
Effect of Substituents on Molecular Reactivity during Lignin Oxidation by Chlorine Dioxide: A Density Functional Theory Study
 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
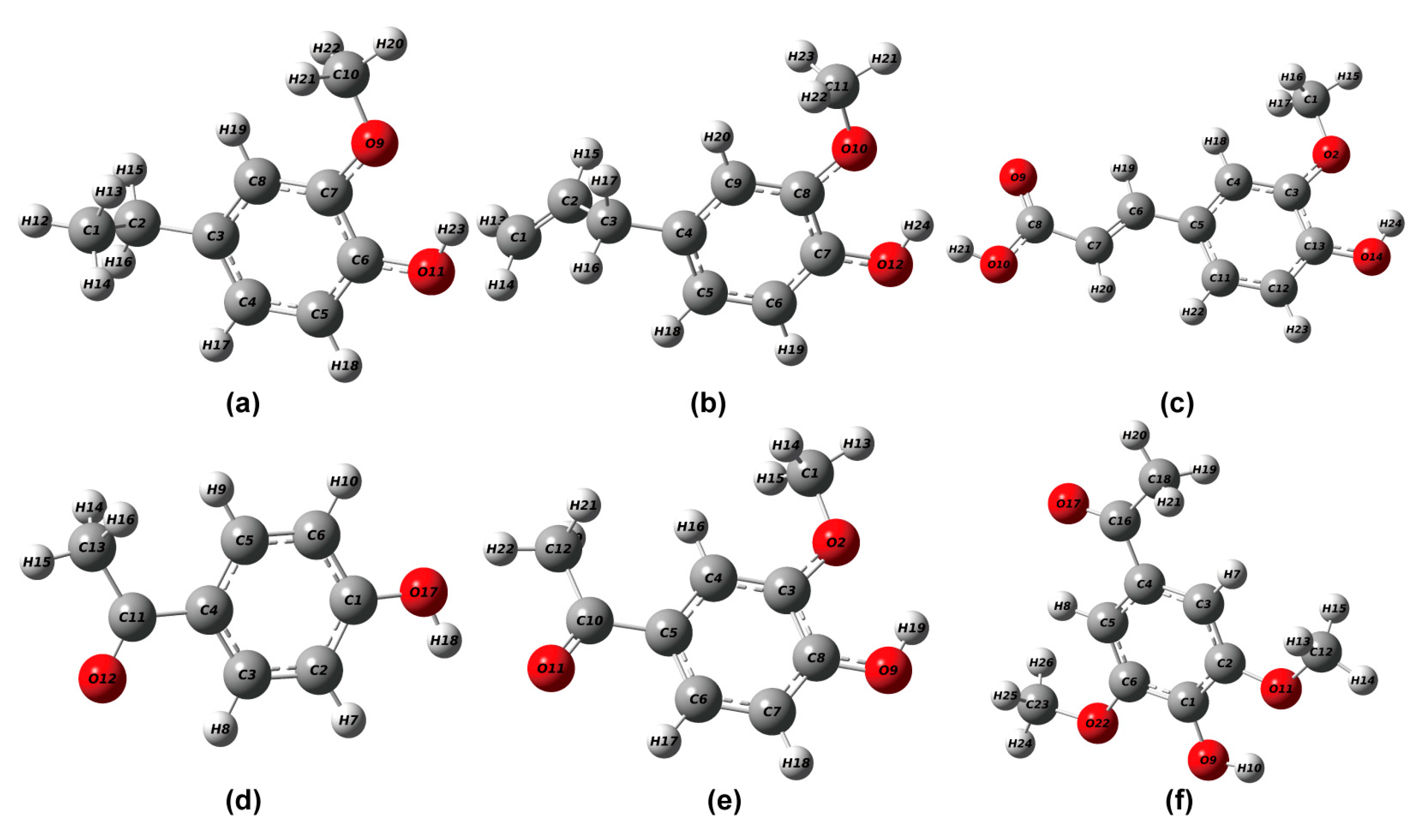
2.1. Optimization of Phenolic Lignin Model Compound Structure
2.2. Effect of Different Benzene Ring Branched Substituents on the Uptake of Lignin Hydroxyl Hydrogen Atoms by ClO2
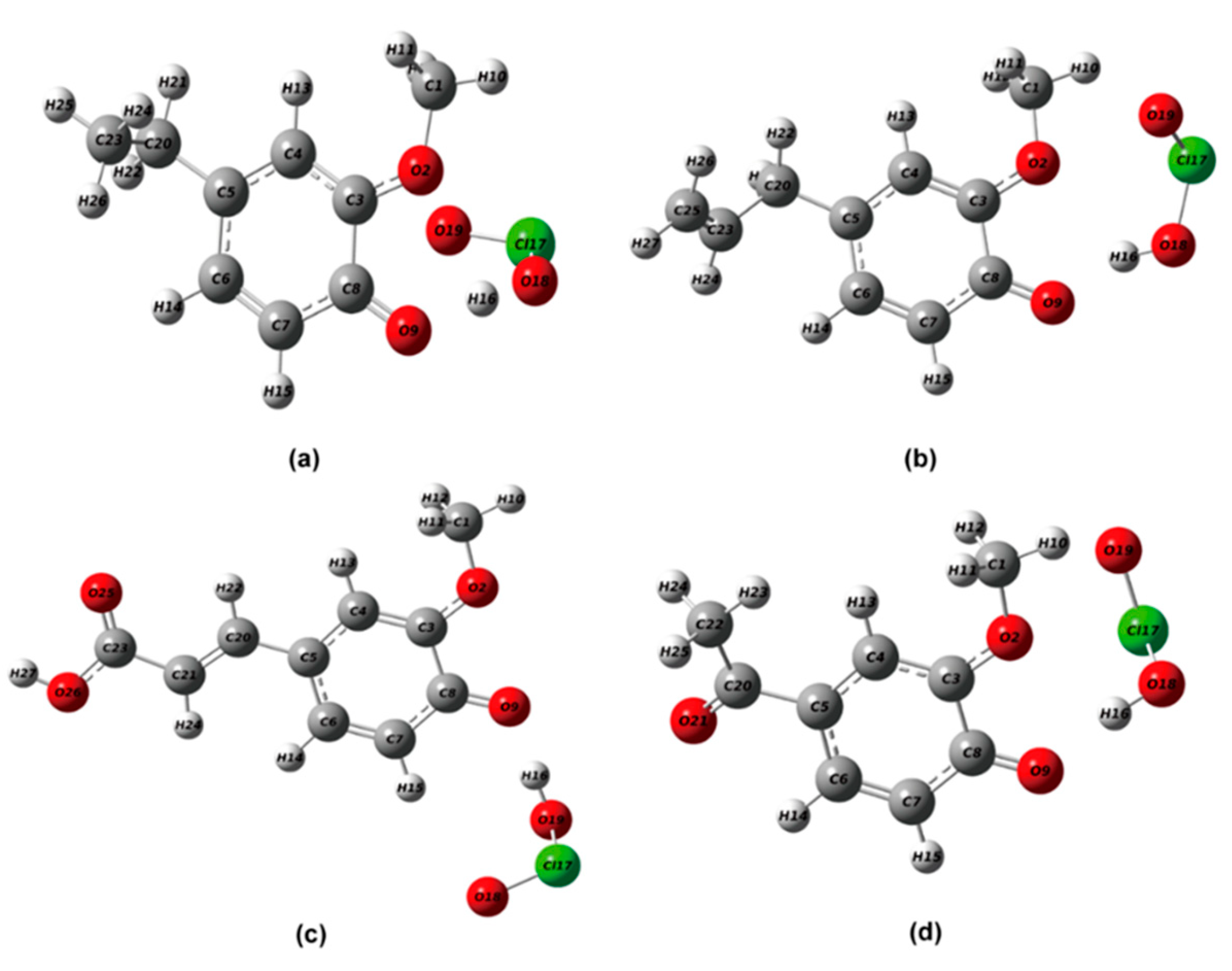
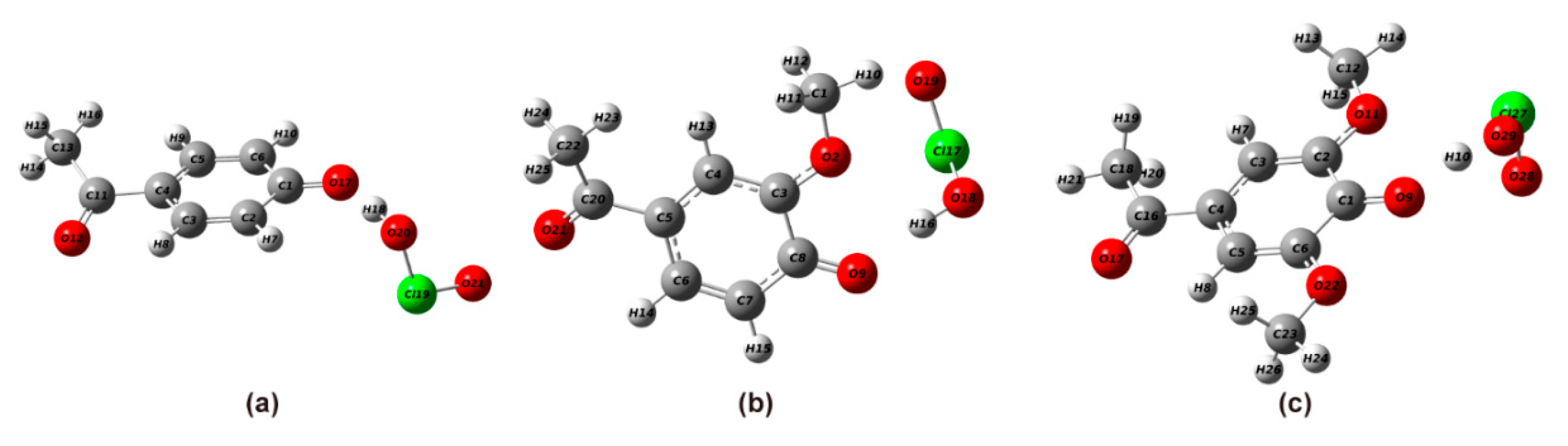
2.2.1. Structural Optimization of Phenolic Lignin-ClO2 Transition State with Different Benzene Ring Branched Substituents
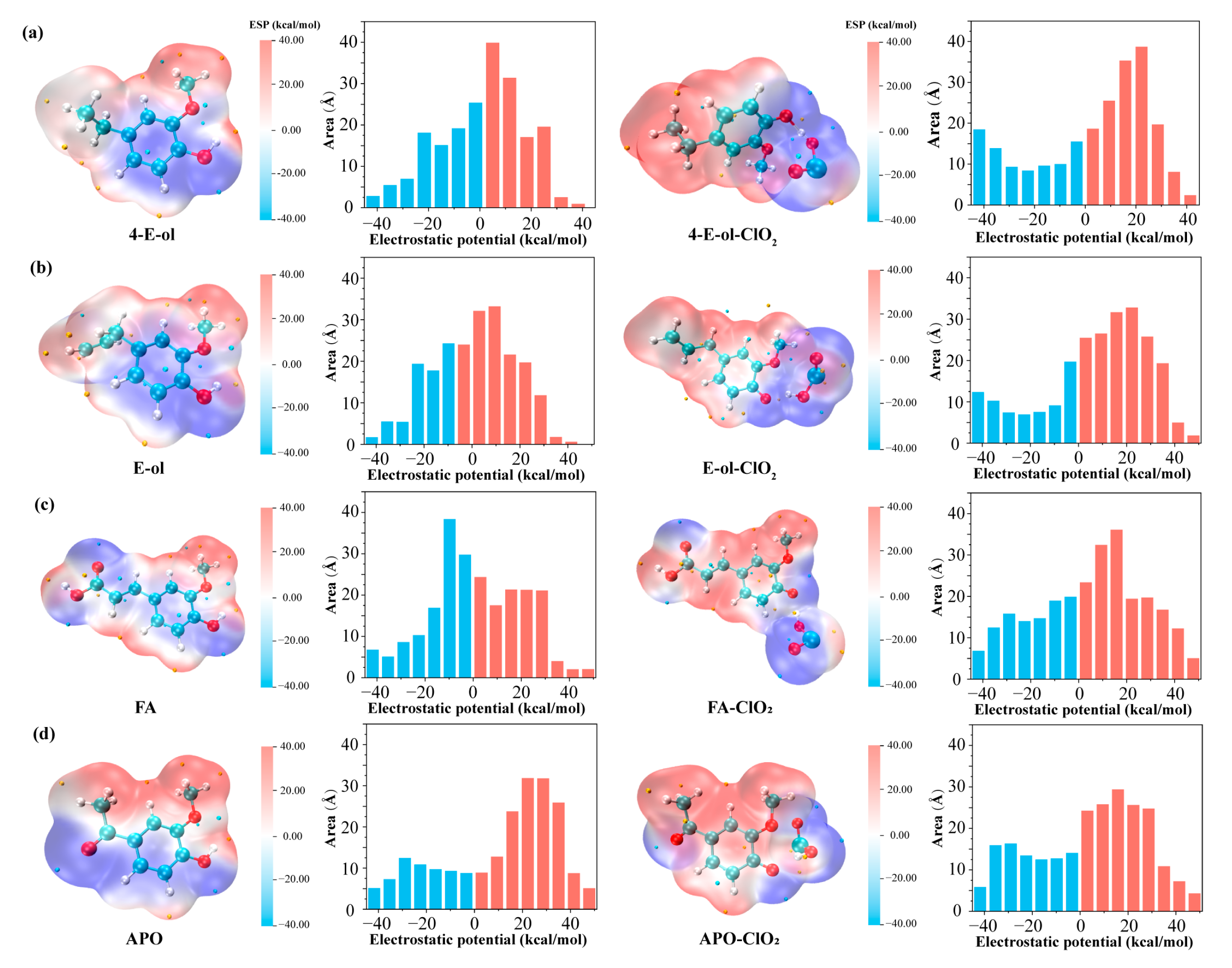
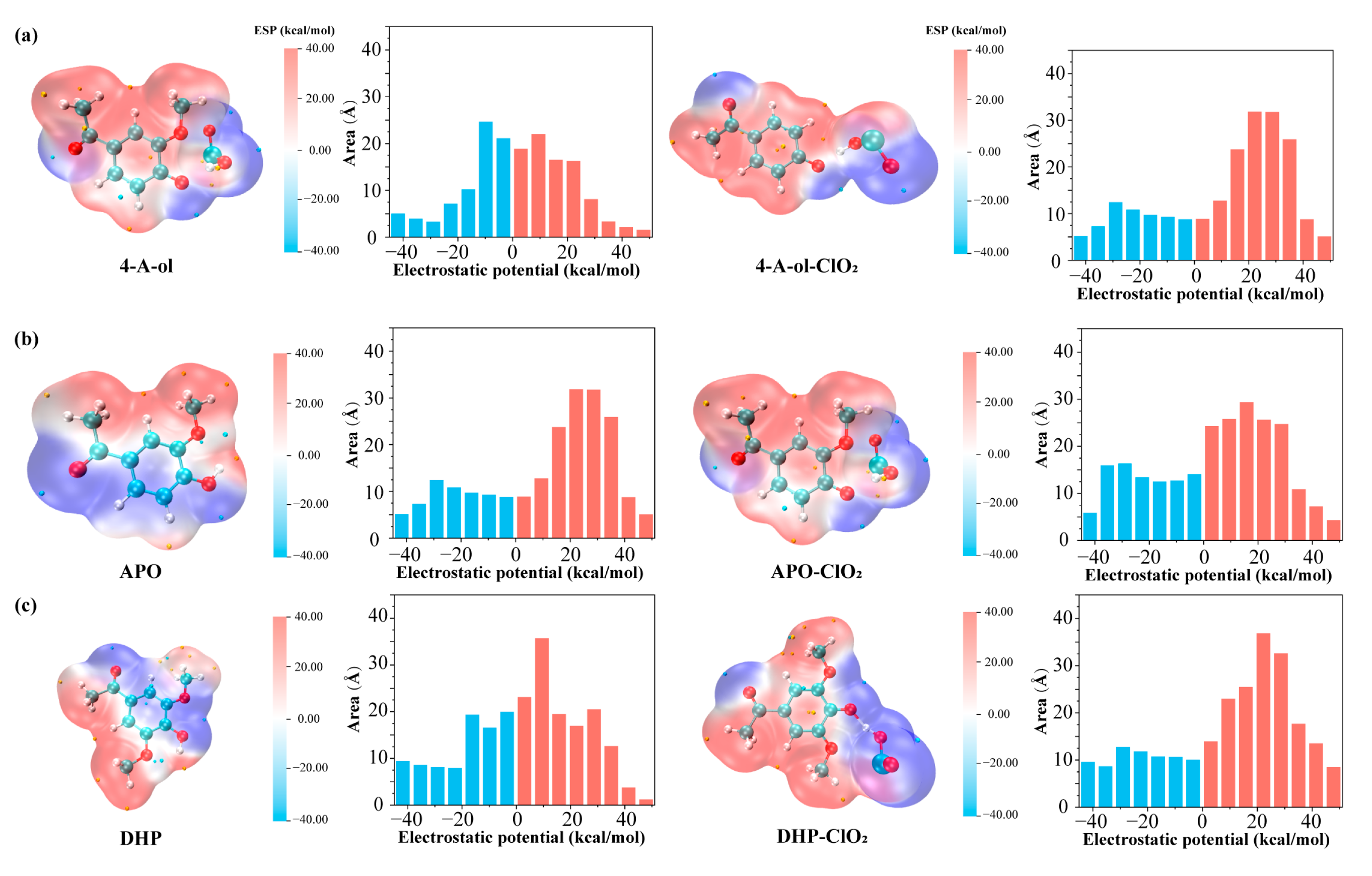
2.2.2. Electrostatic Potential Distribution of Phenolic Lignin-ClO2 Transition State with Different Benzene Ring Branched Substituents
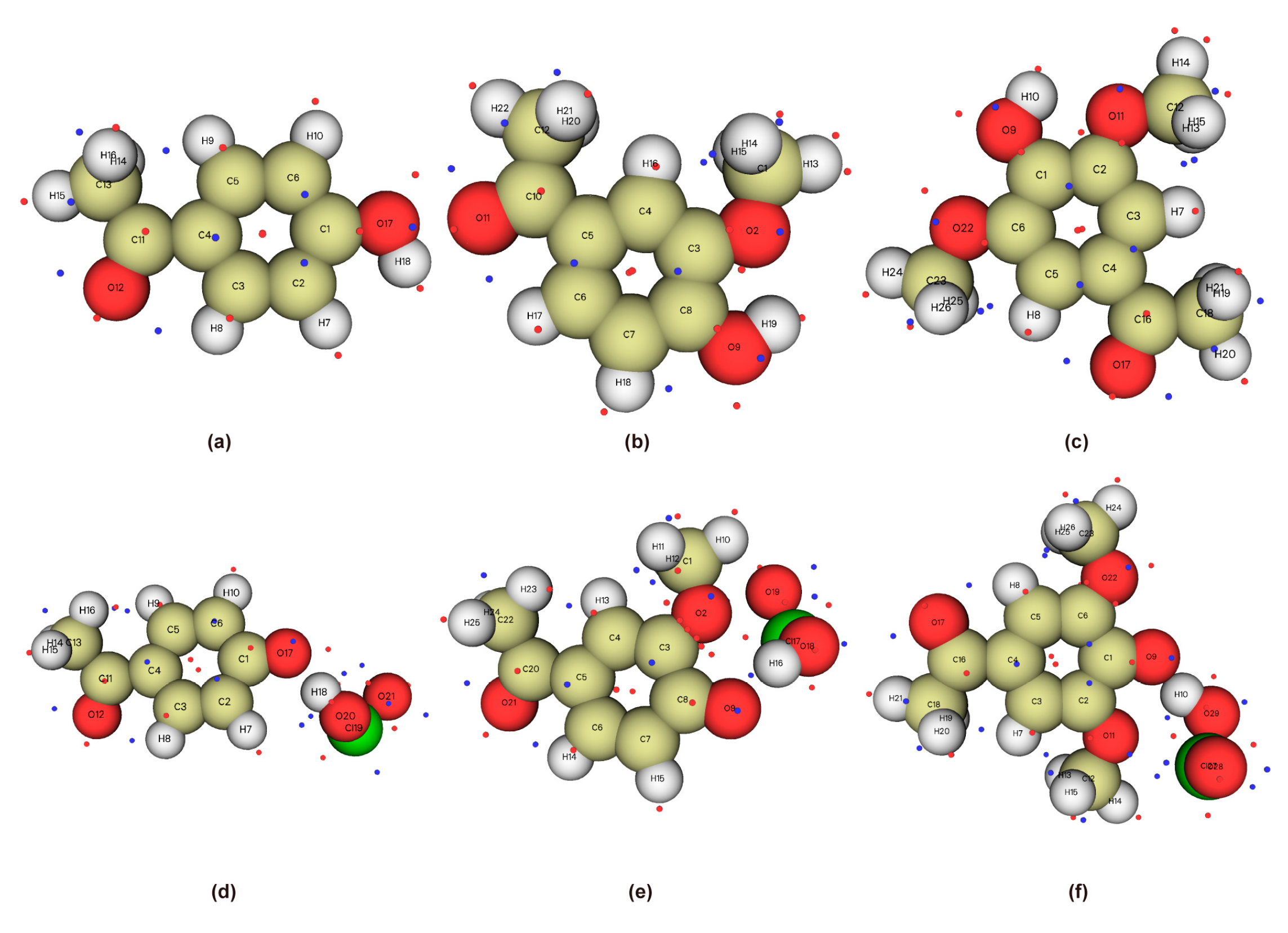
2.2.3. Natural Atomic Charges of Phenolic Lignin-ClO2 Transition States with Different Benzene Ring Branched Substituents
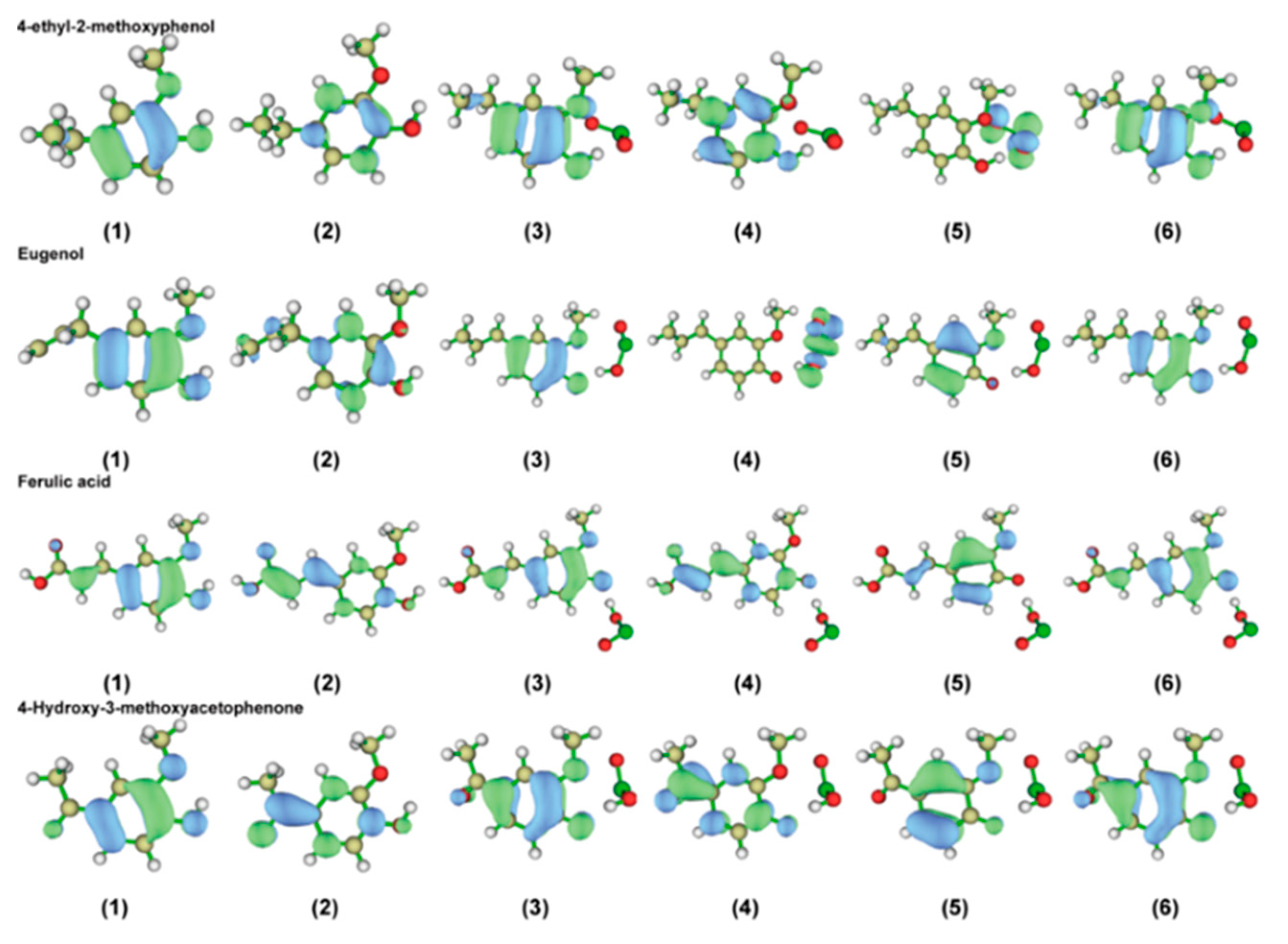
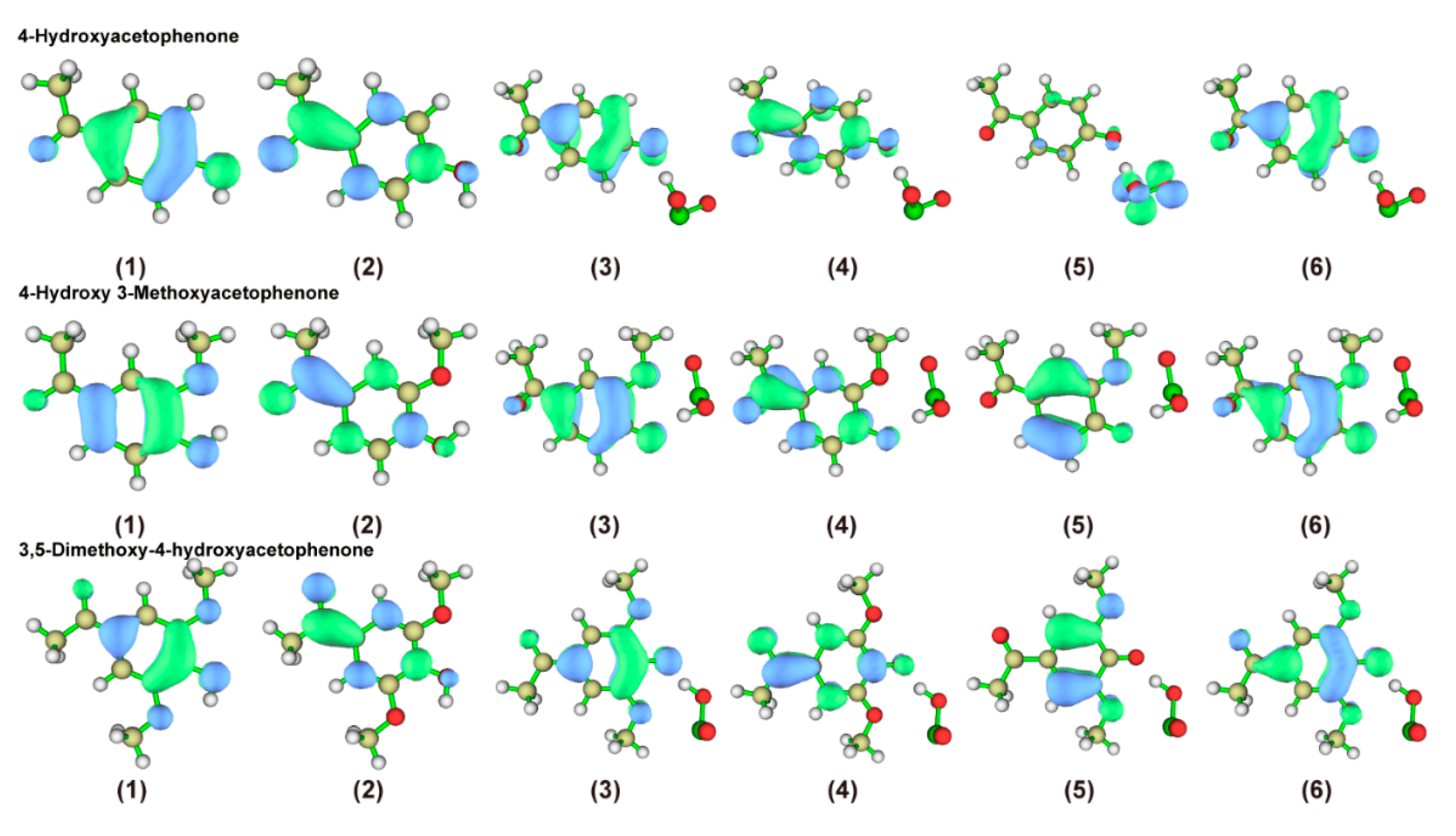
2.2.4. Characterization of the Front-Line Orbital Distribution of Phenolic Lignin-ClO2 Transition States with Different Benzene Ring Branched Substituents
2.2.5. Distribution Characteristics of Fukui Functions and Double Descriptors for Phenolic Lignin-ClO2 Transition States with Different Benzene Ring Branched Substituents
2.2.6. Calculation of Energy Barriers for Phenolic Lignin-ClO2 Transition States with Different Benzene Ring Branched Substituents
2.3. Effect of Methoxy on the Uptake of Lignin Hydroxyl Hydrogen Atoms by ClO2
2.3.1. Structural Optimization of Phenolic Lignin-ClO2 Transition States with Different Methoxy Numbers
2.3.2. Characteristics of Electrostatic Potential Distribution of Phenolic Lignin-ClO2 Transition States with Different Numbers of Methoxy
2.3.3. Natural Atomic Charges of Phenolic Lignin-ClO2 Transition States with Different Methoxy Numbers
2.3.4. Characterization of the Front-Line Orbital Distribution of Phenolic Lignin-ClO2 Transition States with Different Methoxy Numbers
2.3.5. Characteristics of Fukui Functions and Double Descriptor Distribution of Phenolic Lignin-ClO2 Transition States with Different Numbers of Methoxy
2.3.6. Calculation of Energy Barriers for Phenolic Lignin-ClO2 Transition States with Different Numbers of Methoxy
3. Methods and Materials
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lei, M.; Luo, B.; Zhang, Q.T.; Guo, C.Y.; Chi, M.C.; Yun, X.J.; Chen, C.Z.; Min, D.Y.; Wang, S.F. Kinetics of the reaction between a lignin model compound and chlorine dioxide. Chem. Eng. J. 2020, 393, 124783. [Google Scholar] [CrossRef]
- Liu, B.J.; Liu, L.; Deng, B.J.; Huang, C.X.; Zhu, J.T.; Liang, L.L.; He, X.L.; Wei, Y.X.; Qin, C.R.; Liang, C.; et al. Application and prospect of organic acid pretreatment in lignocellulosic biomass separation: A review. Int. J. Biol. Macromol. 2022, 222, 1400–1413. [Google Scholar] [CrossRef]
- Qin, C.R.; Zeng, H.L.; Liu, B.J.; Zhu, J.T.; Wang, F.; Wang, S.; Liang, C.; Huang, C.X.; Ma, J.L.; Yao, S.Q. Efficient removal of residual lignin from eucalyptus pulp via high-concentration chlorine dioxide treatment and its effect on the properties of residual solids. Bioresour. Technol. 2022, 360, 127621. [Google Scholar] [CrossRef] [PubMed]
- Corona-Vasquez, B.; Rennecker, J.L.; Driedger, A.M.; Marinas, B.J. Sequential inactivation of Cryptosporidium parvum oocysts with chlorine dioxide followed by free chlorine or monochloramine. Water Res. 2002, 36, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Deng, B.J.; Liang, J.R.; Li, J.; Liu, B.J.; Wang, F.; Qin, C.R.; Yao, S.Q. Effects of the Preferential Oxidation of Phenolic Lignin Using Chlorine Dioxide on Pulp Bleaching Efficiency. Int. J. Mol. Sci. 2022, 23, 3310. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.H.; Pan, H.; Zhou, Y.H.; Hse, C.Y.; Liu, C.G.; Zhang, B.F.; Xu, B. Chemical Groups and Structural Characterization of Lignin via Thiol-Mediated Demethylation. J. Wood Chem. Technol. 2014, 34, 122–134. [Google Scholar] [CrossRef]
- Weng, S.X.; Zhang, G.X.; Hu, Y.; Bo, C.Y.; Song, F.; Feng, G.D.; Hu, L.H.; Zhou, Y.H.; Jia, P.Y. Lignin Degradation via Chlorine Dioxide at Room Temperature: Chemical Groups and Structural Characterization. Int. J. Mol. Sci. 2023, 24, 1479. [Google Scholar] [CrossRef]
- Liu, B.J.; Qin, C.R.; Zhang, F.Q.; Wang, S.; Liang, C.; Nie, S.X.; Wang, S.F.; Yao, S.Q. Reaction Mechanism of Phenolic Lignin and High Concentration Chlorine Dioxide and Its Application. ACS Omega 2020, 5, 22475–22481. [Google Scholar] [CrossRef]
- Nie, S.X.; Liu, X.L.; Wu, Z.M.; Zhan, L.; Yin, G.D.; Yao, S.Q.; Song, H.N.; Wang, S.F. Kinetics study of oxidation of the lignin model compounds by chlorine dioxide. Chem. Eng. J. 2014, 241, 410–417. [Google Scholar] [CrossRef]
- Lin, M.S.; Yang, L.J.; Zhang, H.; Xia, Y.; He, Y.; Lan, W.; Ren, J.L.; Yue, F.X.; Lu, F.C. Revealing the structure-activity relationship between lignin and anti-UV radiation. Ind. Crops Prod. 2021, 174, 114212. [Google Scholar] [CrossRef]
- Shi, L.S.; Ge, J.Y.; Zhang, F.Q.; Nie, S.X.; Qin, C.R.; Yao, S.Q. Difference in adsorbable organic halogen formation between phenolic and non-phenolic lignin model compounds in chlorine dioxide bleaching. R. Soc. Open Sci. 2019, 6, 191202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alekseev, N.V.; Chernyshev, E.A. Quantum-chemical study of germanium-substituent bonds in tricoordinated germanium compounds. J. Struct. Chem. 2008, 49, 828–836. [Google Scholar] [CrossRef]
- Macedo, G.K.; Haiduke, R.L.A. Quantum Theory Atoms in Molecules Study about the Inductive Effect of Substituents in Methane Derivatives. ACS Omega 2020, 5, 9041–9045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yu, S. Study on the Relationship between Inductive Effect, Conjugative Effect and Activity of Electrophilic Substitution Reaction Based on 13C Chemical Shift. Chemistry 2006, 69, 331–336. [Google Scholar] [CrossRef]
- Zhang, L.X.; Liang, Y.Z.; Ouyang, Y.Z. Prediction of mass spectral characteristic of indole alkaloids utilizing inductive effect index. Chem. J. Chin. Univ.-Chin. 2008, 29, 1740–1743. [Google Scholar] [CrossRef]
- Schultz, T.P.; Fisher, T.H. Alkaline hydrolysis of nonphenolic beta-0-4 lignin models: Substituent effect of the A-ring on the rate. Holzforschung 2002, 56, 592–594. [Google Scholar] [CrossRef]
- Anjalikrishna, P.K.; Gadre, S.R.; Suresh, C.H. Antiaromaticity-Aromaticity Interplay in Fused Benzenoid Systems Using Molecular Electrostatic Potential Topology. J. Phys. Chem. A 2021, 125, 5999–6012. [Google Scholar] [CrossRef]
- Higashioji, T.; Hada, M.; Sugimoto, M.; Nakatsuji, H. Basis set dependence of magnetic shielding constant calculated by the Hartree-Fock finite perturbation method. Chem. Phys. 1996, 203, 159–175. [Google Scholar] [CrossRef]
- Hofmann, J.; Clark, T.; Heinrich, M.R. Strongly Directing Substituents in the Radical Arylation of Substituted Benzenes. J. Org. Chem. 2016, 81, 9785–9791. [Google Scholar] [CrossRef]
- Qin, C.R.; Liu, B.J.; Huang, L.Z.; Liang, C.; Gao, C.; Yao, S.Q. Adsorptive removal of adsorbable organic halogens by activated carbon. R. Soc. Open Sci. 2018, 5, 181507. [Google Scholar] [CrossRef] [Green Version]
- Rice, B.M.; Sahu, S.; Owens, F.J. Density functional calculations of bond dissociation energies for NO2 scission in some nitroaromatic molecules. Theochem.-J. Mol. Struct. 2002, 583, 69–72. [Google Scholar] [CrossRef]
- Olmez-Hanci, T.; Arslan-Alaton, I. Comparison of sulfate and hydroxyl radical based advanced oxidation of phenol. Chem. Eng. J. 2013, 224, 10–16. [Google Scholar] [CrossRef]
- Vandemaele, M.; Franco, J.; Tyaginov, S.; Groeseneken, G.; Kaczer, B. Modeling of Repeated FET Hot-Carrier Stress and Anneal Cycles Using Si-H Bond Dissociation/Passivation Energy Distributions. IEEE Trans. Electron. Dev. 2021, 68, 1454–1460. [Google Scholar] [CrossRef]
- Bach, R.D.; Schlegel, H.B. The Bond Dissociation Energy of the N-O Bond. J. Phys. Chem. A 2021, 125, 5014–5021. [Google Scholar] [CrossRef] [PubMed]
- Michalik, M.; Poliak, P.; Lukes, V.; Klein, E. From phenols to quinones: Thermodynamics of radical scavenging activity of para-substituted phenols. Phytochemistry 2019, 166, 112077. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Y.; Lin, Y.L.; Zhang, T.Y.; Hu, C.Y.; Tang, Y.L.; Deng, J.; Xu, B. Chlorine dioxide-based oxidation processes for water purification: A review. J. Hazard. Mater. 2022, 436, 129195. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Peralta-Inga, Z.; Politzer, P. Conformational dependence of molecular surface electrostatic potentials. Int. J. Quantum Chem. 1999, 75, 267–273. [Google Scholar] [CrossRef]
- Storer, M.C.; Hunter, C.A. The surface site interaction point approach to non-covalent interactions. Chem. Soc. Rev. 2022, 51, 10064–10082. [Google Scholar] [CrossRef]
- Galabov, B.; Nikolova, V.; Cheshmedzhieva, D.; Hadjieva, B.; Schaefer, H.F. Hyperconjugative effects in pi-hydrogen bonding: Theory and experiment. J. Comput. Chem. 2018, 39, 527–534. [Google Scholar] [CrossRef]
- Sosorev, A.; Dominskiy, D.; Chernyshov, I.; Efremov, R. Tuning of Molecular Electrostatic Potential Enables Efficient Charge Transport in Crystalline Azaacenes: A Computational Study. Int. J. Mol. Sci. 2020, 21, 5654. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Comparison of Computational Methods for Atomic Charges. Acta Phys.-Chim. Sin. 2012, 28, 1–18. [Google Scholar] [CrossRef]
- Chen, J.X.; Yang, J.; Ma, L.L.; Li, J.; Shahzad, N.; Kim, C.K. Structure-antioxidant activity relationship of methoxy, phenolic hydroxyl, and carboxylic acid groups of phenolic acids. Sci. Rep. 2020, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Eilmes, A.; Kubisiak, P.; Wrobel, P. Explicit and Hybrid Solvent Models for Estimates of Parameters Relevant to the Reduction Potential of Ethylene Carbonate. Int. J. Mol. Sci. 2022, 23, 15590. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.W.; Hull, E.A.; Windus, T.L. Valence Virtual Orbitals: An Unambiguous ab Initio Quantification of the LUMO Concept. J. Phys. Chem. A 2015, 119, 10408–10427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bultinck, P.; Clarisse, D.; Ayers, P.W.; Carbo-Dorca, R. The Fukui matrix: A simple approach to the analysis of the Fukui function and its positive character. Phys. Chem. Chem. Phys. 2011, 13, 6110–6115. [Google Scholar] [CrossRef] [Green Version]
- Zaklika, J.; Hladyszowski, J.; Ordon, P.; Komorowski, L. From the Electron Density Gradient to the Quantitative Reactivity Indicators: Local Softness and the Fukui Function. ACS Omega 2022, 7, 7745–7758. [Google Scholar] [CrossRef]
- Stuyver, T.; Shaik, S. Promotion Energy Analysis Predicts Reaction Modes: Nucleophilic and Electrophilic Aromatic Substitution Reactions. J. Am. Chem. Soc. 2021, 143, 4367–4378. [Google Scholar] [CrossRef]
- Figueredo, S.; Paez, M.; Torres, F. The electrophilic descriptor. Comput. Theor. Chem. 2019, 1157, 34–39. [Google Scholar] [CrossRef]
- Ekici, O.; Demircioglu, Z.; Ersanli, C.C.; Cukurovali, A. Experimental and theoretical approach: Chemical activity, charge transfer of DNA/ECT, thermodinamic, spectroscopic, structural and electronic properties of N-(4-(3-methyl-3-phenylcyclobutyl)thiazol-2-yl)acetamide molecule. J. Mol. Struct. 2020, 1204, 127513. [Google Scholar] [CrossRef]
- Kogias, I.; Lee, A.R.; Ragy, S.; Adesso, G. Quantification of Gaussian Quantum Steering. Phys. Rev. Lett. 2015, 114, 060403. [Google Scholar] [CrossRef] [Green Version]
- Valdes, H.; Pluhackova, K.; Pitonak, M.; Rezac, J.; Hobza, P. Benchmark database on isolated small peptides containing an aromatic side chain: Comparison between wave function and density functional theory methods and empirical force field. Phys. Chem. Chem. Phys. 2008, 10, 2747–2757. [Google Scholar] [CrossRef] [PubMed]
- Rayne, S.; Forest, K. Gas phase isomerization enthalpies of organic compounds: A semiempirical, density functional theory, and ab initio post-Hartree-Fock theoretical study. Theochem.-J. Mol. Struct. 2010, 948, 102–107. [Google Scholar] [CrossRef]
- Zanuy, D.; Poater, J.; Sola, M.; Hamley, I.W.; Aleman, C. Fmoc-RGDS based fibrils: Atomistic details of their hierarchical assembly. Phys. Chem. Chem. Phys. 2016, 18, 1265–1278. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Vuckovic, S.; Sim, E.; Burke, K. Density-Corrected DFT Explained: Questions and Answers. J. Chem. Theory Comput. 2022, 18, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, J.; Aradilla, D.; Poater, J.; Sola, M.; Estrany, F.; Aleman, C. Properties of poly(3-halidethiophene)s. Phys. Chem. Chem. Phys. 2012, 14, 10050–10062. [Google Scholar] [CrossRef] [PubMed]
- Nandi, A.; Qu, C.; Houston, P.L.; Conte, R.; Bowman, J.M. Delta-machine learning for potential energy surfaces: A PIP approach to bring a DFT-based PES to CCSD(T) level of theory. J. Chem. Phys. 2021, 154, 8301. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Zhu, S.D.; Khan, M.A.; Wang, F.Y.; Bano, Z.; Xia, M.Z. Rapid removal of toxic metals Cu2+ and Pb2+ by amino trimethylene phosphonic acid intercalated layered double hydroxide: A combined experimental and DFT study. Chem. Eng. J. 2020, 392, 123711. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, T. Efficient evaluation of electrostatic potential with computerized optimized code. Phys. Chem. Chem. Phys. 2021, 23, 20323–20328. [Google Scholar] [CrossRef]
- Xu, Y.C.; Li, C.L.; Li, Z.Q.; Wang, Q.Y.; Cai, X.L.; Wei, J.B.; Wang, Y. Constructing Charge-Transfer Excited States Based on Frontier Molecular Orbital Engineering: Narrowband Green Electroluminescence with High Color Purity and Efficiency. Angew. Chem.-Int. Ed. 2020, 59, 17442–17446. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.Y.; Wu, T.; Wang, Q.; van der Spoel, D. Comparison of Implicit and Explicit Solvent Models for the Calculation of Solvation Free Energy in Organic Solvents. J. Chem. Theory Comput. 2017, 13, 1034–1043. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lignin Model Compound | Scientific Name | Substituent Group | ||
|---|---|---|---|---|
| R1 | R2 | R3 | ||
 | 4-ethyl-2-methoxyphenol | -CH2CH3 | H | -OCH3 |
| Eugenol | -CH2-CH-CH2 | H | -OCH3 | |
| Ferulic acid | -CH-CH-OOH | H | -OCH3 | |
| 4-acetylphenol | -O-CH3 | H | H | |
| 4-Hydroxy-3-methoxyacetophenone | -C-OH3 | H | -COH3 | |
| 3,5-Dimethoxy-4-hydroxyacetophenone | -C-OH3 | -OCH3 | -OCH3 | |
| Lignin Model Compound | Dissociation Energy E/(kJ·mol−1) | Bond Length (Å) | |
|---|---|---|---|
| O-H | C-O | O-H | |
| 4-ethyl-2-methoxyphenol | 84.38 | 1.3730 | 0.9679 |
| Eugenol | 86.09 | 1.3721 | 0.9680 |
| Ferulic acid | 87.35 | 1.3619 | 0.9688 |
| 4-acetylphenol | 96.73 | 1.3607 | 0.9668 |
| 4-Hydroxy-3-methoxyacetophenone | 90.36 | 1.3591 | 0.9690 |
| 3,5-Dimethoxy-4-hydroxyaceto- phenone | 86.19 | 1.3583 | 0.9685 |
| Lignin Model Compound | NBO Charge | Hammett Value |
|---|---|---|
| 4-ethyl-2-methoxyphenol | −0.046 | 2.91 |
| Eugenol | −0.049 | 2.86 |
| Ferulic acid | −0.132 | 1.32 |
| 4-acetylphenol | −0.213 | −0.17 |
| 4-Hydroxy-3-methoxyacetophenone | −0.189 | 0.27 |
| 3,5-Dimethoxy-4-hydroxyacetophenone | −0.166 | 0.70 |
| Lignin Model Compound | Bond Length (Å) | Angle (°) | ||
|---|---|---|---|---|
| C-O | CO-H | ClO-H | O-H-O | |
| 4-E-ol | 1.6243 | 1.0743 | 1.0429 | 174.7177 |
| E-ol | 1.6171 | 1.0096 | 1.0096 | 158.9603 |
| FA | 1.5691 | 1.0148 | 1.0148 | 179.6980 |
| APO | 1.6978 | 1.0296 | 0.9983 | 157.3091 |
| Compound | HOMO Energy (eV) | LUMO Energy (eV) | HUMO-LUMO Gap (eV) | |||
|---|---|---|---|---|---|---|
| 4-E-ol | −7.2558 | 0.4027 | 7.6586 | |||
| E-ol | −7.2618 | 0.3840 | 7.6458 | |||
| FA | −7.2889 | −1.1542 | 6.1347 | |||
| APO | −7.6090 | −0.7748 | 6.8342 | |||
| Compound | HOMO Energy (eV) | LUMO Energy (eV) | Gap of Alpha (eV) | HOMO Energy (eV) | LUMO Energy (eV) | Gap of Bate (eV) |
| 4-E-ol + ClO2 | −7.3411 | −0.5542 | 6.7869 | −8.4473 | −3.1274 | 5.3199 |
| E-ol + ClO2 | −7.3431 | −0.4371 | 6.9060 | −8.4203 | −3.1015 | 5.3188 |
| FA + ClO2 | −7.3508 | −1.3546 | 5.9962 | −8.5225 | −3.5028 | 4.5027 |
| APO + ClO2 | −7.6401 | −0.9664 | 6.6737 | −8.6370 | −3.5036 | 5.1334 |
| Index | 4-E-ol | E-ol | FA | APO |
|---|---|---|---|---|
| E/au | −1110.8845 | −1148.9597 | −1298.2664 | −1184.9299 |
| ΔE/au | 0.0311 | 0.0305 | 0.0328 | 0.0342 |
| ΔE/kcal/mol | 19.5153 | 19.1224 | 20.6040 | 21.4906 |
| Lignin Model Compound | Bond Length (d/nm) | Angle (°) | ||
|---|---|---|---|---|
| C-O | CO-H | ClO-H | O-H-O | |
| 4-A-ol | 1.6352 | 1.0041 | 1.0041 | 165.9504 |
| APO | 1.6978 | 1.0296 | 0.9983 | 157.3091 |
| DHP | 1.2499 | 1.4509 | 1.0404 | 162.7035 |
| Compound | HOMO Energy (eV) | LUMO Energy (eV) | HUMO-LUMO Gap (eV) | |||
|---|---|---|---|---|---|---|
| 4-A-ol | −7.9882 | −0.7469 | 7.2413 | |||
| APO | −7.6090 | −0.7748 | 6.8342 | |||
| DHP | −7.5380 | 0.8059 | 6.7321 | |||
| Compound | HOMO Energy (eV) | LUMO Energy (eV) | Gap of Alpha (eV) | HOMO Energy (eV) | LUMO Energy (eV) | Gap of Beta (eV) |
| 4-A-ol + ClO2 | −8.2399 | −0.9407 | 7.3993 | −9.1822 | −3.8501 | 5.3321 |
| APO + ClO2 | −7.6401 | −0.96634 | 6.6737 | −8.6370 | −3.5036 | 5.1334 |
| DHP + ClO2 | −7.6151 | −1.0629 | 6.5522 | −8.1069 | −3.5099 | 4.5970 |
| Index | 4-ol | APO | DHP |
|---|---|---|---|
| E/au | −1070.6150 | −1184.9299 | −1297.6567 |
| ΔE/au | 0.0377 | 0.0342 | 0.0306 |
| ΔE/kcal/mol | 23.6598 | 21.4906 | 19.2268 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Liu, L.; Qin, X.; Liu, Y.; Yang, R.; Mo, X.; Qin, C.; Liang, C.; Yao, S. Effect of Substituents on Molecular Reactivity during Lignin Oxidation by Chlorine Dioxide: A Density Functional Theory Study. Int. J. Mol. Sci. 2023, 24, 11809. https://doi.org/10.3390/ijms241411809
Liu B, Liu L, Qin X, Liu Y, Yang R, Mo X, Qin C, Liang C, Yao S. Effect of Substituents on Molecular Reactivity during Lignin Oxidation by Chlorine Dioxide: A Density Functional Theory Study. International Journal of Molecular Sciences. 2023; 24(14):11809. https://doi.org/10.3390/ijms241411809
Chicago/Turabian StyleLiu, Baojie, Lu Liu, Xin Qin, Yi Liu, Rui Yang, Xiaorong Mo, Chengrong Qin, Chen Liang, and Shuangquan Yao. 2023. "Effect of Substituents on Molecular Reactivity during Lignin Oxidation by Chlorine Dioxide: A Density Functional Theory Study" International Journal of Molecular Sciences 24, no. 14: 11809. https://doi.org/10.3390/ijms241411809