

Challenges of Diagnosing Mendelian Susceptibility to Mycobacterial Diseases in South Africa

 , , , and
, , , and
Abstract
:1. Introduction
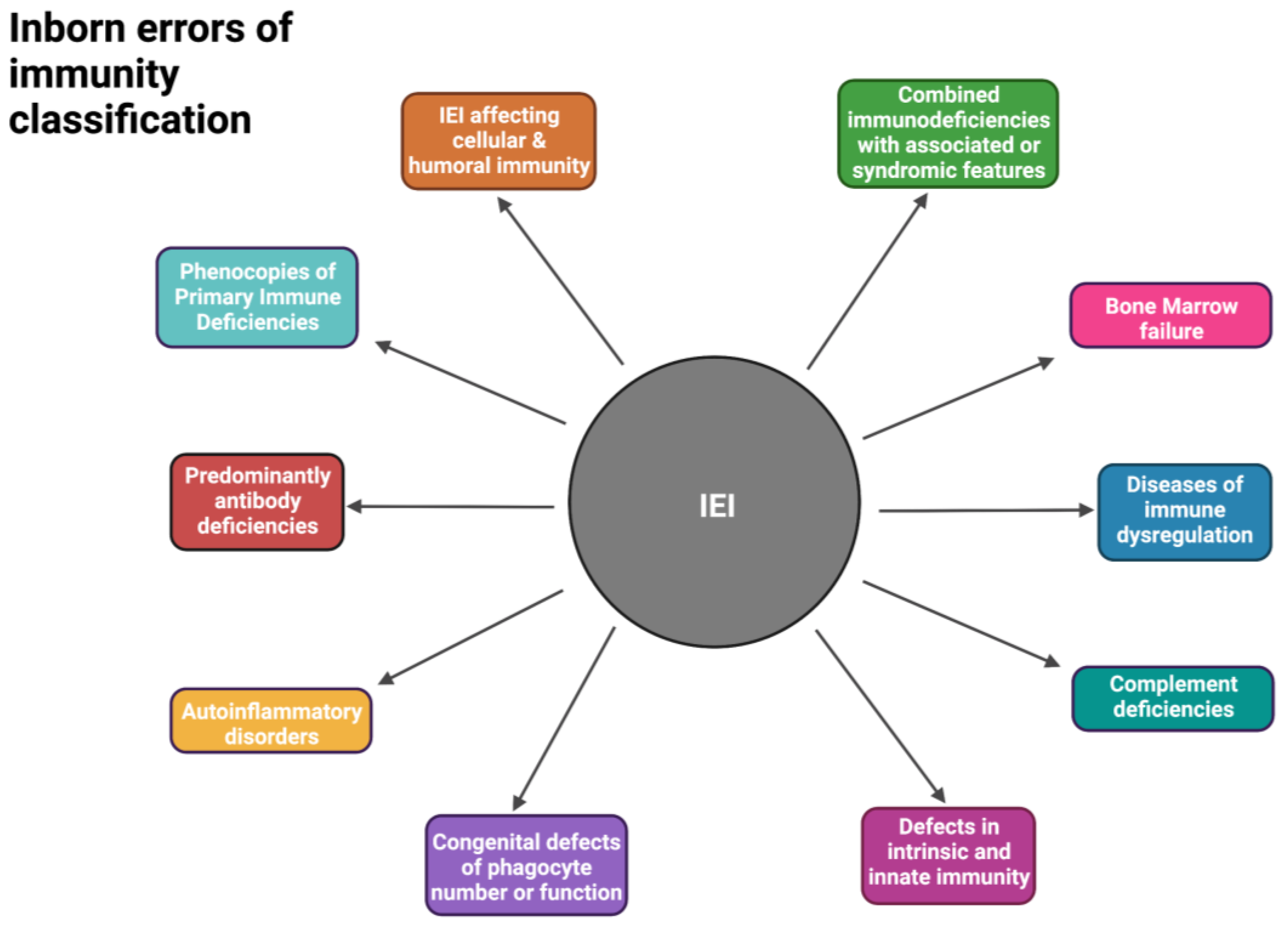
2. Inborn Errors of Immunity in South Africa
2.1. Susceptibility to Mycobacterial Disease
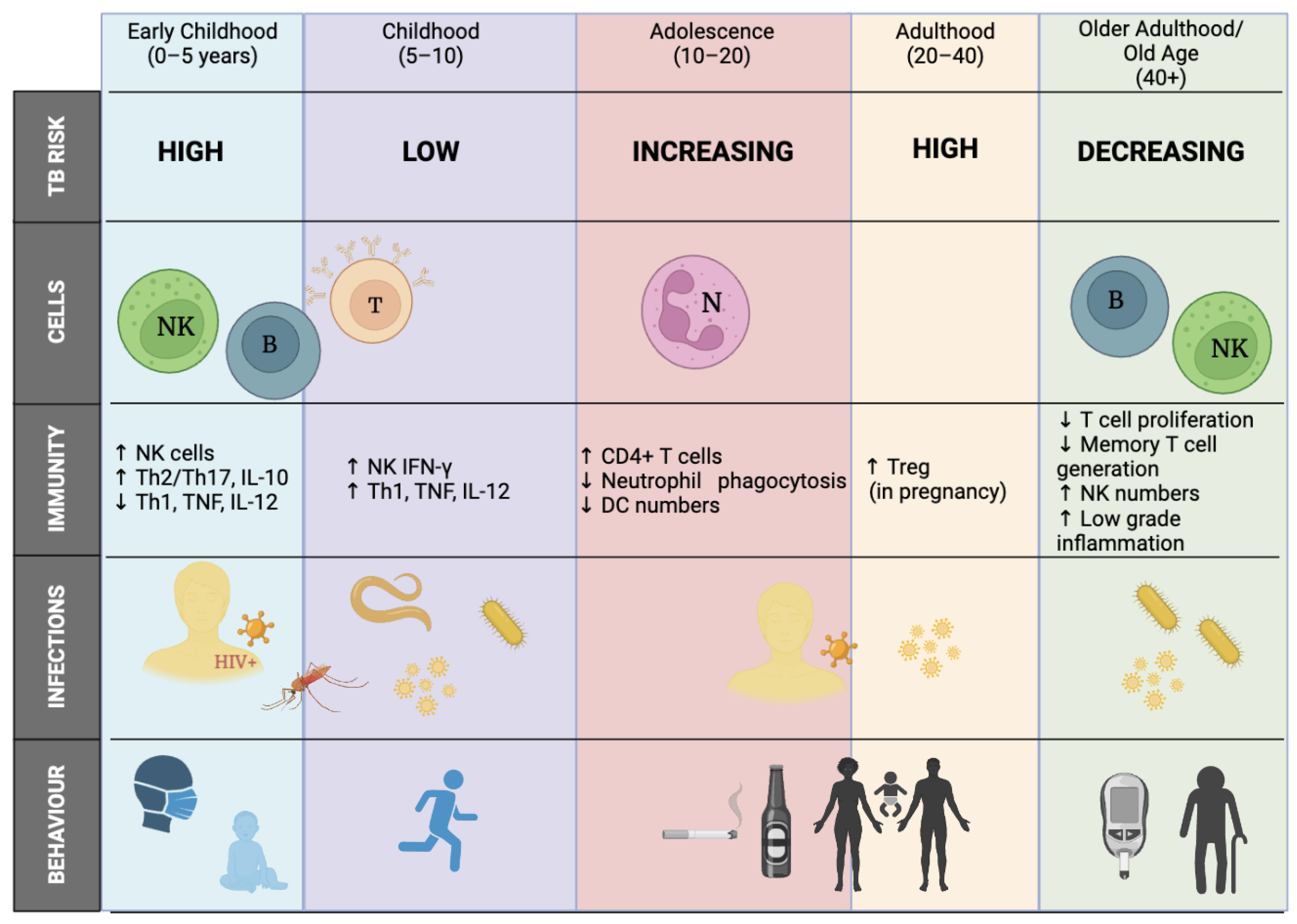
2.1.1. Age/Sex Influence (the Wonder Years)
2.1.2. BCG-Vaccine Strategy in TB-Endemic Countries
3. Mendelian Susceptibility to Mycobacterial Diseases
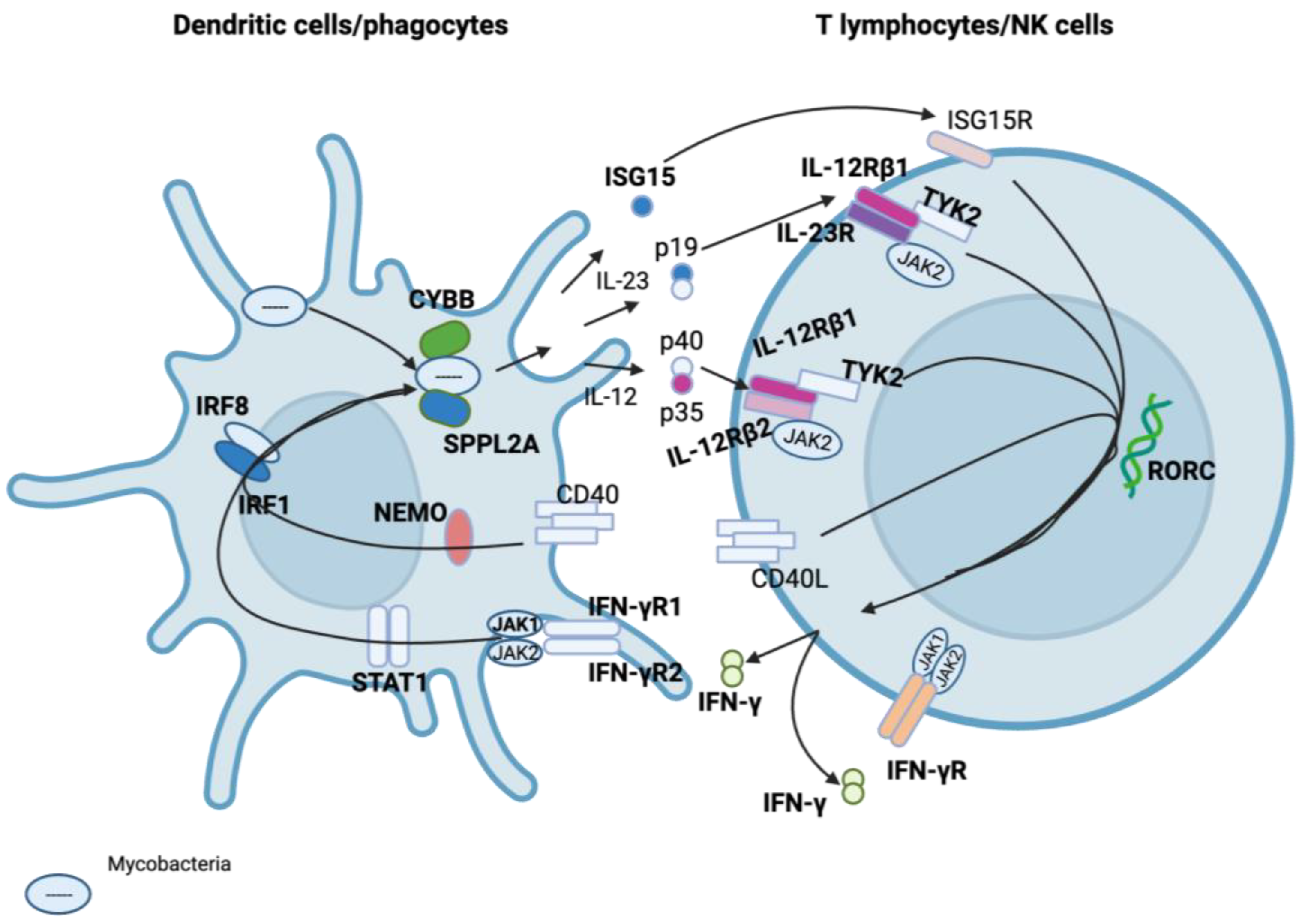
3.1. Genes Involved in MSMD
3.2. Clinical Features and Diagnosis
3.2.1. Consanguinity
3.2.2. Effectiveness of BCG Vaccinations
3.2.3. Inheritance of Maternal Immunity
3.2.4. Challenges of Diagnosing MSMD in an Endemic-TB Setting
3.3. Genetic Approaches to Molecular Diagnosis
3.3.1. NGS and Variants of Unknown Significance (VUS)
3.3.2. Under-Representation in the Human Reference Genome
3.3.3. Lack of Transcriptomics as a Diagnostic Tool
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2020, 40, 24–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eley, B.; Esser, M. Investigation and management of primary immunodeficiency in South African children. S. Afr. Med. J. 2014, 104, 793. [Google Scholar] [CrossRef]
- Delmonte, O.M.; Castagnoli, R.; Calzoni, E.; Notarangelo, L.D. Inborn Errors of Immunity with Immune Dysregulation: From Bench to Bedside. Front. Pediatr. 2019, 1, 353. [Google Scholar] [CrossRef] [Green Version]
- Bigley, T.M.; Cooper, M.A. Monogenic autoimmunity and infectious diseases: The double-edged sword of immune dysregulation. Curr. Opin. Immunol. 2021, 72, 230–238. [Google Scholar] [CrossRef]
- Rey-Jurado, E.; Poli, M.C. Functional genetics in inborn errors of immunity. Future Rare Dis. 2021, 1, FRD11. [Google Scholar] [CrossRef]
- Staels, F.; Collignon, T.; Betrains, A.; Gerbaux, M.; Willemsen, M.; Humblet-Baron, S.; Liston, A.; Vanderschueren, S.; Schrijvers, R. Monogenic Adult-Onset Inborn Errors of Immunity. Front. Immunol. 2021, 12, 753978. [Google Scholar] [CrossRef]
- Van Coller, A.; Glanzmann, B.; Cornelissen, H.; Möller, M.; Kinnear, C.; Esser, M.; Glashoff, R. Phenotypic and immune functional profiling of patients with suspected Mendelian Susceptibility to Mycobacterial Disease in South Africa. BMC Immunol. 2021, 22, 62. [Google Scholar] [CrossRef] [PubMed]
- Glanzmann, E.; Riniker, P. Essential lymphocytophthisis, new clinical aspect of infant pathology. Ann. Paediatr. 1950, 175, 1–32. [Google Scholar]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Cunningham-Rundles, C.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; Oksenhendler, E.; Picard, C.; et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2022, 42, 1473–1507. [Google Scholar] [CrossRef] [PubMed]
- Casanova, J.-L.; Conley, M.E.; Seligman, S.J.; Abel, L.; Notarangelo, L.D. Guidelines for genetic studies in single patients: Lessons from primary immunodeficiencies. J. Exp. Med. 2014, 211, 2137–2149. [Google Scholar] [CrossRef]
- Kwok, A.J.; Mentzer, A.; Knight, J.C. Host genetics and infectious disease: New tools, insights and translational opportunities. Nat. Rev. Genet. 2021, 22, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Boisson-Dupuis, S.; Bustamante, J. Mycobacterial diseases in patients with inborn errors of immunity. Curr. Opin. Immunol. 2021, 72, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Erjaee, A.; Bagherpour, M.; Van Rooyen, C.; van den Berg, S.; Kinnear, C.J.; Green, R.J.; Pepper, M.S. Primary immunodeficiency in Africa—A review. South Afr. Med. J. 2019, 109, 3. [Google Scholar] [CrossRef] [PubMed]
- Glanzmann, B.; Uren, C.; de Villiers, N.; van Coller, A.; Glashoff, R.H.; Urban, M.; Hoal, E.G.; Esser, M.M.; Möller, M.; Kinnear, C.J. Primary immunodeficiency diseases in a tuberculosis endemic region: Challenges and opportunities. Genes Immun. 2019, 20, 447–454. [Google Scholar] [CrossRef]
- Prokofjeva, T.; Lucane, Z.; Kovalova, Z.; Kurjane, N. Inborn Errors of Immunity in Latvia: Analysis of Data from 1994 to 2020. J. Clin. Immunol. 2022, 42, 876–879. [Google Scholar] [CrossRef]
- Bousfiha, A.A.; Jeddane, L.; Ailal, F.; Benhsaien, I.; Mahlaoui, N.; Casanova, J.-L.; Abel, L. Primary Immunodeficiency Diseases Worldwide: More Common than Generally Thought. J. Clin. Immunol. 2013, 33, 1–7. [Google Scholar] [CrossRef]
- Joshi, A.Y.; Iyer, V.N.; Hagan, J.B.; Sauver, J.L.S.; Boyce, T.G. Incidence and Temporal Trends of Primary Immunodeficiency: A Population-Based Cohort Study. Mayo Clin. Proc. 2009, 84, 16–22. [Google Scholar] [CrossRef]
- Boyle, J.M.; Buckley, R.H. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J. Clin. Immunol. 2007, 27, 497–502. [Google Scholar] [CrossRef] [Green Version]
- Moodley, S.; Goddard, E.; Levin, M.; Scott, C.; van Eyssen, A.; Davidson, A.; de Decker, R.; Wilmshurst, J.M.; Spitaels, A.; Eley, B. A retrospective description of primary immunodeficiency diseases at Red Cross War Memorial Children’s Hospital, Cape Town, South Africa, 1975–2017. S. Afr. Med. J. 2020, 110, 197. [Google Scholar] [CrossRef]
- Goda, R.; Sobh, A.; Nermeen, G.; Radwan, N.; Barbouche, M.R.; Ben-Mustapha, I.; Bousfiha, A.; Jeddane, L.; Mahlaoui, N.; Elfeky, R. African Society for Immunodeciency (Asid) Guidelines for Diagnosis and Management of Inborn Errors of Immunity in Africa: Core Concept, Development and Initial Results. 2022. Available online: https://www.researchsquare.com/article/rs-2235434/v1 (accessed on 25 June 2023).
- Aghamohammadi, A.; Rezaei, N.; Yazdani, R.; Delavari, S.; Kutukculer, N.; Topyildiz, E.; Ozen, A.; Baris, S.; Karakoc-Aydiner, E.; Kilic, S.S.; et al. Consensus Middle East and North Africa Registry on Inborn Errors of Immunity. J. Clin. Immunol. 2021, 41, 1339–1351. [Google Scholar] [CrossRef]
- Abolhassani, H.; Azizi, G.; Sharifi, L.; Yazdani, R.; Mohsenzadegan, M.; Delavari, S.; Sohani, M.; Shirmast, P.; Chavoshzadeh, Z.; Mahdaviani, S.A.; et al. Global systematic review of primary immunodeficiency registries. Expert. Rev. Clin. Immunol. 2020, 16, 717–732. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Global TB Report; WHO: Geneva, Switzerland, 2020; Volume 2020. [Google Scholar]
- Boutayeb, A. The Impact of Infectious Diseases on the Development of Africa. In Handbook of Disease Burdens and Quality of Life Measures; Springer: New York, NY, USA, 2010; pp. 1171–1188. [Google Scholar]
- Nkengasong, J.N.; Tessema, S.K. Africa Needs a New Public Health Order to Tackle Infectious Disease Threats. Cell 2020, 183, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Organization WHO. Global Tuberculosis Report; WHO: Geneva, Switzerland, 2022. [Google Scholar]
- Campbell, M.C.; Tishkoff, S.A. African Genetic Diversity: Implications for Human Demographic History, Modern Human Origins, and Complex Disease Mapping. Annu. Rev. Genomics Hum. Genet. 2008, 9, 403–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, D.C.; Steyl, C.; Scholtz, D.; Baker, B.; Abdullah, I.; Uren, C.; Möller, M. African Genetic Representation in the Context of SARS-CoV-2 Infection and COVID-19 Severity. Front. Genet. 2022, 13, 909117. [Google Scholar] [CrossRef]
- Uren, C.; Möller, M.; van Helden, P.D.; Henn, B.M.; Hoal, E.G. Population structure and infectious disease risk in southern Africa. Mol. Genet. Genomics 2017, 292, 499–509. [Google Scholar] [CrossRef]
- Wiener, E.K.; Buchanan, J.; Krause, A.; Lombard, Z. Retrospective file review shows limited genetic services fail most patients—An argument for the implementation of exome sequencing as a first-tier test in resource-constrained settings. Orphanet J. Rare Dis. 2023, 18, 81. [Google Scholar] [CrossRef]
- Bousfiha, A.A.; Jeddane, L.; El Hafidi, N.; Benajiba, N.; Rada, N.; El Bakkouri, J.; Kili, A.; Benmiloud, S.; Benhsaien, I.; Faiz, I.; et al. First report on the Moroccan registry of primary immunodeficiencies: 15 Years of experience (1998–2012). J. Clin. Immunol. 2014, 34, 459–468. [Google Scholar] [CrossRef]
- Cornelissen, H.M.; Glanzmann, B.; Van Coller, A.; Engelbrecht, C.; Abraham, D.R.; Reddy, K.; Möller, M.; Kinnear, C.; Glashoff, R.H.; Esser, M. Mendelian susceptibility to mycobacterial disease in tuberculosis-hyperendemic South Africa. S. Afr. Med. J. 2021, 111, 998. [Google Scholar] [CrossRef]
- Quinn, J.; Modell, V.; Orange, J.S.; Modell, F. Growth in diagnosis and treatment of primary immunodeficiency within the global Jeffrey Modell Centers Network. Allergy Asthma Clin. Immunol. 2022, 18, 19. [Google Scholar] [CrossRef]
- Leiding, J.W.; Holland, S.M. Chronic Granulomatous Disease; Academic Press: Cambridge, MA, USA, 1993. [Google Scholar]
- Carranza, C.; Pedraza-Sanchez, S.; de Oyarzabal-Mendez, E.; Torres, M. Diagnosis for Latent Tuberculosis Infection: New Alternatives. Front. Immunol. 2020, 11, 2006. [Google Scholar] [CrossRef]
- Menzies, N.A.; Swartwood, N.; Testa, C.; Malyuta, Y.; Hill, A.N.; Marks, S.M.; Cohen, T.; Salomon, J.A. Time Since Infection and Risks of Future Disease for Individuals with Mycobacterium tuberculosis Infection in the United States. Epidemiology 2021, 32, 70–78. [Google Scholar] [CrossRef]
- Roya-Pabon, C.L.; Perez-Velez, C.M. Tuberculosis exposure, infection and disease in children: A systematic diagnostic approach. Pneumonia 2016, 8, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, L.; Fellay, J.; Haas, D.W.; Schurr, E.; Srikrishna, G.; Urbanowski, M.; Chaturvedi, N.; Srinivasan, S.; Johnson, D.H.; Bishai, W.R. Genetics of human susceptibility to active and latent tuberculosis: Present knowledge and future perspectives HHS Public Access. Lancet Infect. Dis. 2018, 18, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, G.; Murray, M.; Winthrop, K.; Centis, R.; Sotgiu, G.; Migliori, G.B.; Maeurer, M.; Zumla, A. Risk factors associated with pulmonary tuberculosis: Smoking, diabetes and anti-TNFα drugs. Curr. Opin. Pulm. Med. 2012, 18, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.A.; Chiang, S.S.; Esmail, H.; Coussens, A.K. The Wonder Years: What Can Primary School Children Teach Us About Immunity to Mycobacterium tuberculosis? Front. Immunol. 2018, 9, 2946. [Google Scholar] [CrossRef] [PubMed]
- Middelkoop, K.; Bekker, L.-G.; Myer, L.; Dawson, R.; Wood, R. Rates of tuberculosis transmission to children and adolescents in a community with high adult HIV prevalence. Clin. Infect. Dis. 2008, 47, 349–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcaïs, A.; Fieschi, C.; Abel, L.; Casanova, J.-L. Tuberculosis in children and adults. J. Exp. Med. 2005, 202, 1617–1621. [Google Scholar] [CrossRef]
- Snow, K.J.; Sismanidis, C.; Denholm, J.; Sawyer, S.M.; Graham, S. The incidence of tuberculosis among adolescents and young adults: A global estimate. Eur. Respir. J. 2018, 51, 1702352. [Google Scholar] [CrossRef] [Green Version]
- Upton, C.M.; Van Wijk, R.C.; Mockeliunas, L.; Simonsson, U.S.; McHarry, K.; van den Hoogen, G.; Muller, C.; von Delft, A.; van der Westhuizen, H.M.; Van Crevel, R.; et al. Safety and efficacy of BCG re-vaccination in relation to COVID-19 morbidity in healthcare workers: A double-blind, randomised, controlled, phase 3 trial. EClinicalMedicine 2022, 48, 101414. [Google Scholar] [CrossRef]
- Hesseling, A.C.; Caldwell, J.; Cotton, M.F.; Eley, B.S.; Jaspan, H.B.; Jennings, K.; Marais, B.J.; Nuttall, J.; Rabie, H.; Roux, P.; et al. BCG vaccination in South African HIV-exposed infants—Risks and benefits. South Afr. Med. J. 2009, 99, 88–91. [Google Scholar]
- Clark, M.; Cameron, D.W. The benefits and risks of bacille Calmette-Guérin vaccination among infants at high risk for both tuberculosis and severe combined immunodeficiency: Assessment by Markov model. BMC Pediatr. 2006, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Marciano, B.E.; Huang, C.Y.; Joshi, G.; Rezaei, N.; Carvalho, B.C.; Allwood, Z.; Ikinciogullari, A.; Reda, S.M.; Gennery, A.; Thon, V.; et al. BCG vaccination in patients with severe combined immunodeficiency: Complications, risks, and vaccination policies. J. Allergy Clin. Immunol. 2014, 133, 1134–1141. [Google Scholar] [CrossRef] [Green Version]
- Bustamante, J.; Boisson-Dupuis, S.; Abel, L.; Casanova, J.L. Mendelian susceptibility to mycobacterial disease: Genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin. Immunol. 2014, 26, 454–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adebamowo, S.N.; Francis, V.; Tambo, E.; Diallo, S.H.; Landouré, G.; Nembaware, V.; Dareng, E.; Muhamed, B.; Odutola, M.; Akeredolu, T.; et al. Implementation of genomics research in Africa: Challenges and recommendations. Glob. Health Action. 2018, 11, 1419033. [Google Scholar] [CrossRef] [Green Version]
- Mahdaviani, S.A.; Mansouri, D.; Jamee, M.; Zaki-Dizaji, M.; Aghdam, K.R.; Mortaz, E.; Khorasanizadeh, M.; Eskian, M.; Movahedi, M.; Ghaffaripour, H.; et al. Mendelian Susceptibility to Mycobacterial Disease (MSMD): Clinical and Genetic Features of 32 Iranian Patients. J. Clin. Immunol. 2020, 40, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Liu, X.-H.; Yuan, Y.; Lowrie, D.B.; Fan, X.-Y.; Li, T.; Hu, Z.-D.; Lu, S.-H. An Updated Review on MSMD Research Globally and A Literature Review on the Molecular Findings, Clinical Manifestations, and Treatment Approaches in China. Front. Immunol. 2022, 13, 926781. [Google Scholar] [CrossRef]
- Bandari, A.K.; Muthusamy, B.; Bhat, S.; Govindaraj, P.; Rajagopalan, P.; Dalvi, A.; Shankar, S.; Raja, R.; Reddy, K.S.; Madkaikar, M.; et al. A Novel Splice Site Mutation in IFNGR2 in Patients with Primary Immunodeficiency Exhibiting Susceptibility to Mycobacterial Diseases. Front. Immunol. 2019, 10, 1964. [Google Scholar] [CrossRef]
- Markle, J.G.; Martĺnez-Barricarte, R.; Ma, C.S.; Deenick, E.K.; Ramírez-Alejo, N.; Mele, F.; Latorre, D.; Mahdaviani, S.A.; Aytekin, C.; Mansouri, D.; et al. Human IFN-γ immunity to mycobacteria is governed by both IL-12 and IL-23. Sci. Immunol. 2018, 3, eaau6759. [Google Scholar]
- Rosain, J.; Kong, X.-F.; Martinez-Barricarte, R.; Oleaga-Quintas, C.; Ramirez-Alejo, N.; Markle, J.; Okada, S.; Boisson-Dupuis, S.; Casanova, J.; Bustamante, J. Mendelian susceptibility to mycobacterial disease: 2014–2018 update. Immunol. Cell Biol. 2019, 97, 360–367. [Google Scholar] [CrossRef]
- Casanova, J.-L. Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc. Natl. Acad. Sci. USA 2015, 112, E7128–E7137. [Google Scholar] [CrossRef]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef] [Green Version]
- Esteve-Solé, A.; Sologuren, I.; Martínez-Saavedra, M.T.; Deyà-Martínez, A.; Oleaga-Quintas, C.; Martinez-Barricarte, R.; Martin-Nalda, A.; Juan, M.; Casanova, J.-L.; Rodriguez-Gallego, C.; et al. Laboratory evaluation of the IFN-γ circuit for the molecular diagnosis of Mendelian susceptibility to mycobacterial disease. Crit. Rev. Clin. Lab. Sci. 2018, 55, 184–204. [Google Scholar] [CrossRef]
- Errami, A.; El Baghdadi, J.; Ailal, F.; Benhsaien, I.; Ouazahrou, K.; Abel, L.; Casanova, J.-L.; Boisson-Dupuis, S.; Bustamante, J.; Bousfiha, A.A. Mendelian susceptibility to mycobacterial disease: An overview. Egypt. J. Med. Hum. Genet. 2023, 24, 7. [Google Scholar] [CrossRef]
- Bousfiha, A.; Jeddane, L.; Picard, C.; Al-Herz, W.; Ailal, F.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J. Clin. Immunol. 2020, 40, 66–81. [Google Scholar] [CrossRef] [Green Version]
- Errami, A.; Baghdadi, J.E.; Ailal, F.; Benhsaien, I.; Bakkouri, J.E.; Jeddane, L.; Rada, N.; Benajiba, N.; Mokhantar, K.; Ouazahrou, K.; et al. Mendelian Susceptibility to Mycobacterial Disease (MSMD): Clinical, immunological and genetic features of 22 Patients from 15 Moroccan kindreds. J. Clin. Immunol. 2022, 43, 728–740. [Google Scholar] [CrossRef]
- Angelidou, A.; Conti, M.-G.; Diray-Arce, J.; Benn, C.S.; Shann, F.; Netea, M.G.; Liu, M.; Potluri, L.P.; Sanchez-Schmitz, G.; Husson, R.; et al. Licensed Bacille Calmette-Guérin (BCG) formulations differ markedly in bacterial viability, RNA content and innate immune activation. Vaccine 2020, 38, 2229–2240. [Google Scholar] [CrossRef]
- Mahomed, H.; Kibel, M.; Hawkridge, T.; Schaaf, H.S.; Hanekom, W.A.; Iloni, K.; Michaels, D.; Workman, L.; Verver, S.; Geiter, L.; et al. The Impact of a Change in Bacille Calmette-Guérin Vaccine Policy on Tuberculosis Incidence in Children in Cape Town, South Africa. Pediatr. Infect. Dis. J. 2006, 25, 1167–1172. [Google Scholar] [CrossRef]
- Jethwa, H.; Wong, R.; Abraham, S. COVID-19 vaccine trials: Ethnic diversity and immunogenicity. Vaccine 2021, 39, 3541–3543. [Google Scholar] [CrossRef]
- Edwards, K.M. Maternal antibodies and infant immune responses to vaccines. Vaccine 2015, 33, 6469–6472. [Google Scholar] [CrossRef] [Green Version]
- Cinicola, B.; Conti, M.G.; Terrin, G.; Sgrulletti, M.; Elfeky, R.; Carsetti, R.; Salinas, A.F.; Mortari, E.P.; Brindisi, G.; de Curtis, M.; et al. The Protective Role of Maternal Immunization in Early Life. Front. Pediatr. 2021, 9, 638871. [Google Scholar] [CrossRef]
- Esser, M.M.; Potter, P.; Nortje, R. Meeting the needs of primary immunodeficiency patients in South Africa—Some findings from the South African registry summary. Curr. Allergy Clin. Immunol. 2016, 29, 56–61. [Google Scholar]
- Kampmann, B.; Hemingway, C.; Stephens, A.; Davidson, R.; Goodsall, A.; Anderson, S.; Nicol, M.; Schölvinck, E.; Relman, D.; Waddell, S.; et al. Acquired predisposition to mycobacterial disease due to autoantibodies to IFN-γ. J. Clin. Investig. 2005, 115, 2480–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lye, J.J.; Williams, A.; Baralle, D. Exploring the RNA Gap for Improving Diagnostic Yield in Primary Immunodeficiencies. Front. Genet. 2019, 10, 1204. [Google Scholar] [CrossRef] [PubMed]
- Meyts, I.; Bosch, B.; Bolze, A.; Boisson, B.; Itan, Y.; Belkadi, A.; Pedergnana, V.; Moens, L.; Picard, C.; Cobat, A.; et al. Exome and genome sequencing for inborn errors of immunity. J. Allergy Clin. Immunol. 2016, 138, 957–969. [Google Scholar] [CrossRef] [Green Version]
- Schwarze, K.; Buchanan, J.; Taylor, J.C.; Wordsworth, S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet. Med. 2018, 20, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Hansen, M.E.B.; Tishkoff, S.A. Advances in integrative African genomics. Trends Genet. 2022, 38, 152–168. [Google Scholar] [CrossRef]
- Omotoso, O.E.; Teibo, J.O.; Atiba, F.A.; Oladimeji, T.; Adebesin, A.O.; Babalghith, A.O. Bridging the genomic data gap in Africa: Implications for global disease burdens. Glob. Health 2022, 18, 103. [Google Scholar] [CrossRef]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef]
- Swart, Y.; van Eeden, G.; Sparks, A.; Uren, C.; Möller, M. Prospective avenues for human population genomics and disease mapping in southern Africa. Mol. Genet. Genomics 2020, 295, 1079–1089. [Google Scholar] [CrossRef]
- Gurdasani, D.; Carstensen, T.; Tekola-Ayele, F.; Pagani, L.; Tachmazidou, I.; Hatzikotoulas, K.; Karthikeyan, S.; Iles, L.; Pollard, M.O.; Choudhury, A.; et al. The African Genome Variation Project shapes medical genetics in Africa. Nature 2015, 517, 327–332. [Google Scholar] [CrossRef] [Green Version]
- Tetikol, H.S.; Turgut, D.; Narci, K.; Budak, G.; Kalay, O.; Arslan, E.; Demirkaya-Budak, S.; Dolgoborodov, A.; Kabakci-Zorlu, D.; Semenyuk, V.; et al. Pan-African genome demonstrates how population-specific genome graphs improve high-throughput sequencing data analysis. Nat. Commun. 2022, 13, 4384. [Google Scholar] [CrossRef]
- Adepoju, P. Tackling Africa’s underrepresentation in genomics studies. Nat. Afr. 2022. Available online: https://www.nature.com/articles/d44148-022-00051-6 (accessed on 23 June 2023).
- Nica, A.C.; Dermitzakis, E.T. Expression quantitative trait loci: Present and future. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairfax, B.P.; Humburg, P.; Makino, S.; Naranbhai, V.; Wong, D.; Lau, E.; Jostins, L.; Plant, K.; Andrews, R.; McGee, C.; et al. Innate Immune Activity Conditions the Effect of Regulatory Variants upon Monocyte Gene Expression. Science 2014, 343, 1246949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piasecka, B.; Duffy, D.; Urrutia, A.; Quach, H.; Patin, E.; Posseme, C.; Bergstedt, J.; Charbit, B.; Rouilly, V.; MacPherson, C.R.; et al. Distinctive roles of age, sex, and genetics in shaping transcriptional variation of human immune responses to microbial challenges. Proc. Natl. Acad. Sci. USA 2018, 115, E488–E497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Both, U.; Kaforou, M.; Levin, M.; Newton, S.M. Understanding immune protection against tuberculosis using RNA expression profiling. Vaccine 2015, 33, 5289–5293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billington, C.; Toles, O.; Ebens, C.; Johnson, A.; Pozos, T.; Binstadt, B.; Albert, F.; Thielen, B. Transcriptomic Approaches to Diagnosis of Inborn Errors of Immunity. Clin. Immunol. 2023, 250. [Google Scholar] [CrossRef]
- Bougarn, S.; Boughorbel, S.; Chaussabel, D.; Marr, N. A curated transcriptome dataset collection to investigate inborn errors of immunity. F1000Research 2019, 8, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | MSMD-Causing Mutation Effect |
|---|---|
| IL12RB1 | Impaired IFN-γ production |
| IL12RB2 | Impaired IFN-γ production |
| IL12B | Impaired IFN-γ production |
| IFNGR1 | Impaired antimicrobial, antiviral, and anti-tumour responses, impaired cellular response to IFN-γ |
| IFNGR2 | Impaired antimicrobial, antiviral, and anti-tumour responses, impaired cellular response to IFN-γ |
| STAT1 | Defective type I and type II IFN responses |
| CYBB | Oxidative burst defects in macrophages |
| IRF8 | Loss of myeloid dendritic cells |
| TYK2 | Dysregulation of essential signalling for Type I and Type II cytokines |
| ISG15 | Impaired IFN-γ production |
| RORC | Disruption of IL-27/IFN-γ immunity |
| NEMO | Impaired T-cell response and CD40-dependent production of IL-12 |
| SPPL2A | Dysfunction of antigen-presenting cells |
| JAK1 | Dysfunctional INF-α, IL-6/10, and IL-12 signalling |
| IL23R | Dysfunction in the receptor of Type I and Type II cytokines |
| TBX21 | Th1 cell-specific transcription factor that functions as a regulator of IFN-γ expression |
| ZNFX1 | Can initiate IFN expression following exposure to viruses |
| PDCD1 | Immune-inhibitory receptor expressed in activated T cells |
| IFNG | Codes for IFN-γ used for communication between cells to trigger the protective defences of the immune system that help eradicate pathogens |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scholtz, D.; Jooste, T.; Möller, M.; van Coller, A.; Kinnear, C.; Glanzmann, B. Challenges of Diagnosing Mendelian Susceptibility to Mycobacterial Diseases in South Africa. Int. J. Mol. Sci. 2023, 24, 12119. https://doi.org/10.3390/ijms241512119
Scholtz D, Jooste T, Möller M, van Coller A, Kinnear C, Glanzmann B. Challenges of Diagnosing Mendelian Susceptibility to Mycobacterial Diseases in South Africa. International Journal of Molecular Sciences. 2023; 24(15):12119. https://doi.org/10.3390/ijms241512119
Chicago/Turabian StyleScholtz, Denise, Tracey Jooste, Marlo Möller, Ansia van Coller, Craig Kinnear, and Brigitte Glanzmann. 2023. "Challenges of Diagnosing Mendelian Susceptibility to Mycobacterial Diseases in South Africa" International Journal of Molecular Sciences 24, no. 15: 12119. https://doi.org/10.3390/ijms241512119