
Inhibition of Topoisomerases by Metal Thiosemicarbazone Complexes

Abstract
:1. Introduction
2. Thiosemicarbazones as Inhibitors of Topoisomerases
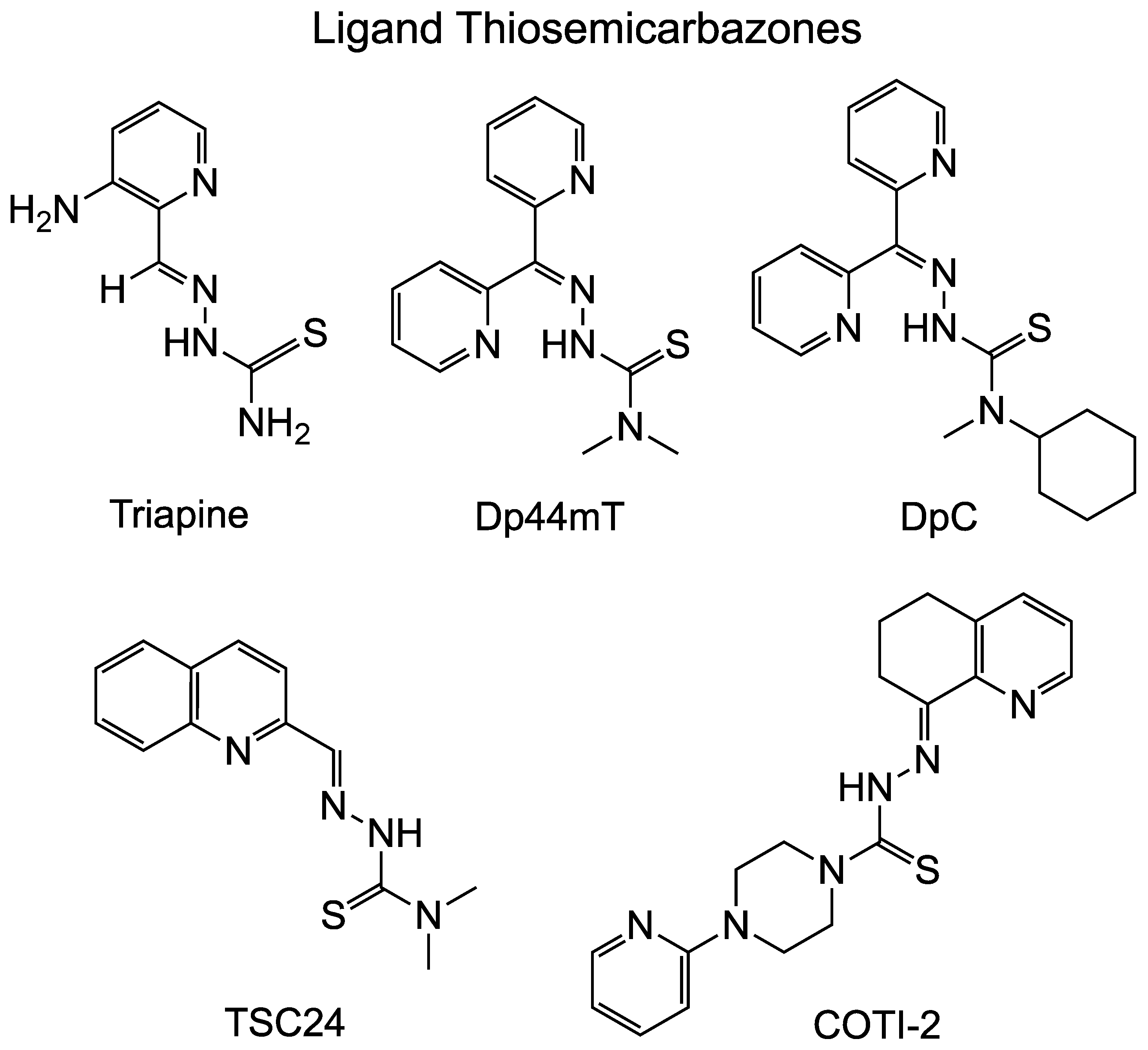
2.1. TSC Ligand or bis(TSC) Ligand Inhibition of Topoisomerases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Inhibition of Top | Reference |
|---|---|---|
| TSC24 | Inhibits Top2α DNA relaxation at 25 µM Inhibits Top2 at decatenation at 100 µM | [27] |
| Dp44mT | Inhibits human Top2α in relaxation assay with 5′ labeled 161-bp fragment from pBluescript SK phagemid DNA Does not inhibit human Top2β or human TopoI | [28] |
| Dp44mT | Does not inhibit Top2α in decatenation Does not increase cleavage complex by human Top2α | [26] |
| Triapine | ||
| Compound 2b | Inhibits human Top2α weakly at 100 µM | [29] |
| Compound 3a | Inhibit human Top2α-mediated DNA relaxation at 100 µM | [30] |
| Compound 3h | ||
| Triapine | Do not inhibit Top2α-mediated DNA relaxation at 50 µM | [25] |
| Dp44mT | ||
| Compound 24 | Do not inhibit isolated Top2 from L1210 | [12] |
| Compound 36 | ||
| NQTS | Does not inhibit Top2α | [36] |
| HFp4mT, HFp4pyrrT | Inhibit Top2α at 100 µM | [37] |
| HFp4eT, HFp4ipT, HFp4alT, HAp4mT, HAp4-eT, HFp4bzT, HFpT, HAp4alT, and HApz4mT | Do not inhibit Top2α at 100 µM | [37] |
| BZP–TSC ligands series | Inhibits Top2α slightly at 50 µM | [20] |
| ATZ ligand series | Do not inhibit Top2α at 10 µM, do not increase cleavage complex | [33] |
| BZP ligands series | ||
| APY | Does not inhibit Top2α at 100 µM | [32] |
| APY | Does not inhibit Top2β at 200 µM | [34] |
| APZ | Inhibits Top2α at 100 µM | [32] |
| BZP | Inhibits Top2β at 200 µM weakly | [34] |
| HPyCT4BrPh | Does not inhibit human Top1B at 50 µM | [35] |
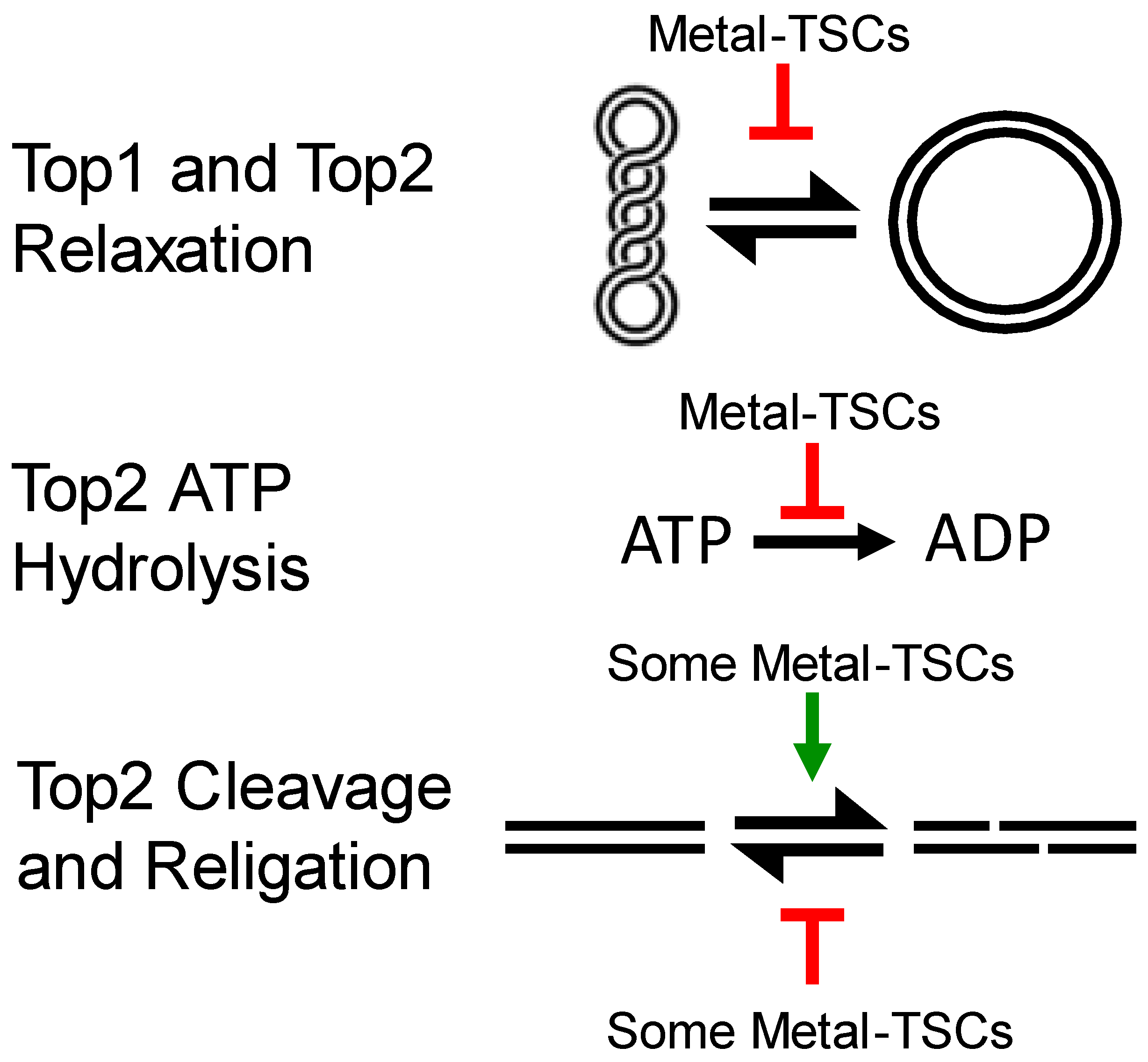
2.2. Metal–TSC or Metal–Bis(TSC) Inhibition of Topoisomerases
2.2.1. Inhibition of Type I Top
2.2.2. Inhibition of Type II Top
- Ni chelated with bis(TSC)
- 2.
- Cu-chelated TSCs
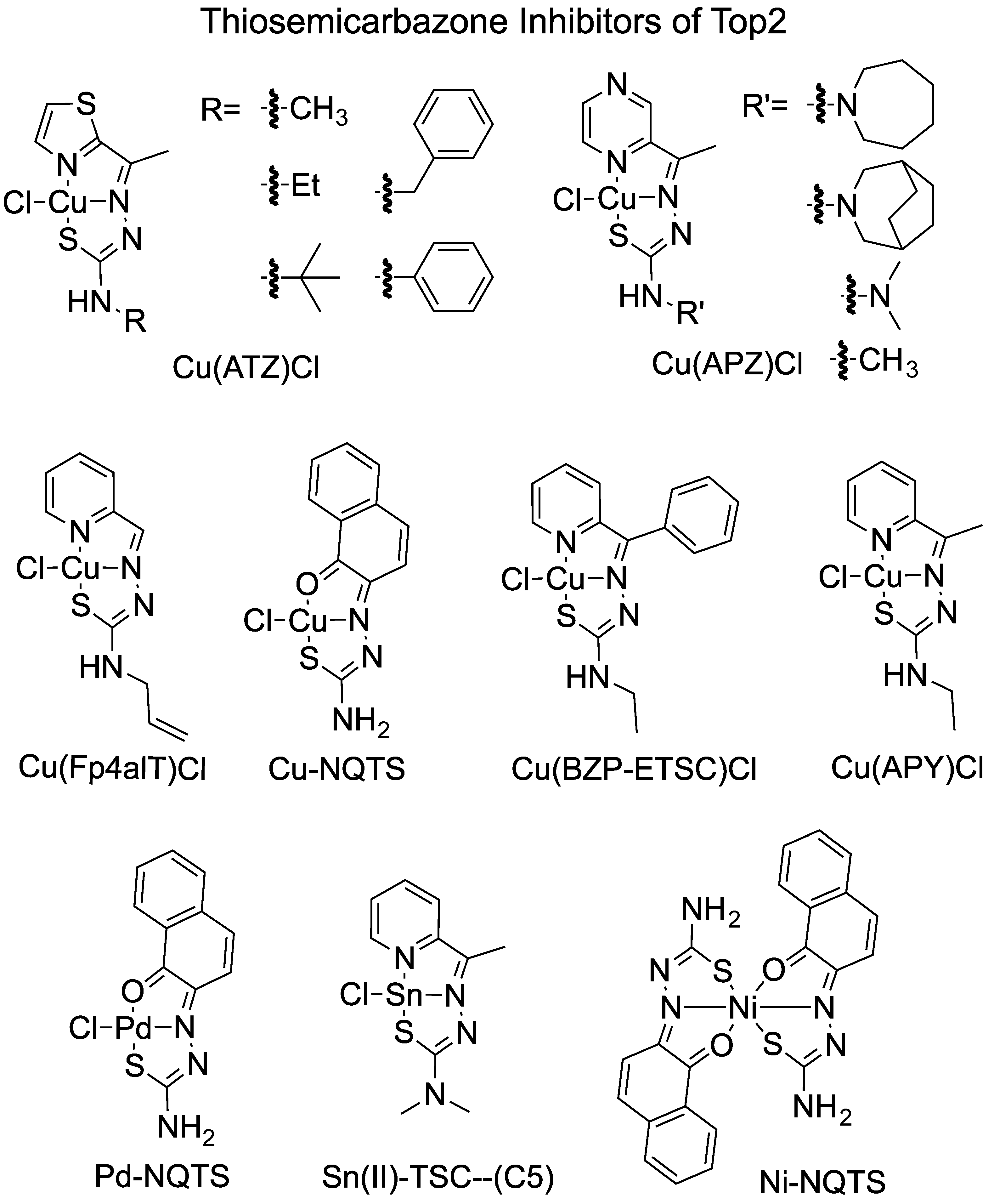
| Name | Inhibition of Top2 | Reference |
|---|---|---|
| Nine compounds and their copper complexes | Inhibit human Top2α | [45] |
| Cobalt (III) chelated with TSC ligand Complex 4 | Inhibits human Topo2α-induced DNA relaxation | [41] |
| Ni–bis(TSC) Complex 1 | Does not inhibit isolated Top2 from L1210 cells at 100 µM | [38] |
| Cu–TSCs (Compound 1 and 2) | Inhibits isolated Top2 from L1210 cells | [12] |
| Copper TSC | Inhibits Top2 of L1210 cells with IC50 value of 6.25–12.2 µM. Antagonizes the DNA break affect by etoposide. | [48] |
| Ni-NQTS | Inhibits DNA relaxation | [36] |
| Cu-NQTS | Inhibits DNA relaxation (TOPOGEN kit) | |
| Pd-NQTS | ||
| Cu(Fp4alT)Cl and its family of Cu(TSC)Cl | Inhibits relaxation by Top2 at 10 µM Completely inhibits Top2α without promoting the formation of linear DNA products | [37] |
| Ni–bis(TSC) | No effect in stabilizing DNA breaks | [51] |
| Cu(TSC) | Stabilizes DNA breaks | [51] |
| Ni(L1)(H1)Cl, Ni(HL2)2Cl2, Ni(L3)2,Ni(L4)2 Ni(L5)2Cl2 | No inhibition of Top2 (TopoGEN) | [47] |
| Cu(L1)Cl, Cu(L2)Cl, Cu(L3)Cl, Cu(L4)Cl, Cu(L5)Cl2 | Inhibit Top2 (TopoGEN) | [47] |
| Complex 1 (CuTSC cation) | Inhibits DNA relaxation and increase DNA cleavage | [49] |
| Sn(II)–TSC—(C5) | Inhibits Topo2α at 20 µM | [40] |
| Cu(S,R)L and Cu(R,S)L | Inhibit Top2α relaxation at 300 µM | [50] |
| [Cu(APY)Cl] and [Cu(APZ)Cl] | Inhibit Top2α from 0.5 µM Increase DNA cleavage Inhibit Top2α ATP hydrolysis No inhibition of ligation by Top2α Pre-incubating compounds with Top2α inactivated the enzyme | [32] |
| [Cu(APY)Cl] and [Cu(BZP)Cl] | Inhibit Top2β at 5 µM Increase DNA cleavage by Top2β Inhibit Top2β ATP hydrolysis Inhibit ligation by Top2β Pre-incubating compounds with Top2β inactivate the enzyme Stabilized closure of N-terminal Top2α and Top2β clamp | [34] |
| Cu(BZP)Cl series and | Inhibit Top2α relaxation and increase DNA cleavage | [33] |
| Cu(ATZ)Cl series | ||
| Pd(BZP)Cl series | Inhibit Top2α relaxation and increase DNA cleavage | [33] |
2.2.3. Inhibition of Type I and Type II Top
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Pommier, Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef] [PubMed]
- McKie, S.J.; Desai, P.R.; Seol, Y.; Allen, A.M.; Maxwell, A.; Neuman, K.C. Topoisomerase VI is a chirally-selective, preferential DNA decatenase. Elife 2022, 11, e67021. [Google Scholar] [CrossRef]
- Vann, K.R.; Oviatt, A.A.; Osheroff, N. Topoisomerase II Poisons: Converting Essential Enzymes into Molecular Scissors. Biochemistry 2021, 60, 1630–1641. [Google Scholar] [CrossRef] [PubMed]
- Deweese, J.E.; Osheroff, N. The DNA cleavage reaction of topoisomerase II: Wolf in sheep’s clothing. Nucleic Acids Res. 2009, 37, 738–749. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.B.; Mercer, S.L.; Deweese, J.E. Inhibitors and Poisons of Mammalian Type II Topoisomerases. In Advances in Molecular Toxicology; Fishbein, J.C., Heilman, J., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 11, pp. 203–240. [Google Scholar]
- Gibson, E.G.; Deweese, J.E. Covalent poisons of topoisomerase II. Curr. Top. Pharm. 2013, 17, 1–12. [Google Scholar]
- Moorthy, N.S.; Cerqueira, N.M.; Ramos, M.J.; Fernandes, P.A. Aryl- and heteroaryl-thiosemicarbazone derivatives and their metal complexes: A pharmacological template. Recent Pat. Anticancer Drug Discov. 2013, 8, 168–182. [Google Scholar] [CrossRef]
- Bai, X.G.; Zheng, Y.; Qi, J. Advances in thiosemicarbazone metal complexes as anti-lung cancer agents. Front. Pharmacol. 2022, 13, 1018951. [Google Scholar] [CrossRef]
- Matesanz, A.I.; Herrero, J.M.; Quiroga, A.G. Chemical and Biological Evaluation of Thiosemicarbazone-Bearing Heterocyclic Metal Complexes. Curr. Top. Med. Chem. 2021, 21, 59–72. [Google Scholar] [CrossRef]
- Liberta, A.E.; West, D.X. Antifungal and antitumor activity of heterocyclic thiosemicarbazones and their metal complexes: Current status. Biometals 1992, 5, 121–126. [Google Scholar] [CrossRef]
- Miller, M.C., 3rd; Stineman, C.N.; Vance, J.R.; West, D.X.; Hall, I.H. The cytotoxicity of copper(II) complexes of 2-acetyl-pyridyl-4N-substituted thiosemicarbazones. Anticancer Res. 1998, 18, 4131–4139. [Google Scholar] [PubMed]
- Baruffini, E.; Ruotolo, R.; Bisceglie, F.; Montalbano, S.; Ottonello, S.; Pelosi, G.; Buschini, A.; Lodi, T. Mechanistic insights on the mode of action of an antiproliferative thiosemicarbazone-nickel complex revealed by an integrated chemogenomic profiling study. Sci. Rep. 2020, 10, 10524. [Google Scholar] [CrossRef] [PubMed]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Novel chelators for cancer treatment: Where are we now? Antioxid. Redox Signal. 2013, 18, 973–1006. [Google Scholar] [CrossRef] [PubMed]
- Whitnall, M.; Howard, J.; Ponka, P.; Richardson, D.R. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc. Natl. Acad. Sci. USA 2006, 103, 14901–14906. [Google Scholar] [CrossRef]
- Jansson, P.J.; Kalinowski, D.S.; Lane, D.J.; Kovacevic, Z.; Seebacher, N.A.; Fouani, L.; Sahni, S.; Merlot, A.M.; Richardson, D.R. The renaissance of polypharmacology in the development of anti-cancer therapeutics: Inhibition of the “Triad of Death” in cancer by Di-2-pyridylketone thiosemicarbazones. Pharmacol. Res. 2015, 100, 255–260. [Google Scholar] [CrossRef]
- Salim, K.Y.; Maleki Vareki, S.; Danter, W.R.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef] [Green Version]
- Lindemann, A.; Patel, A.A.; Silver, N.L.; Tang, L.; Liu, Z.; Wang, L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, A Novel Thiosemicarbazone Derivative, Exhibits Antitumor Activity in HNSCC through p53-dependent and -independent Mechanisms. Clin. Cancer Res. 2019, 25, 5650–5662. [Google Scholar] [CrossRef] [Green Version]
- McKenzie-Nickson, S.; Bush, A.I.; Barnham, K.J. Bis(thiosemicarbazone) Metal Complexes as Therapeutics for Neurodegenerative Diseases. Curr. Top. Med. Chem. 2016, 16, 3058–3068. [Google Scholar] [CrossRef]
- Karp, J.E.; Giles, F.J.; Gojo, I.; Morris, L.; Greer, J.; Johnson, B.; Thein, M.; Sznol, M.; Low, J. A phase I study of the novel ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) in combination with the nucleoside analog fludarabine for patients with refractory acute leukemias and aggressive myeloproliferative disorders. Leuk. Res. 2008, 32, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Goh, B.C.; Tan, E.H.; Lam, K.C.; Soo, R.; Leong, S.S.; Wang, L.Z.; Mo, F.; Chan, A.T.; Zee, B.; et al. A multicenter phase II trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) and gemcitabine in advanced non-small-cell lung cancer with pharmacokinetic evaluation using peripheral blood mononuclear cells. Investig. New Drugs 2008, 26, 169–173. [Google Scholar] [CrossRef]
- Nutting, C.M.; van Herpen, C.M.; Miah, A.B.; Bhide, S.A.; Machiels, J.P.; Buter, J.; Kelly, C.; de Raucourt, D.; Harrington, K.J. Phase II study of 3-AP Triapine in patients with recurrent or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2009, 20, 1275–1279. [Google Scholar] [CrossRef]
- Finch, R.A.; Liu, M.C.; Cory, A.H.; Cory, J.G.; Sartorelli, A.C. Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone; 3-AP): An inhibitor of ribonucleotide reductase with antineoplastic activity. Adv. Enzyme Regul. 1999, 39, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Finch, R.A.; Liu, M.; Grill, S.P.; Rose, W.C.; Loomis, R.; Vasquez, K.M.; Cheng, Y.; Sartorelli, A.C. Triapine (3-aminopyridine-2-carboxaldehyde- thiosemicarbazone): A potent inhibitor of ribonucleotide reductase activity with broad spectrum antitumor activity. Biochem. Pharmacol. 2000, 59, 983–991. [Google Scholar] [CrossRef]
- Posa, V.; Hajdu, B.; Toth, G.; Domotor, O.; Kowol, C.R.; Keppler, B.K.; Spengler, G.; Gyurcsik, B.; Enyedy, E.A. The coordination modes of (thio)semicarbazone copper(II) complexes strongly modulate the solution chemical properties and mechanism of anticancer activity. J. Inorg. Biochem. 2022, 231, 111786. [Google Scholar] [CrossRef] [PubMed]
- Yalowich, J.C.; Wu, X.; Zhang, R.; Kanagasabai, R.; Hornbaker, M.; Hasinoff, B.B. The anticancer thiosemicarbazones Dp44mT and triapine lack inhibitory effects as catalytic inhibitors or poisons of DNA topoisomerase IIα. Biochem. Pharmacol. 2012, 84, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Chen, Q.; Ku, X.; Meng, L.; Lin, L.; Wang, X.; Zhu, C.X.; Wang, Y.; Chen, Z.; Li, M.; et al. A series of α-heterocyclic carboxaldehyde thiosemicarbazones inhibit topoisomerase IIα catalytic activity. J. Med. Chem. 2010, 53, 3048–3064. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.A.; Klein, S.R.; Agama, K.K.; Toyoda, E.; Adachi, N.; Pommier, Y.; Shacter, E.B. The iron chelator Dp44mT causes DNA damage and selective inhibition of topoisomerase IIalpha in breast cancer cells. Cancer Res. 2009, 69, 948–957. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, J.F.; Lima, T.S.; Vendramini-Costa, D.B.; de Lacerda Pedrosa, S.C.B.; Lafayette, E.A.; da Silva, R.M.F.; de Almeida, S.M.V.; de Moura, R.O.; Ruiz, A.; de Carvalho, J.E.; et al. Thiosemicarbazones and 4-thiazolidinones indole-based derivatives: Synthesis, evaluation of antiproliferative activity, cell death mechanisms and topoisomerase inhibition assay. Eur. J. Med. Chem. 2017, 136, 305–314. [Google Scholar] [CrossRef]
- da Silva Filho, F.A.; de Freitas Souza, T.; Ribeiro, A.G.; Alves, J.E.F.; de Oliveira, J.F.; de Lima Souza, T.R.C.; de Moura, R.O.; do Carmo Alves de Lima, M.; de Carvalho Junior, L.B.; de Almeida, S.M.V. Topoisomerase inhibition and albumin interaction studies of acridine-thiosemicarbazone derivatives. Int. J. Biol. Macromol. 2019, 138, 582–589. [Google Scholar] [CrossRef]
- Samia, L.B.; Parrilha, G.L.; Da Silva, J.G.; Ramos, J.P.; Souza-Fagundes, E.M.; Castelli, S.; Vutey, V.; Desideri, A.; Beraldo, H. Metal complexes of 3-(4-bromophenyl)-1-pyridin-2-ylprop-2-en-1-one thiosemicarbazone: Cytotoxic activity and investigation on the mode of action of the gold(III) complex. Biometals 2016, 29, 515–526. [Google Scholar] [CrossRef]
- Wilson, J.T.; Jiang, X.; McGill, B.C.; Lisic, E.C.; Deweese, J.E. Examination of the Impact of Copper(II) alpha-(N)-Heterocyclic Thiosemicarbazone Complexes on DNA Topoisomerase IIalpha. Chem. Res. Toxicol. 2016, 29, 649–658. [Google Scholar] [CrossRef]
- Morris, W.H.; Ngo, L.; Wilson, J.T.; Medawala, W.; Brown, A.R.; Conner, J.D.; Fabunmi, F.; Cashman, D.J.; Lisic, E.C.; Yu, T.; et al. Structural and Metal Ion Effects on Human Topoisomerase IIalpha Inhibition by alpha-(N)-Heterocyclic Thiosemicarbazones. Chem. Res. Toxicol. 2019, 32, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Keck, J.M.; Conner, J.D.; Wilson, J.T.; Jiang, X.; Lisic, E.C.; Deweese, J.E. Clarifying the Mechanism of Copper(II) alpha-(N)-Heterocyclic Thiosemicarbazone Complexes on DNA Topoisomerase IIalpha and IIbeta. Chem. Res. Toxicol. 2019, 32, 2135–2143. [Google Scholar] [CrossRef] [PubMed]
- Vutey, V.; Castelli, S.; D’Annessa, I.; Samia, L.B.; Souza-Fagundes, E.M.; Beraldo, H.; Desideri, A. Human topoisomerase IB is a target of a thiosemicarbazone copper(II) complex. Arch. Biochem. Biophys. 2016, 606, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, Y.; Liu, G.; Afrasiabi, Z.; Sinn, E.; Padhye, S.; Ma, Y. The cytotoxicity and mechanisms of 1,2-naphthoquinone thiosemicarbazone and its metal derivatives against MCF-7 human breast cancer cells. Tox. App. Pharm. 2004, 197, 40–48. [Google Scholar] [CrossRef]
- Zeglis, B.M.; Divilov, V.; Lewis, J.S. Role of metalation in the topoisomerase IIα inhibition and antiproliferation activity of a series of α-heterocyclic-N4-substituted thiosemicarbazones and their Cu(II) complexes. J. Med. Chem. 2011, 54, 2391–2398. [Google Scholar] [CrossRef]
- Hall, I.H.; Miller, M.C.; West, D.X. Antineoplastic and Cytotoxic Activities of Nickel(II) Complexes of Thiosemicarbazones. Met. Based Drugs 1997, 4, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Beckford, F.; Thessing, J.; Woods, J.; Didion, J.; Gerasimchuk, N.; Gonzalez-Sarrias, A.; Seeram, N.P. Synthesis and structure of [(eta(6)-p-cymene)Ru(2-anthracen-9-ylmethylene-N-ethylhydrazinecarbothioamide)Cl]Cl; biological evaluation, topoisomerase II inhibition and reaction with DNA and human serum albumin. Metallomics 2011, 3, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Yang, T.; Wang, X.; Li, W.; Pang, M.; Sun, H.; Liang, H.; Yang, F. Development of a multi-target anticancer Sn(ii) pyridine-2-carboxaldehyde thiosemicarbazone complex. Dalton Trans. 2021, 50, 10909–10921. [Google Scholar] [CrossRef] [PubMed]
- Beebe, S.J.; Celestine, M.J.; Bullock, J.L.; Sandhaus, S.; Arca, J.F.; Cropek, D.M.; Ludvig, T.A.; Foster, S.R.; Clark, J.S.; Beckford, F.A.; et al. Synthesis, characterization, DNA binding, topoisomerase inhibition, and apoptosis induction studies of a novel cobalt(III) complex with a thiosemicarbazone ligand. J. Inorg. Biochem. 2020, 203, 110907. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Zheng, Y.; Qian, K.; Tian, L.; Zhang, G.X.; Cheng, Z.; Wang, Y. Synthesis, crystal structure and antiproliferative mechanisms of 2-acetylpyridine-thiosemicarbazones Ga(III) with a greater selectivity against tumor cells. J. Inorg. Biochem. 2017, 177, 110–117. [Google Scholar] [CrossRef]
- Oliveira, C.G.; Romero-Canelon, I.; Silva, M.M.; Coverdale, J.P.C.; Maia, P.I.S.; Batista, A.A.; Castelli, S.; Desideri, A.; Sadler, P.J.; Deflon, V.M. Palladium(ii) complexes with thiosemicarbazones derived from pyrene as topoisomerase IB inhibitors. Dalton Trans. 2019, 48, 16509–16517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heng, M.P.; Sinniah, S.K.; Teoh, W.Y.; Sim, K.S.; Ng, S.W.; Cheah, Y.K.; Tan, K.W. Synthesis of a DNA-targeting nickel (II) complex with testosterone thiosemicarbazone which exhibits selective cytotoxicity towards human prostate cancer cells (LNCaP). Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 150, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Zheng, Y.; Li, B.; Ai, Y.; Chen, M.; Zheng, X. Pyridoxal hydrochloride thiosemicarbazones with copper ions inhibit cell division via Topo-I and Topo-IIa. J. Inorg. Biochem. 2022, 232, 111816. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A. Next generation topoisomerase I inhibitors: Rationale and biomarker strategies. Biochem. Pharmacol. 2008, 75, 1262–1271. [Google Scholar] [CrossRef]
- Bisceglie, F.; Musiari, A.; Pinelli, S.; Alinovi, R.; Menozzi, I.; Polverini, E.; Tarasconi, P.; Tavone, M.; Pelosi, G. Quinoline-2-carboxaldehyde thiosemicarbazones and their Cu(II) and Ni(II) complexes as topoisomerase IIα inhibitors. J. Inorg. Biochem. 2015, 152, 10–19. [Google Scholar] [CrossRef]
- Miller III, M.C.; Stineman, C.N.; Vance, J.R.; West, D.X.; Hall, I.H. Multiple mechanisms for cytotoxicity induced by copper(II) complexes of 2-acetylpyrazine-N-substituted thiosemicarbazones. Appl. Organomet. Chem. 1999, 13, 9–19. [Google Scholar] [CrossRef]
- Sandhaus, S.; Taylor, R.; Edwards, T.; Huddleston, A.; Wooten, Y.; Venkatraman, R.; Weber, R.T.; Gonzalez-Sarrias, A.; Martin, P.M.; Cagle, P.; et al. A novel copper(II) complex identified as a potent drug against colorectal and breast cancer cells and as a poison inhibitor for human topoisomerase IIalpha. Inorg. Chem. Commun. 2016, 64, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Bacher, F.; Enyedy, E.; Nagy, N.V.; Rockenbauer, A.; Bognar, G.M.; Trondl, R.; Novak, M.S.; Klapproth, E.; Kiss, T.; Arion, V.B. Copper(II) complexes with highly water-soluble L- and D-proline-thiosemicarbazone conjugates as potential inhibitors of Topoisomerase IIalpha. Inorg. Chem. 2013, 52, 8895–8908. [Google Scholar] [CrossRef] [Green Version]
- Bisceglie, F.; Pinelli, S.; Alinovi, R.; Goldoni, M.; Mutti, A.; Camerini, A.; Piola, L.; Tarasconi, P.; Pelosi, G. Cinnamaldehyde and cuminaldehyde thiosemicarbazones and their copper(II) and nickel(II) complexes: A study to understand their biological activity. J. Inorg. Biochem. 2014, 140, 111–125. [Google Scholar] [CrossRef]
- Lindsey, R.H.; Bender, R.P.; Osheroff, N. Stimulation of topoisomerase II-mediated DNA cleavage by benzene metabolites. Chem. Biol. Interact. 2005, 153-154, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, R.H., Jr.; Bender, R.P.; Osheroff, N. Effects of benzene metabolites on DNA cleavage mediated by human topoisomerase IIα: 1,4-hydroquinone is a topoisomerase II poison. Chem. Res. Toxicol. 2005, 18, 761–770. [Google Scholar] [CrossRef]
- Lindsey, R.H., Jr.; Bromberg, K.D.; Felix, C.A.; Osheroff, N. 1,4-Benzoquinone is a topoisomerase II poison. Biochemistry 2004, 43, 7563–7574. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Inouye, M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem. Sci. 2000, 25, 24–28. [Google Scholar] [CrossRef] [PubMed]




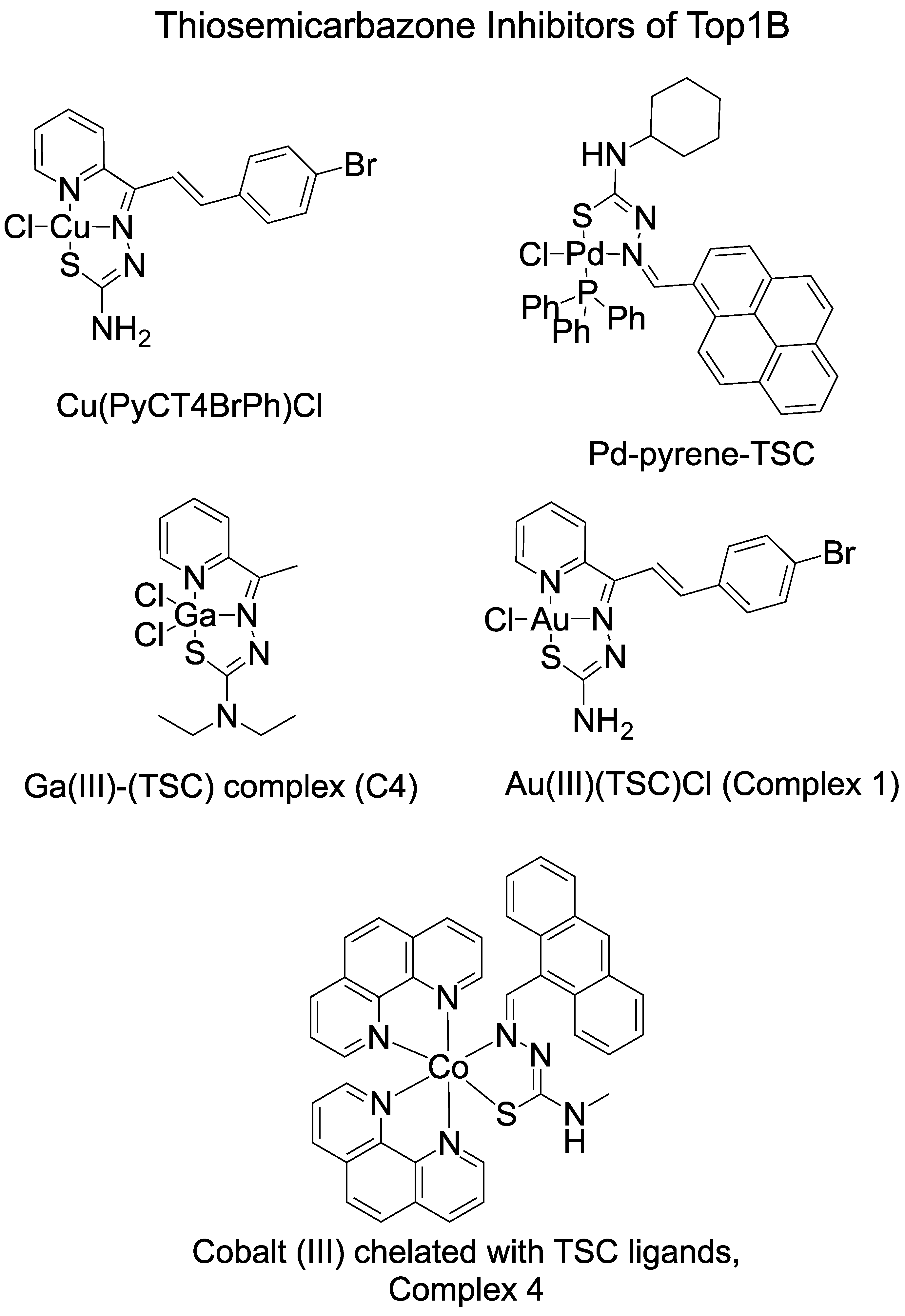
| Name | Inhibition of TopI | Reference |
|---|---|---|
| Cu(PyCT4BrPh)Cl | Inhibits TopI The inhibition is severe with pre-incubation of the compound with TopI Inhibited the cleavage step and partially inhibited religation | [35] |
| Pd–pyrene–TSC | Inhibits human Top1B at 12.5 µM | [43] |
| Ga(III)–TSC complex (C4) | Inhibits TopI | [42] |
| Au(III)(TSC)Cl (complex 1) | Inhibits human Top1B activity starting at 1.5 μM Pre-incubation of Top1B with Complex 1 increased the inhibition | [31] |
| Ni-bis(TSC) | No inhibition of E. coli TopI | [44] |
| Nine copper complexes | Inhibits TopI | [45] |
| Cobalt (III)–TSC (Complex 4) | Inhibits TopI-induced DNA relaxation | [41] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, X.; Fielding, L.A.; Davis, H.; Carroll, W.; Lisic, E.C.; Deweese, J.E. Inhibition of Topoisomerases by Metal Thiosemicarbazone Complexes. Int. J. Mol. Sci. 2023, 24, 12010. https://doi.org/10.3390/ijms241512010
Jiang X, Fielding LA, Davis H, Carroll W, Lisic EC, Deweese JE. Inhibition of Topoisomerases by Metal Thiosemicarbazone Complexes. International Journal of Molecular Sciences. 2023; 24(15):12010. https://doi.org/10.3390/ijms241512010
Chicago/Turabian StyleJiang, Xiaohua, Lauren A. Fielding, Hunter Davis, William Carroll, Edward C. Lisic, and Joseph E. Deweese. 2023. "Inhibition of Topoisomerases by Metal Thiosemicarbazone Complexes" International Journal of Molecular Sciences 24, no. 15: 12010. https://doi.org/10.3390/ijms241512010