
Emerging Roles of Cells and Molecules of Innate Immunity in Alzheimer’s Disease

 ,
,  , ,
, ,  , and
, and 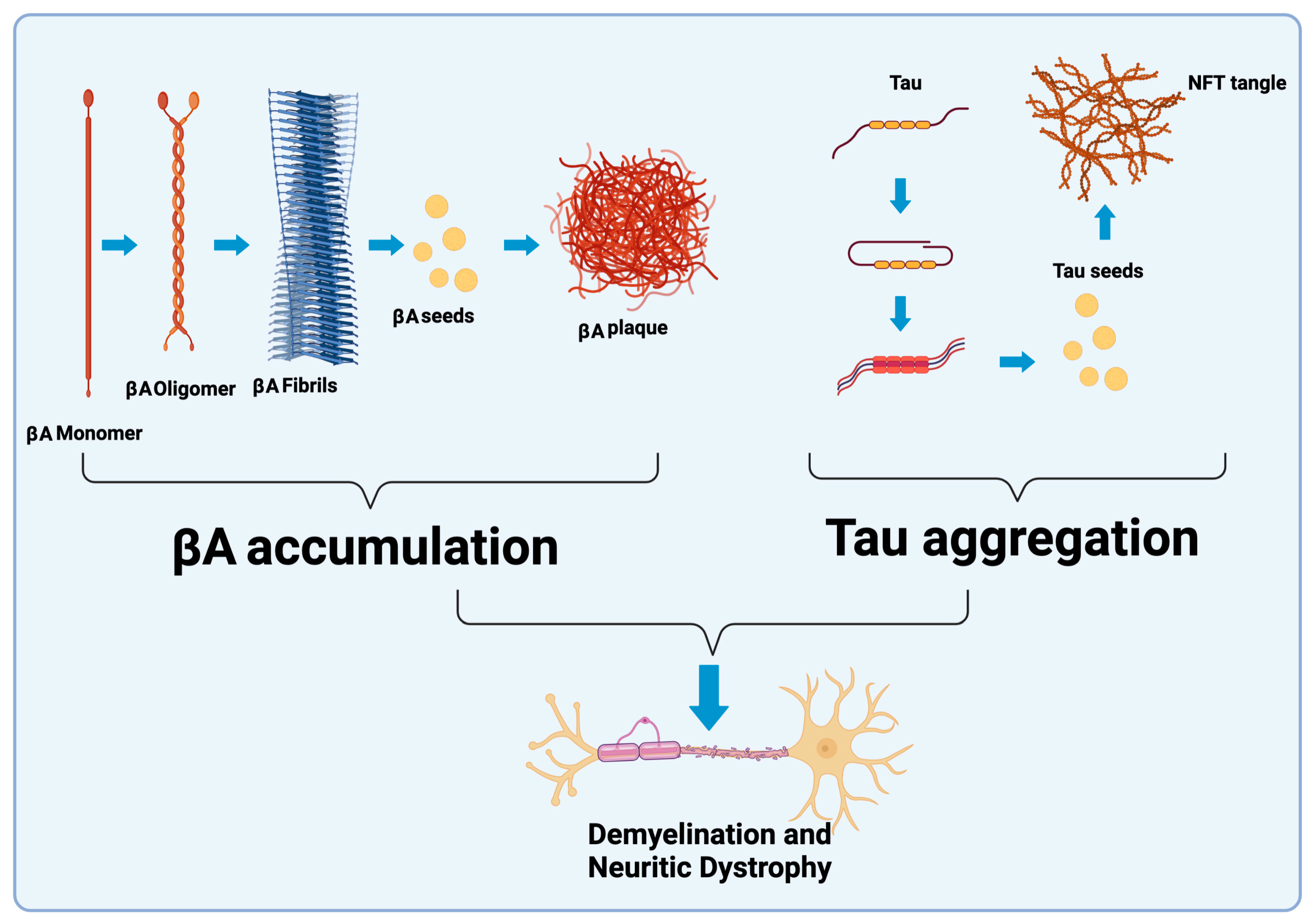{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Role of Microglia in Alzheimer’s Disease
3. The Role of Astrocytes in Alzheimer’s Disease
4. The Role of Oligodendrocytes in Alzheimer’s Disease
5. Tau and βA Proteins
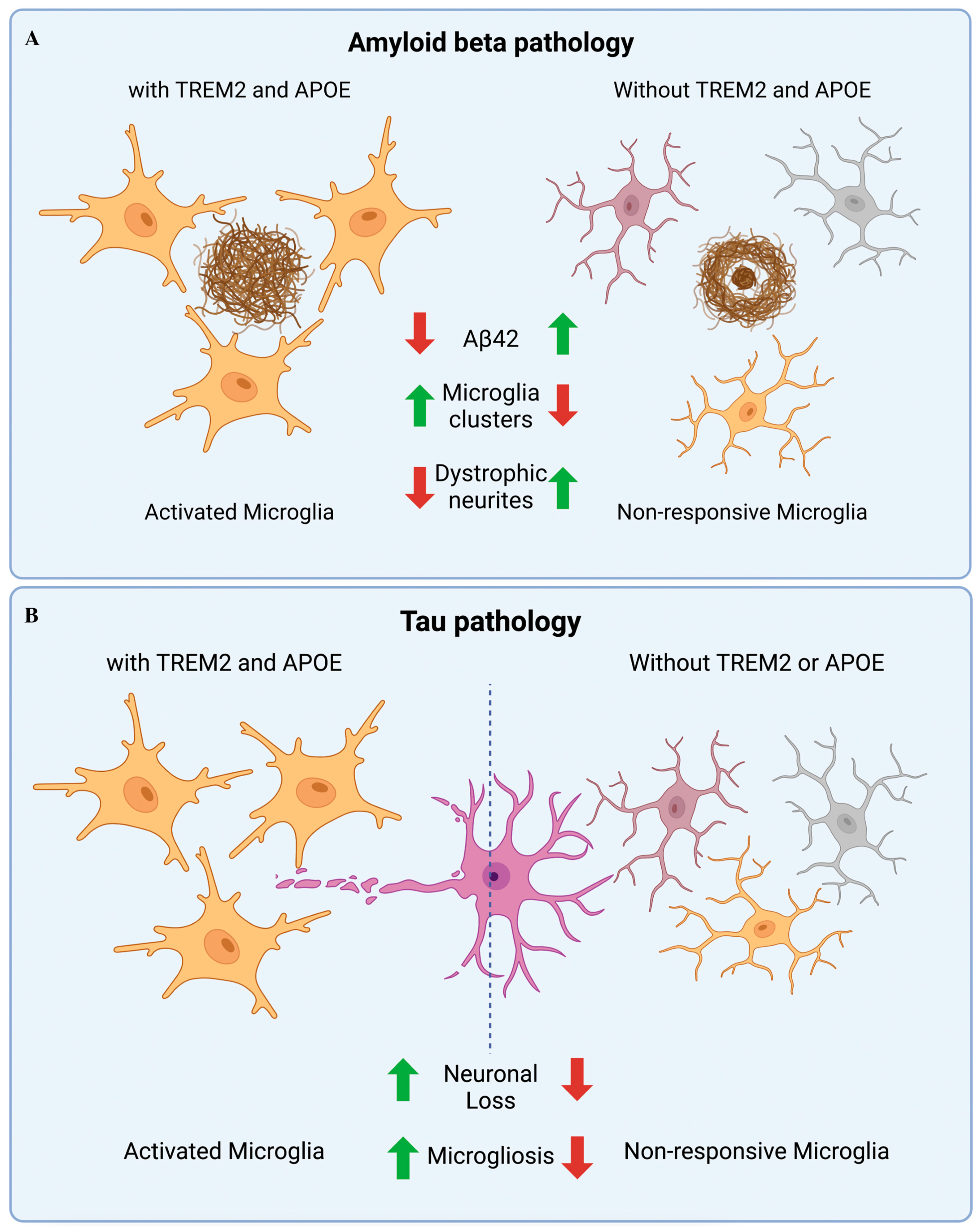
6. TREM1 and TREM2
7. Illuminating the Immune Landscape of Alzheimer’s Disease: Insights from Bulk and Single-Cell RNA Sequencing
8. Possible Treatment Strategies
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2022 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2022, 18, 700–789. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef]
- Armstrong, R.A. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019, 57, 87–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Friker, L.L.; Scheiblich, H.; Hochheiser, I.V.; Brinkschulte, R.; Riedel, D.; Latz, E.; Geyer, M.; Heneka, M.T. β-Amyloid Clustering around ASC Fibrils Boosts Its Toxicity in Microglia. Cell Rep. 2020, 30, 3743–3754.e6. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Rubio-Perez, J.M.; Morillas-Ruiz, J.M. A Review: Inflammatory Process in Alzheimer’s Disease, Role of Cytokines. Sci. World J. 2012, 2012, 756357. [Google Scholar] [CrossRef]
- Stancu, I.-C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.-N.; et al. Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer Disease—A Brief Review of the Basic Science and Clinical Literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef]
- Hanzel, C.E.; Pichet-Binette, A.; Pimentel, L.S.B.; Iulita, M.F.; Allard, S.; Ducatenzeiler, A.; Do Carmo, S.; Cuello, A.C. Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2249–2262. [Google Scholar] [CrossRef]
- Okello, A.; Edison, P.; Archer, H.A.; Turkheimer, F.E.; Kennedy, J.; Bullock, R.; Walker, Z.; Kennedy, A.; Fox, N.; Rossor, M.; et al. Microglial activation and amyloid deposition in mild cognitive impairment. Neurology 2009, 72, 56. [Google Scholar] [CrossRef] [Green Version]
- Grazia Daniela, F.; Melanie, D.; Melanie, W.; Zhen, F.; Valeria, C.; Rebecca, A.; Trudi, E.; Rainer, H.; David, J.B.; Paul, E. Microglial activation in early Alzheimer trajectory is associated with higher gray matter volume. Neurology 2019, 92, e1331. [Google Scholar] [CrossRef] [Green Version]
- Dani, M.; Wood, M.; Mizoguchi, R.; Fan, Z.; Edginton, T.; Hinz, R.; Win, Z.; Brooks, D.J.; Edison, P. Tau Aggregation Correlates with Amyloid Deposition in Both Mild Cognitive Impairment and Alzheimer’s Disease Subjects. J. Alzheimer’s Dis. 2019, 70, 455–465. [Google Scholar] [CrossRef]
- Hamelin, L.; Lagarde, J.; Dorothée, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18 F-DPA-714 PET imaging. Brain 2016, 139, 1252–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreisl, W.C.; Henter, I.D.; Innis, R.B. Chapter Eight—Imaging Translocator Protein as a Biomarker of Neuroinflammation in Dementia. In Advances in Pharmacology; Pasternak, G.W., Coyle, J.T., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 82, pp. 163–185. [Google Scholar]
- Kreisl, W.C.; Lyoo, C.H.; Liow, J.-S.; Wei, M.; Snow, J.; Page, E.; Jenko, K.J.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; et al. 11C-PBR28 binding to translocator protein increases with progression of Alzheimer’s disease. Neurobiol. Aging 2016, 44, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Rogers, J.; Luber-Narod, J.; Styren, S.D.; Civin, W.H. Expression of immune system-associated antigens by cells of the human central nervous system: Relationship to the pathology of Alzheimer’s disease. Neurobiol. Aging 1988, 9, 339–349. [Google Scholar] [CrossRef]
- McGeer, P.L.; Akiyama, H.; Itagaki, S.; McGeer, E.G. Immune System Response in Alzheimer’s Disease. Can. J. Neurol. Sci. 1989, 16, 516–527. [Google Scholar] [CrossRef] [Green Version]
- Togo, T.; Akiyama, H.; Iseki, E.; Kondo, H.; Ikeda, K.; Kato, M.; Oda, T.; Tsuchiya, K.; Kosaka, K. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J. Neuroimmunol. 2002, 124, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Parachikova, A.; Agadjanyan, M.G.; Cribbs, D.H.; Blurton-Jones, M.; Perreau, V.; Rogers, J.; Beach, T.G.; Cotman, C.W. Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol. Aging 2007, 28, 1821–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaitanya, J.; Karthigayini, S.; Scott, C.; Sara, I.; Jacqueline, R.; Wei, Z.; Danielle, S.; Rebecca, L.; Doris, L.-W.; Roger, R.; et al. CSF-Derived CD4+ T-Cell Diversity Is Reduced in Patients with Alzheimer Clinical Syndrome. Neurol.-Neuroimmunol. Neuroinflamm. 2022, 9, e1106. [Google Scholar] [CrossRef]
- Stowe, A.M.; Ireland, S.J.; Ortega, S.B.; Chen, D.; Huebinger, R.M.; Tarumi, T.; Harris, T.S.; Cullum, C.M.; Rosenberg, R.; Monson, N.L.; et al. Adaptive lymphocyte profiles correlate to brain Aβ burden in patients with mild cognitive impairment. J. Neuroinflamm. 2017, 14, 149. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; He, X.; Li, Y.; Wang, T. Cerebrospinal fluid CD4+ T lymphocyte-derived miRNA-let-7b can enhances the diagnostic performance of Alzheimer’s disease biomarkers. Biochem. Biophys. Res. Commun. 2018, 495, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Lueg, G.; Gross, C.C.; Lohmann, H.; Johnen, A.; Kemmling, A.; Deppe, M.; Groger, J.; Minnerup, J.; Wiendl, H.; Meuth, S.G.; et al. Clinical relevance of specific T-cell activation in the blood and cerebrospinal fluid of patients with mild Alzheimer’s disease. Neurobiol. Aging 2015, 36, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; Calabrese, E.; Marventano, I.; Piancone, F.; Gatti, A.; Alberoni, M.; Nemni, R.; Clerici, M. Increased activity of Th-17 and Th-9 lymphocytes and a skewing of the post-thymic differentiation pathway are seen in Alzheimer’s disease. Brain Behav. Immun. 2011, 25, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L. The CD14+ CD16+ blood monocytes: Their role in infection and inflammation. J. Leukoc. Biol. 2007, 81, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.L.; Brooks, C.L. Tailoring the structural integrity process to meet the challenges of aging aircraft. Int. J. Fatigue 1999, 21, S1–S14. [Google Scholar] [CrossRef]
- Kuhla, A.; Ludwig, S.C.; Kuhla, B.; Münch, G.; Vollmar, B. Advanced glycation end products are mitogenic signals and trigger cell cycle reentry of neurons in Alzheimer’s disease brain. Neurobiol. Aging 2015, 36, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; De Los Santos, M.B.; et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 2020, 577, 399–404. [Google Scholar] [CrossRef]
- Lee, S.; Cho, H.-J.; Ryu, J.-H. Innate Immunity and Cell Death in Alzheimer’s Disease. ASN Neuro 2021, 13, 17590914211051908. [Google Scholar] [CrossRef] [PubMed]
- Dulken, B.W.; Buckley, M.T.; Navarro Negredo, P.; Saligrama, N.; Cayrol, R.; Leeman, D.S.; George, B.M.; Boutet, S.C.; Hebestreit, K.; Pluvinage, J.V.; et al. Single-cell analysis reveals T cell infiltration in old neurogenic niches. Nature 2019, 571, 205–210. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Cowan, M.; Petri, W.A. Microglia: Immune Regulators of Neurodevelopment. Front. Immunol. 2018, 9, 2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef] [Green Version]
- Venegas, C.; Heneka, M.T. Danger-associated molecular patterns in Alzheimer’s disease. J. Leukoc. Biol. 2017, 101, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Li, S.; Xu, H.; Walsh, D.M.; Selkoe, D.J. Large Soluble Oligomers of Amyloid β-Protein from Alzheimer Brain Are Far Less Neuroactive Than the Smaller Oligomers to Which They Dissociate. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Simon, E.; Obst, J.; Gomez-Nicola, D. The Evolving Dialogue of Microglia and Neurons in Alzheimer’s Disease: Microglia as Necessary Transducers of Pathology. Neuroscience 2019, 405, 24–34. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Khoury, J.E. Microglia in Health and Disease. Cold Spring Harb. Perspect. Biol. 2016, 8, a020560. [Google Scholar] [CrossRef] [Green Version]
- Perea, J.R.; Bolós, M.; Avila, J. Microglia in Alzheimer’s disease in the context of tau pathology. Biomolecules 2020, 10, 1439. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 Mediates the Innate Host Response to β-Amyloid. J. Exp. Med. 2003, 197, 1657–1666. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Ewers, M.; Franzmeier, N.; Suárez-Calvet, M.; Morenas-Rodriguez, E.; Caballero, M.A.A.; Kleinberger, G.; Piccio, L.; Cruchaga, C.; Deming, Y.; Dichgans, M.; et al. Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci. Transl. Med. 2019, 11, eaav6221. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, E.; Sankowski, R.; Dietrich, H.; Oikonomidi, A.; Huerta, P.T.; Popp, J.; Al-Abed, Y.; Bacher, M. Key role of MIF-related neuroinflammation in neurodegeneration and cognitive impairment in Alzheimer’s disease. Mol. Med. 2020, 26, 34. [Google Scholar] [CrossRef] [Green Version]
- Popp, J.; Bacher, M.; Kölsch, H.; Noelker, C.; Deuster, O.; Dodel, R.; Jessen, F. Macrophage migration inhibitory factor in mild cognitive impairment and Alzheimer’s disease. J. Psychiatr. Res. 2009, 43, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Oikonomidi, A.; Tautvydaitė, D.; Gholamrezaee, M.M.; Henry, H.; Bacher, M.; Popp, J. Macrophage Migration Inhibitory Factor is Associated with Biomarkers of Alzheimer’s Disease Pathology and Predicts Cognitive Decline in Mild Cognitive Impairment and Mild Dementia. J. Alzheimer’s Dis. 2017, 60, 273–281. [Google Scholar] [CrossRef]
- Franco Bocanegra, D.K.; Nicoll, J.A.R.; Boche, D. Innate immunity in Alzheimer’s disease: The relevance of animal models? J. Neural Transm. 2018, 125, 827–846. [Google Scholar] [CrossRef] [Green Version]
- Karran, E.; Mercken, M.; Strooper, B.D. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Hefendehl, J.K.; Wegenast-Braun, B.M.; Liebig, C.; Eicke, D.; Milford, D.; Calhoun, M.E.; Kohsaka, S.; Eichner, M.; Jucker, M. Long-term in vivo imaging of β-amyloid plaque appearance and growth in a mouse model of cerebral β-amyloidosis. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 624–629. [Google Scholar] [CrossRef] [Green Version]
- Zotova, E.; Bharambe, V.; Cheaveau, M.; Morgan, W.; Holmes, C.; Harris, S.; Neal, J.W.; Love, S.; Nicoll, J.A.R.; Boche, D. Inflammatory components in human Alzheimer’s disease and after active amyloid-β42 immunization. Brain 2013, 136, 2677–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Pozo, A.; Muzikansky, A.; Gómez-Isla, T.; Growdon, J.H.; Betensky, R.A.; Frosch, M.P.; Hyman, B.T. Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2013, 72, 462–471. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, C.; Yang, E.-J.; Shin, J.; Park, J.; Kim, S.-H.; Park, S.-W.; Chang, W.S.; Lee, C.-H.; Kim, H.; Kim, H.-S. Enhanced delivery of a low dose of aducanumab via FUS in 5× FAD mice, an AD model. Transl. Neurodegener. 2022, 11, 57. [Google Scholar] [CrossRef]
- Xia, D.; Lianoglou, S.; Sandmann, T.; Calvert, M.; Suh, J.H.; Thomsen, E.; Dugas, J.; Pizzo, M.E.; DeVos, S.L.; Earr, T.K. Novel App knock-in mouse model shows key features of amyloid pathology and reveals profound metabolic dysregulation of microglia. Mol. Neurodegener. 2022, 17, 41. [Google Scholar] [CrossRef] [PubMed]
- Clayton, K.; Delpech, J.C.; Herron, S.; Iwahara, N.; Ericsson, M.; Saito, T.; Saido, T.C.; Ikezu, S.; Ikezu, T. Plaque associated microglia hyper-secrete extracellular vesicles and accelerate tau propagation in a humanized APP mouse model. Mol. Neurodegener. 2021, 16, 18. [Google Scholar] [CrossRef]
- Sastre, M.; Klockgether, T.; Heneka, M.T. Contribution of inflammatory processes to Alzheimer’s disease: Molecular mechanisms. Int. J. Dev. Neurosci. 2006, 24, 167–176. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Streit, W.J.; Braak, H.; Xue, Q.S. Bechmann, I2737117: Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janelsins, M.C.; Mastrangelo, M.A.; Oddo, S.; LaFerla, F.M.; Federoff, H.J.; Bowers, W.J. Early correlation of microglial activation with enhanced tumor necrosis factor-alpha and monocyte chemoattractant protein-1 expression specifically within the entorhinal cortex of triple transgenic Alzheimer’s disease mice. J. Neuroinflamm. 2005, 2, 23. [Google Scholar] [CrossRef] [Green Version]
- Caruso, D.; Barron, A.M.; Brown, M.A.; Abbiati, F.; Carrero, P.; Pike, C.J.; Garcia-Segura, L.M.; Melcangi, R.C. Age-related changes in neuroactive steroid levels in 3xTg-AD mice. Neurobiol. Aging 2013, 34, 1080–1089. [Google Scholar] [CrossRef] [Green Version]
- Colton, C.A.; Wilcock, D.M.; Wink, D.A.; Davis, J.; Van Nostrand, W.E.; Vitek, M.P. The effects of NOS2 gene deletion on mice expressing mutated human AβPP. J. Alzheimer’s Dis. 2008, 15, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Stephan, A.H.; Madison, D.V.; Mateos, J.M.; Fraser, D.A.; Lovelett, E.A.; Coutellier, L.; Kim, L.; Tsai, H.-H.; Huang, E.J.; Rowitch, D.H. A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 2013, 33, 13460–13474. [Google Scholar] [CrossRef] [Green Version]
- Buffo, A.; Rolando, C.; Ceruti, S. Astrocytes in the damaged brain: Molecular and cellular insights into their reactive response and healing potential. Biochem. Pharmacol. 2010, 79, 77–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar]
- Maccioni, R.B.; Rojo, L.E.; Fernández, J.A.; Kuljis, R.O. The Role of Neuroimmunomodulation in Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 2009, 1153, 240–246. [Google Scholar] [CrossRef]
- Cortés, N.; Andrade, V.; Guzmán-Martínez, L.; Estrella, M.; Maccioni, R.B. Neuroimmune Tau Mechanisms: Their Role in the Progression of Neuronal Degeneration. Int. J. Mol. Sci. 2018, 19, 956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapiola, T.; Alafuzoff, I.; Herukka, S.-K.; Parkkinen, L.; Hartikainen, P.; Soininen, H.; Pirttilä, T. Cerebrospinal Fluid β-Amyloid 42 and Tau Proteins as Biomarkers of Alzheimer-Type Pathologic Changes in the Brain. Arch. Neurol. 2009, 66, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Sunderland, T.; Linker, G.; Mirza, N.; Putnam, K.T.; Friedman, D.L.; Kimmel, L.H.; Bergeson, J.; Manetti, G.J.; Zimmermann, M.; Tang, B.; et al. Decreased β-Amyloid1-42 and Increased Tau Levels in Cerebrospinal Fluid of Patients with Alzheimer Disease. JAMA 2003, 289, 2094–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of Tau Pathology in a Model of Early Alzheimer’s Disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Ye, Y. Filamentous recombinant human Tau activates primary astrocytes via an integrin receptor complex. Nat. Commun. 2021, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Yue, Q.; Hoi, M.P.M. Emerging roles of astrocytes in blood-brain barrier disruption upon amyloid-beta insults in Alzheimer’s disease. Neural Regen. Res. 2023, 18, 1890. [Google Scholar] [PubMed]
- Ugolini, F.; Lana, D.; Nardiello, P.; Nosi, D.; Pantano, D.; Casamenti, F.; Giovannini, M.G. Different patterns of neurodegeneration and glia activation in CA1 and CA3 hippocampal regions of TgCRND8 mice. Front. Aging Neurosci. 2018, 10, 372. [Google Scholar] [CrossRef] [PubMed]
- López-Sánchez, N.; Garrido-García, A.; Ramón-Landreau, M.; Cano-Daganzo, V.; Frade, J.M. E2F4-based gene therapy mitigates the phenotype of the Alzheimer’s disease mouse model 5xFAD. Neurotherapeutics 2021, 18, 2484–2503. [Google Scholar] [CrossRef]
- López-Sánchez, N.; Ramón-Landreau, M.; Trujillo, C.; Garrido-García, A.; Frade, J.M. A mutant variant of E2F4 triggers multifactorial therapeutic effects in 5xFAD mice. Mol. Neurobiol. 2022, 59, 3016–3039. [Google Scholar] [CrossRef] [PubMed]
- Ramón-Landreau, M.; Sánchez-Puelles, C.; López-Sánchez, N.; Lozano-Ureña, A.; Llabrés-Mas, A.M.; Frade, J.M. E2F4DN transgenic mice: A tool for the evaluation of E2F4 as a therapeutic target in neuropathology and brain aging. Int. J. Mol. Sci. 2022, 23, 12093. [Google Scholar] [CrossRef] [PubMed]
- Muraleedharan, A.; Rotem-Dai, N.; Strominger, I.; Anto, N.P.; Isakov, N.; Monsonego, A.; Livneh, E. Protein kinase C eta is activated in reactive astrocytes of an Alzheimer’s disease mouse model: Evidence for its immunoregulatory function in primary astrocytes. Glia 2021, 69, 697–714. [Google Scholar] [CrossRef]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Baumann, N.; Pham-Dinh, D. Biology of Oligodendrocyte and Myelin in the Mammalian Central Nervous System. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef]
- Pak, K.; Chan, S.L.; Mattson, M.P. Presenilin-1 mutation sensitizes oligodendrocytes to glutamate and amyloid toxicities, and exacerbates white matter damage and memory impairment in mice. NeuroMolecular Med. 2003, 3, 53–64. [Google Scholar] [CrossRef]
- Gagyi, E.; Kormos, B.; Castellanos, K.J.; Valyi-Nagy, K.; Korneff, D.; LoPresti, P.; Woltjer, R.; Valyi-Nagy, T. Decreased Oligodendrocyte Nuclear Diameter in Alzheimer’s Disease and Lewy Body Dementia. Brain Pathol. 2012, 22, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, G.; Baer, K.; Buffo, A.; Curtis, M.A.; Faull, R.L.; Rees, M.I.; Götz, M.; Dimou, L.J.G. Dynamic changes in myelin aberrations and oligodendrocyte generation in chronic amyloidosis in mice and men. Glia 2013, 61, 273–286. [Google Scholar] [CrossRef]
- Simpson, J.E.; Fernando, M.S.; Clark, L.; Ince, P.G.; Matthews, F.; Forster, G.; O’Brien, J.T.; Barber, R.; Kalaria, R.N.; Brayne, C.; et al. White matter lesions in an unselected cohort of the elderly: Astrocytic, microglial and oligodendrocyte precursor cell responses. Neuropathol. Appl. Neurobiol. 2007, 33, 410–419. [Google Scholar] [CrossRef]
- Desai, M.K.; Mastrangelo, M.A.; Ryan, D.A.; Sudol, K.L.; Narrow, W.C.; Bowers, W.J. Early Oligodendrocyte/Myelin Pathology in Alzheimer’s Disease Mice Constitutes a Novel Therapeutic Target. Am. J. Pathol. 2010, 177, 1422–1435. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, S.; Ahmed, S.H.; Chen, H.; Ku, G.; Goldberg, M.P.; Hsu, C.Y. Amyloid-β Peptides Are Cytotoxic to Oligodendrocytes. J. Neurosci. 2001, 21, RC118. [Google Scholar] [CrossRef] [Green Version]
- Morishima-Kawashima, M.; Ihara, Y. Alzheimer’s disease: β-Amyloid protein and tau. J. Neurosci. Res. 2002, 70, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 22–35. [Google Scholar] [CrossRef]
- Ayyubova, G. Dysfunctional microglia and tau pathology in Alzheimer’s disease. Rev. Neurosci. 2023, 34, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef]
- Xia, Y.; Prokop, S.; Gorion, K.-M.M.; Kim, J.D.; Sorrentino, Z.A.; Bell, B.M.; Manaois, A.N.; Chakrabarty, P.; Davies, P.; Giasson, B.I. Tau Ser208 phosphorylation promotes aggregation and reveals neuropathologic diversity in Alzheimer’s disease and other tauopathies. Acta Neuropathol. Commun. 2020, 8, 88. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Overk, C.R.; Masliah, E. Pathogenesis of synaptic degeneration in Alzheimer’s disease and Lewy body disease. Biochem. Pharmacol. 2014, 88, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Galimberti, D.; Scarpini, E. Disease-modifying treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2011, 4, 203–216. [Google Scholar] [CrossRef] [Green Version]
- Coughlin, D.G.; Litvan, I. Progressive supranuclear palsy: Advances in diagnosis and management. Park. Relat. Disord. 2020, 73, 105–116. [Google Scholar] [CrossRef]
- Pernègre, C.; Duquette, A.; Leclerc, N. Tau Secretion: Good and Bad for Neurons. Front. Neurosci. 2019, 13, 649. [Google Scholar] [CrossRef] [Green Version]
- Han, P.; Serrano, G.; Beach, T.G.; Caselli, R.J.; Yin, J.; Zhuang, N.; Shi, J. A Quantitative Analysis of Brain Soluble Tau and the Tau Secretion Factor. J. Neuropathol. Exp. Neurol. 2017, 76, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Hampel, H. CSF markers for incipient Alzheimer’s disease. Lancet Neurol. 2003, 2, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Barthélemy, N.R.; Bateman, R.J.; Hirtz, C.; Marin, P.; Becher, F.; Sato, C.; Gabelle, A.; Lehmann, S. Cerebrospinal fluid phospho-tau T217 outperforms T181 as a biomarker for the differential diagnosis of Alzheimer’s disease and PET amyloid-positive patient identification. Alzheimer’s Res. Ther. 2020, 12, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Wells, E.A.; Robinson, A.S. Critical Molecular and Cellular Contributors to Tau Pathology. Biomedicines 2021, 9, 190. [Google Scholar] [CrossRef] [PubMed]
- Mirbaha, H.; Holmes, B.B.; Sanders, D.W.; Bieschke, J.; Diamond, M.I. Tau Trimers Are the Minimal Propagation Unit Spontaneously Internalized to Seed Intracellular Aggregation. J. Biol. Chem. 2015, 290, 14893–14903. [Google Scholar] [CrossRef] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [Green Version]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- D‘Errico, P.; Meyer-Luehmann, M. Mechanisms of Pathogenic Tau and Aβ Protein Spreading in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 265. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Colonna, M. Two-faced behavior of microglia in Alzheimer’s disease. Nat. Neurosci. 2022, 25, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchon, A.; Dietrich, J.; Colonna, M. Cutting Edge: Inflammatory Responses Can Be Triggered by TREM-1, a Novel Receptor Expressed on Neutrophils and Monocytes1. J. Immunol. 2000, 164, 4991–4995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchon, A.; Hernández-Munain, C.; Cella, M.; Colonna, M. A Dap12-Mediated Pathway Regulates Expression of Cc Chemokine Receptor 7 and Maturation of Human Dendritic Cells. J. Exp. Med. 2001, 194, 1111–1122. [Google Scholar] [CrossRef]
- Bouchon, A.; Facchetti, F.; Weigand, M.A.; Colonna, M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature 2001, 410, 1103–1107. [Google Scholar] [CrossRef]
- Lucas, M.; Daniel, L.; Tomasello, E.; Guia, S.; Horschowski, N.; Aoki, N.; Figarella-Branger, D.; Gomez, S.; Vivier, E. Massive inflammatory syndrome and lymphocytic immunodeficiency in KARAP/DAP12-transgenic mice. Eur. J. Immunol. 2002, 32, 2653–2663. [Google Scholar] [CrossRef]
- Bernhagen, J.; Calandra, T.; Mitchell, R.A.; Martin, S.B.; Tracey, K.J.; Voelter, W.; Manogue, K.R.; Cerami, A.; Bucala, R. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature 1993, 365, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Bleharski, J.R.; Kiessler, V.; Buonsanti, C.; Sieling, P.A.; Stenger, S.; Colonna, M.; Modlin, R.L. A Role for Triggering Receptor Expressed on Myeloid Cells-1 in Host Defense During the Early-Induced and Adaptive Phases of the Immune Response1. J. Immunol. 2003, 170, 3812–3818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paloneva, J.; Manninen, T.; Christman, G.; Hovanes, K.; Mandelin, J.; Adolfsson, R.; Bianchin, M.; Bird, T.; Miranda, R.; Salmaggi, A.; et al. Mutations in Two Genes Encoding Different Subunits of a Receptor Signaling Complex Result in an Identical Disease Phenotype. Am. J. Hum. Genet. 2002, 71, 656–662. [Google Scholar] [CrossRef] [Green Version]
- Schmid, C.D.; Sautkulis, L.N.; Danielson, P.E.; Cooper, J.; Hasel, K.W.; Hilbush, B.S.; Sutcliffe, J.G.; Carson, M.J. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem. 2002, 83, 1309–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, N.; Kimura, S.; Takiyama, Y.; Atsuta, Y.; Abe, A.; Sato, K.; Katagiri, M. The Role of the DAP12 Signal in Mouse Myeloid Differentiation1. J. Immunol. 2000, 165, 3790–3796. [Google Scholar] [CrossRef]
- Kaifu, T.; Nakahara, J.; Inui, M.; Mishima, K.; Momiyama, T.; Kaji, M.; Sugahara, A.; Koito, H.; Ujike-Asai, A.; Nakamura, A.; et al. Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J. Clin. Investig. 2003, 111, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Piccio, L.; Buonsanti, C.; Cella, M.; Tassi, I.; Schmidt, R.E.; Fenoglio, C.; Rinker, J., II; Naismith, R.T.; Panina-Bordignon, P.; Passini, N.; et al. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 2008, 131, 3081–3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Lucena, D.; Kruse, N.; Thüne, K.; Schmitz, M.; Villar-Piqué, A.; da Cunha, J.E.G.; Hermann, P.; López-Pérez, Ó.; Andrés-Benito, P.; Ladogana, A.; et al. TREM2 expression in the brain and biological fluids in prion diseases. Acta Neuropathol. 2021, 141, 841–859. [Google Scholar] [CrossRef] [PubMed]
- Piccio, L.; Deming, Y.; Del-Águila, J.L.; Ghezzi, L.; Holtzman, D.M.; Fagan, A.M.; Fenoglio, C.; Galimberti, D.; Borroni, B.; Cruchaga, C. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016, 131, 925–933. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Calvet, M.; Araque Caballero, M.Á.; Kleinberger, G.; Bateman, R.J.; Fagan, A.M.; Morris, J.C.; Levin, J.; Danek, A.; Ewers, M.; Haass, C.; et al. Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci. Transl. Med. 2016, 8, ra178–ra369. [Google Scholar] [CrossRef] [Green Version]
- Henjum, K.; Almdahl, I.S.; Årskog, V.; Minthon, L.; Hansson, O.; Fladby, T.; Nilsson, L.N.G. Cerebrospinal fluid soluble TREM2 in aging and Alzheimer’s disease. Alzheimer’s Res. Ther. 2016, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Katsumoto, A.; Kokiko-Cochran, O.N.; Bemiller, S.M.; Xu, G.; Ransohoff, R.M.; Lamb, B.T. Triggering receptor expressed on myeloid cells 2 deficiency exacerbates injury-induced inflammation in a mouse model of tauopathy. Front. Immunol. 2022, 13, 978423. [Google Scholar] [CrossRef] [PubMed]
- Guennewig, B.; Lim, J.; Marshall, L.; McCorkindale, A.N.; Paasila, P.J.; Patrick, E.; Kril, J.J.; Halliday, G.M.; Cooper, A.A.; Sutherland, G.T. Defining early changes in Alzheimer’s disease from RNA sequencing of brain regions differentially affected by pathology. Sci. Rep. 2021, 11, 4865. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, C.; Qi, R.; Fu, H.; Ma, Q. scREAD: A Single-Cell RNA-Seq Database for Alzheimer’s Disease. iScience 2020, 23, 101769. [Google Scholar] [CrossRef]
- Walker, D.G.; Lue, L.-F. Immune phenotypes of microglia in human neurodegenerative disease: Challenges to detecting microglial polarization in human brains. Alzheimer’s Res. Ther. 2015, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, D.M. A changing perspective on the role of neuroinflammation in Alzheimer’s disease. Int. J. Alzheimer’s Dis. 2012, 2012, 495243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudduth, T.L.; Schmitt, F.A.; Nelson, P.T.; Wilcock, D.M. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1051–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.M.H.; Kumasaka, N.; Calvert, F.; Hammond, T.R.; Knights, A.; Panousis, N.; Park, J.S.; Schwartzentruber, J.; Liu, J.; Kundu, K.; et al. A map of transcriptional heterogeneity and regulatory variation in human microglia. Nat. Genet. 2021, 53, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization from M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef]
- Monterey, M.D.; Wei, H.; Wu, X.; Wu, J.Q. The Many Faces of Astrocytes in Alzheimer’s Disease. Front. Neurol. 2021, 12, 619626. [Google Scholar] [CrossRef]
- Soreq, L.; Bird, H.; Mohamed, W.; Hardy, J. Single-cell RNA sequencing analysis of human Alzheimer’s disease brain samples reveals neuronal and glial specific cells differential expression. PLoS ONE 2023, 18, e0277630. [Google Scholar] [CrossRef]
- Casali, B.T.; MacPherson, K.P.; Reed-Geaghan, E.G.; Landreth, G.E. Microglia depletion rapidly and reversibly alters amyloid pathology by modification of plaque compaction and morphologies. Neurobiol. Dis. 2020, 142, 104956. [Google Scholar] [CrossRef]
- Oosterhof, N.; Chang, I.J.; Karimiani, E.G.; Kuil, L.E.; Jensen, D.M.; Daza, R.; Young, E.; Astle, L.; van der Linde, H.C.; Shivaram, G.M. Homozygous mutations in CSF1R cause a pediatric-onset leukoencephalopathy and can result in congenital absence of microglia. Am. J. Hum. Genet. 2019, 104, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- Scheiblich, H.; Trombly, M.; Ramirez, A.; Heneka, M.T. Neuroimmune connections in aging and neurodegenerative diseases. Trends Immunol. 2020, 41, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Thawkar, B.S.; Kaur, G. Inhibitors of NF-κB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J. Neuroimmunol. 2019, 326, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Sperlagh, B.; Illes, P. P2X7 receptor: An emerging target in central nervous system diseases. Trends Pharmacol. Sci. 2014, 35, 537–547. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Jones, D.N.C. Emerging role of the P2X7-NLRP3-IL1β pathway in mood disorders. Psychoneuroendocrinology 2018, 98, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Knight, G.E. The potential of P2X7 receptors as a therapeutic target, including inflammation and tumour progression. Purinergic Signal. 2018, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhu, Z.; Klebe, D.; Bian, H.; Krafft, P.R.; Tang, J.; Zhang, J.; Zhang, J.H. Role of P2X purinoceptor 7 in neurogenic pulmonary edema after subarachnoid hemorrhage in rats. PLoS ONE 2014, 9, e89042. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; McLarnon, J.G. Thalidomide inhibition of perturbed vasculature and glial-derived tumor necrosis factor-α in an animal model of inflamed Alzheimer’s disease brain. Neurobiol. Dis. 2008, 29, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-W.; Lee, M.-S. Mitochondria and the NLRP3 inflammasome: Physiological and pathological relevance. Arch. Pharmacal Res. 2016, 39, 1503–1518. [Google Scholar] [CrossRef]
- Li, Q.; Yang, Y.; Reis, C.; Tao, T.; Li, W.; Li, X.; Zhang, J.H. Cerebral Small Vessel Disease. Cell Transplant. 2018, 27, 1711–1722. [Google Scholar] [CrossRef]
- Yin, J.; Zhao, F.; Chojnacki, J.E.; Fulp, J.; Klein, W.L.; Zhang, S.; Zhu, X. NLRP3 inflammasome inhibitor ameliorates amyloid pathology in a mouse model of Alzheimer’s disease. Mol. Neurobiol. 2018, 55, 1977–1987. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.-Y.; Zhao, D.-Q.; Wang, H.-B.; Feng, S.; Liu, S.-B.; Xing, J.-H.; Qu, Y.; Gao, P.; Sun, X.-L.; Zhao, M.-G. A New Chiral Pyrrolyl α-Nitronyl Nitroxide Radical Attenuates β-Amyloid Deposition and Rescues Memory Deficits in a Mouse Model of Alzheimer Disease. Neurotherapeutics 2013, 10, 340–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, L.; Liao, Y.-T.; He, J.-C.; Xie, C.-L.; Chen, S.-Y.; Fan, H.-H.; Su, Z.-P.; Wang, Z. Plasma long non-coding RNA BACE1 as a novel biomarker for diagnosis of Alzheimer disease. BMC Neurol. 2018, 18, 4. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, V.; Dye, R.; Pakavathkumar, P.; Foveau, B.; Flores, J.; Hyman, B.; Ghetti, B.; Koller, B.H.; LeBlanc, A.C. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 2015, 22, 1676–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemetchek, M.D.; Stierle, A.A.; Stierle, D.B.; Lurie, D.I. The Ayurvedic plant Bacopa monnieri inhibits inflammatory pathways in the brain. J. Ethnopharmacol. 2017, 197, 92–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, J.; Noël, A.; Foveau, B.; Lynham, J.; Lecrux, C.; LeBlanc, A.C. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat. Commun. 2018, 9, 3916. [Google Scholar] [CrossRef] [Green Version]
- Cignarella, F.; Filipello, F.; Bollman, B.; Cantoni, C.; Locca, A.; Mikesell, R.; Manis, M.; Ibrahim, A.; Deng, L.; Benitez, B.A. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 2020, 140, 513–534. [Google Scholar] [CrossRef]
- Price, B.R.; Sudduth, T.L.; Weekman, E.M.; Johnson, S.; Hawthorne, D.; Woolums, A.; Wilcock, D.M. Therapeutic Trem2 activation ameliorates amyloid-beta deposition and improves cognition in the 5XFAD model of amyloid deposition. J. Neuroinflammation 2020, 17, 238. [Google Scholar] [CrossRef] [PubMed]
- Schlepckow, K.; Monroe, K.M.; Kleinberger, G.; Cantuti-Castelvetri, L.; Parhizkar, S.; Xia, D.; Willem, M.; Werner, G.; Pettkus, N.; Brunner, B. Enhancing protective microglial activities with a dual function TREM 2 antibody to the stalk region. EMBO Mol. Med. 2020, 12, e11227. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20200785. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Danao, J.; Talreja, S.; Wen, P.; Yin, J.; Sun, N.; Li, C.-M.; Chui, D.; Tran, D.; Koirala, S. TREM2-activating antibodies abrogate the negative pleiotropic effects of the Alzheimer’s disease variant Trem2R47H on murine myeloid cell function. J. Biol. Chem. 2018, 293, 12620–12633. [Google Scholar] [CrossRef] [Green Version]
- Fassler, M.; Rappaport, M.S.; Cuño, C.B.; George, J. Engagement of TREM2 by a novel monoclonal antibody induces activation of microglia and improves cognitive function in Alzheimer’s disease models. J. Neuroinflamm. 2021, 18, 19. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, P.; Glebov, K.; Kemmerling, N.; Tien, N.T.; Neumann, H.; Walter, J. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and γ-secretase-dependent intramembranous cleavage. J. Biol. Chem. 2013, 288, 33027–33036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, P.-H.; Colombo, A.V.; Schusser, B.; Dreymueller, D.; Wetzel, S.; Schepers, U.; Herber, J.; Ludwig, A.; Kremmer, E.; Montag, D. Systematic substrate identification indicates a central role for the metalloprotease ADAM10 in axon targeting and synapse function. Elife 2016, 5, e12748. [Google Scholar] [CrossRef]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Asghar, S.; Gao, S.; Su, Z.; Song, J.; Huo, M.; Meng, W.; Ping, Q.; Xiao, Y. A novel LDL-mimic nanocarrier for the targeted delivery of curcumin into the brain to treat Alzheimer’s disease. Colloids Surf. B Biointerfaces 2015, 134, 88–97. [Google Scholar] [CrossRef]
- Li, W.; Guo, Q.; Zhao, H.; Zhang, L.; Li, J.; Gao, J.; Qian, W.; Li, B.; Chen, H.; Wang, H. Novel dual-control poly (N-isopropylacrylamide-co-chlorophyllin) nanogels for improving drug release. Nanomedicine 2012, 7, 383–392. [Google Scholar] [CrossRef]
- Silveira, C.A.; Dias, P.J.; Santos, M.V.; Oliveira, F.P.; Alves, G.M.; Rato, L.; Silva, M.B. The Action of Polyphenols in Diabetes Mellitus and Alzheimer’s Disease: A Common Agent for Overlapping Pathologies. Curr. Neuropharmacol. 2019, 17, 590–613. [Google Scholar] [CrossRef] [Green Version]
- Picone, P.; Ditta, L.A.; Sabatino, M.A.; Militello, V.; San Biagio, P.L.; Di Giacinto, M.L.; Cristaldi, L.; Nuzzo, D.; Dispenza, C.; Giacomazza, D. Ionizing radiation-engineered nanogels as insulin nanocarriers for the development of a new strategy for the treatment of Alzheimer’s disease. Biomaterials 2016, 80, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamburini, B.; Badami, G.D.; La Manna, M.P.; Shekarkar Azgomi, M.; Caccamo, N.; Dieli, F. Emerging Roles of Cells and Molecules of Innate Immunity in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 11922. https://doi.org/10.3390/ijms241511922
Tamburini B, Badami GD, La Manna MP, Shekarkar Azgomi M, Caccamo N, Dieli F. Emerging Roles of Cells and Molecules of Innate Immunity in Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(15):11922. https://doi.org/10.3390/ijms241511922
Chicago/Turabian StyleTamburini, Bartolo, Giusto Davide Badami, Marco Pio La Manna, Mojtaba Shekarkar Azgomi, Nadia Caccamo, and Francesco Dieli. 2023. "Emerging Roles of Cells and Molecules of Innate Immunity in Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 15: 11922. https://doi.org/10.3390/ijms241511922