AMPK/DDiT4/mTOR Axis in HT22 Cells Under Oxygen and Glucose Deprivation/Reoxygenation. ACS Omega 2023, 8, 9221–9229. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Shin, Y.-K.; Yang, X.; Zehr, B.; Chakrabarti, P.; Kandror, K.-V. 4E-BPs Control Fat Storage by Regulating the Expression of Egr1 and ATGL. J. Biol. Chem. 2015, 290, 17331–17338. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.-A. mTOR signaling: New networks for ALL. Blood 2016, 127, 2658–2659. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhou, J.-Y.; Kho, D.; Reiners, J.-J., Jr.; Wu, G.S. Role for DUSP1 (dual-specificity protein phosphatase 1) in the regulation of autophagy. Autophagy 2016, 12, 1791–1803. [Google Scholar] [CrossRef]
- Pinno, J.; Bongartz, H.; Klepsch, O.; Wundrack, N.; Poli, V.; Schaper, F.; Dittrich, A. Interleukin-6 influences stress-signalling by reducing the expression of the mTOR-inhibitor REDD1 in a STAT3-dependent manner. Cell Signal 2016, 28, 907–916. [Google Scholar] [CrossRef]
- Mahajan, S.; Saini, A.; Chandra, V.; Nanduri, R.; Kalra, R.; Bhagyaraj, E.; Khatri, N.; Gupta, P. Nuclear Receptor Nr4a2 Promotes Alternative Polarization of Macrophages and Confers Protection in Sepsis. J. Biol. Chem. 2015, 290, 18304–18314. [Google Scholar] [CrossRef] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usenko, T.; Bezrukova, A.; Rudenok, M.M.; Basharova, K.; Shadrina, M.I.; Slominsky, P.A.; Zakharova, E.; Pchelina, S. Whole Transcriptome Analysis of Substantia Nigra in Mice with MPTP-Induced Parkinsonism Bearing Defective Glucocerebrosidase Activity. Int. J. Mol. Sci. 2023, 24, 12164. https://doi.org/10.3390/ijms241512164
Usenko T, Bezrukova A, Rudenok MM, Basharova K, Shadrina MI, Slominsky PA, Zakharova E, Pchelina S. Whole Transcriptome Analysis of Substantia Nigra in Mice with MPTP-Induced Parkinsonism Bearing Defective Glucocerebrosidase Activity. International Journal of Molecular Sciences. 2023; 24(15):12164. https://doi.org/10.3390/ijms241512164
Chicago/Turabian StyleUsenko, Tatiana, Anastasia Bezrukova, Margarita M. Rudenok, Katerina Basharova, Maria I. Shadrina, Petr A. Slominsky, Ekaterina Zakharova, and Sofya Pchelina. 2023. "Whole Transcriptome Analysis of Substantia Nigra in Mice with MPTP-Induced Parkinsonism Bearing Defective Glucocerebrosidase Activity" International Journal of Molecular Sciences 24, no. 15: 12164. https://doi.org/10.3390/ijms241512164