
Synthesis and Properties of (1R(S),5R(S),7R(S),8R(S))-1,8-Bis(hydroxymethyl)-6-azadispiro[4.1.4.2]tridecane-6-oxyl: Reduction-Resistant Spin Labels with High Spin Relaxation Times
 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
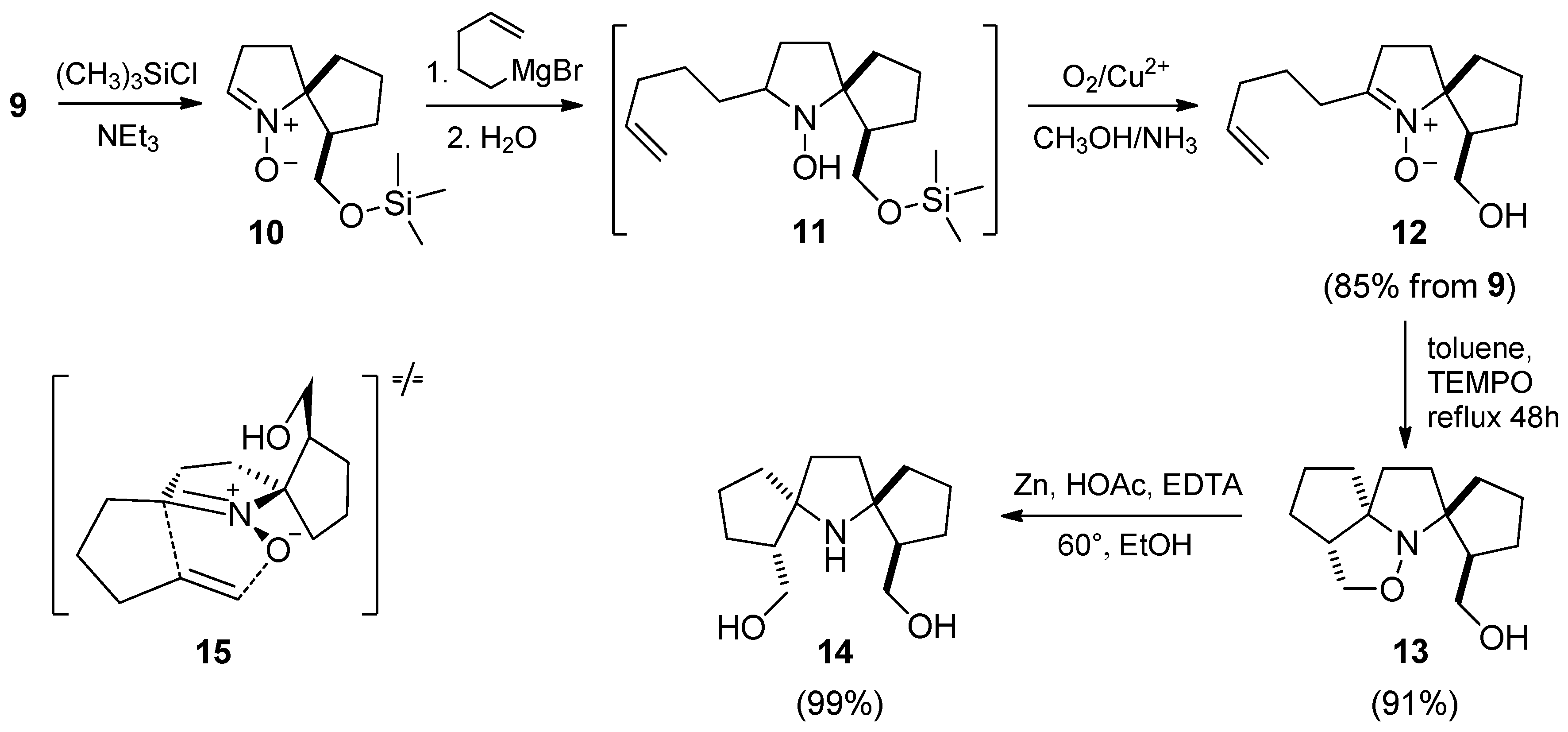
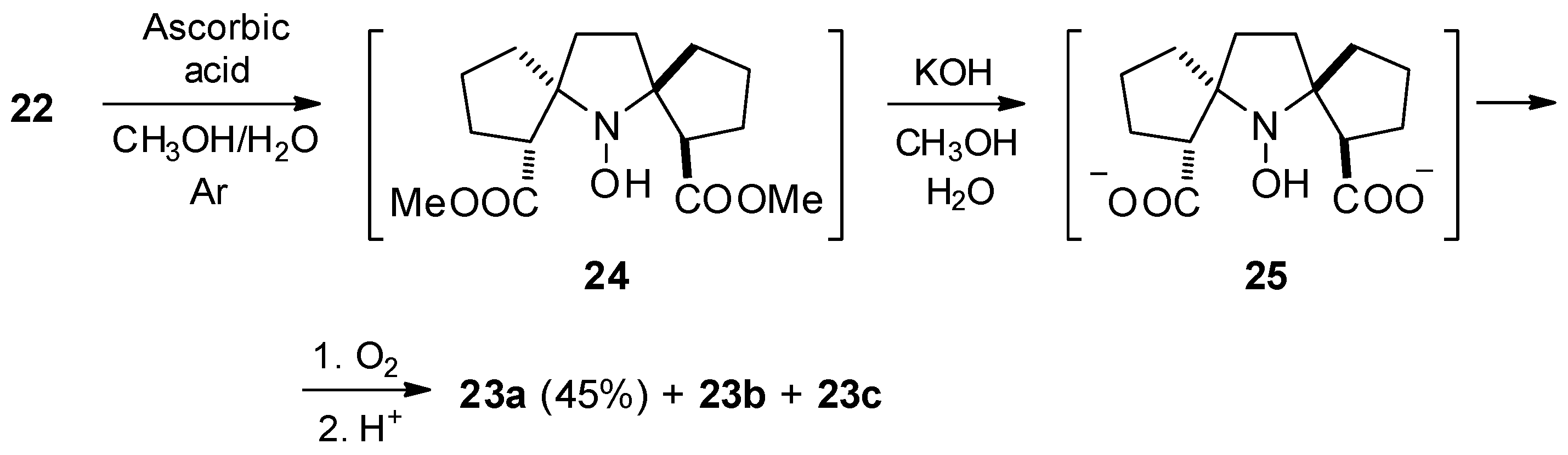
2.1. Synthesis
2.2. CW EPR Spectra
2.3. Electron Spin Relaxation Time
2.4. Reduction Rate Constants
3. Materials and Methods
3.1. General
3.2. EPR
3.3. Kinetic Measurements and Partition Coefficients
3.4. Synthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pierro, A.; Drescher, M. Dance with Spins: Site-Directed Spin Labeling Coupled to Electron Paramagnetic Resonance Spectroscopy Directly inside Cells. Chem. Commun. 2023, 59, 1274–1284. [Google Scholar] [CrossRef]
- Klug, C.S.; Lerch, M.T.; Feix, J.B. Chapter 10—Applications of Nitroxide Spin Labels to Structural Biology. In Nitroxides: Synthesis, Properties and Applications; Ouari, O., Gigmes, D., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2021; pp. 392–419. [Google Scholar] [CrossRef]
- Sahu, I.D.; Lorigan, G.A. Site-Directed Spin Labeling EPR for Studying Membrane Proteins. BioMed Res. Int. 2018, 2018, 3248289. [Google Scholar] [CrossRef]
- Klare, J.P. Site-directed spin labeling EPR spectroscopy in protein research. Biol. Chem. 2013, 394, 1281–1300. [Google Scholar] [CrossRef]
- Bonucci, A.; Ouari, O.; Guigliarelli, B.; Belle, V.; Mileo, E. In-Cell EPR: Progress towards Structural Studies Inside Cells. ChemBioChem 2020, 21, 451–460. [Google Scholar] [CrossRef]
- Galazzo, L.; Bordignon, E. Electron paramagnetic resonance spectroscopy in structural-dynamic studies of large protein complexes. Prog. Nucl. Magn. Reson. Spectrosc. 2023, 134–135, 1–19. [Google Scholar] [CrossRef]
- Haugland, M.M.; Anderson, E.A.; Lovett, J.E. Tuning the properties of nitroxide spin labels for use in electron paramagnetic resonance spectroscopy through chemical modification of the nitroxide framework. In Electron Paramagnetic Resonance; Chechik, V., Murphy, D.M., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2017; Volume 25, pp. 1–34. [Google Scholar] [CrossRef] [Green Version]
- Nilaweera, T.D.; Nyenhuis, D.A.; Nakamoto, R.K.; Cafiso, D.S. Disulfide Chaperone Knockouts Enable In Vivo Double Spin Labeling of an Outer Membrane Transporter. Biophys. J. 2019, 117, 1476–1484. [Google Scholar] [CrossRef]
- Wang, Y.; Sarkar, M.; Smith, A.E.; Krois, A.S.; Pielak, G.J. Macromolecular Crowding and Protein Stability. J. Am. Chem. Soc. 2012, 134, 16614–16618. [Google Scholar] [CrossRef] [PubMed]
- Kyne, C.; Crowley, P.B. Grasping the nature of the cell interior: From Physiological Chemistry to Chemical Biology. FEBS J. 2016, 283, 3016–3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singewald, K.; Lawless, M.J.; Saxena, S. Increasing nitroxide lifetime in cells to enable in-cell protein structure and dynamics measurements by electron spin resonance spectroscopy. J. Magn. Res. 2019, 299, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, A.P.; Krstic, I.; Kunjir, N.C.; Hänsel, R.; Prisner, T.F.; Sigurdsson, S.T. Sterically Shielded Spin Labels for In-Cell EPR Spectroscopy: Analysis of Stability in Reducing Environment. Free Radic. Res. 2015, 49, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Evans, E.G.B.; Jang, H.S.; Zhang, S.; Zhang, H.; Rajca, A.; Gordon, S.E.; Zagotta, W.N.; Stoll, S.; Mehl, R.A. Ultra-Fast Bioorthogonal Spin-Labeling and Distance Measurements in Mammalian Cells Using Small, Genetically Encoded Tetrazine Amino Acids. bioRxiv 2023. [Google Scholar] [CrossRef]
- Karthikeyan, G.; Bonucci, A.; Casano, G.; Gerbaud, G.; Abel, S.; Thomé, V.; Kodjabachian, L.; Magalon, A.; Guigliarelli, B.; Belle, V.; et al. Bioresistant Nitroxide Spin Label for In-Cell EPR Spectroscopy: In Vitro and In Oocytes Protein Structural Dynamics Studies. Angew. Chem. Int. Ed. Engl. 2018, 57, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Jagtap, A.P.; Sigurdsson, S.T. Site-directed spin labeling of 20-amino groups in RNA with isoindoline nitroxides that are resistant to reduction. Chem. Commun. 2015, 51, 13142. [Google Scholar] [CrossRef] [PubMed]
- Bleicken, S.; Assafa, T.E.; Zhang, H.; Elsner, C.; Ritsch, I.; Pink, M.; Rajca, S.; Jeschke, G.; Rajca, A.; Bordignon, E. gem-Diethyl Pyrroline Nitroxide Spin Labels: Synthesis, EPR Characterization, Rotamer Libraries and Biocompatibility. ChemistryOpen 2019, 8, 1035. [Google Scholar] [CrossRef] [Green Version]
- Braun, T.S.; Widder, P.; Osswald, U.; Groß, L.; Williams, L.; Schmidt, M.; Helmle, I.; Summerer, D.; Drescher, M. Isoindoline-Based Nitroxides as Bioresistant Spin Labels for Protein Labeling through Cysteines and Alkyne-Bearing Noncanonical Amino Acids. Chembiochem 2020, 21, 958–962. [Google Scholar] [CrossRef]
- Ketter, S.; Dajka, M.; Rogozhnikova, O.; Dobrynin, S.A.; Tormyshev, V.M.; Bagryanskaya, E.G.; Joseph, B. In situ distance measurements in a membrane transporter using maleimide functionalized orthogonal spin labels and 5-pulse electron-electron double resonance spectroscopy. J. Magn. Reson. Open (Companion Title J. Magn. Reson.) 2022, 10–11, 100041. [Google Scholar] [CrossRef]
- Dobrynin, S.A.; Usatov, M.S.; Zhurko, I.F.; Morozov, D.A.; Polienko, Y.F.; Glazachev, Y.I.; Parkhomenko, D.A.; Tyumentsev, M.A.; Gatilov, Y.V.; Chernyak, E.I.; et al. A simple method of synthesis of 3-carboxy-2,2,5,5-tetraethylpyrrolidine-1-oxyl and preparation of reduction-resistant spin labels and probes of pyrrolidine series. Molecules 2021, 26, 5761. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, G. The contribution of modern EPR to structural biology. Emerg. Top. Life Sci. 2018, 2, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Eaton, S.S.; Eaton, G.R. Measurement of Distances Between Electron Spins Using Pulsed EPR. In Biomedical EPR, Part B: Methodology, Instrumentation, and Dynamics; Eaton, S.R., Eaton, G.R., Berliner, L.J., Eds.; Biological Magnetic Resonance Series; Springer: Boston, MA, USA, 2005; Volume 24/B, pp. 223–236. [Google Scholar] [CrossRef]
- Rajca, A.; Kathirvelu, V.; Roy, S.K.; Pink, M.; Rajca, S.; Sarkar, S.; Eaton, S.S.; Eaton, G.R. A spirocyclohexyl nitroxide amino acid spin label for pulsed EPR spectroscopy distance measurements. Chemistry 2010, 16, 5778–5782. [Google Scholar] [CrossRef] [Green Version]
- Meyer, V.; Swanson, M.A.; Clouston, L.J.; Boratyński, P.J.; Stein, R.A.; Mchaourab, H.S.; Rajca, A.; Eaton, S.S.; Eaton, G.R. Room-temperature distance measurements of immobilized spin-labeled protein by DEER/PELDOR. Biophys. J. 2015, 108, 1213–1219. [Google Scholar] [CrossRef] [Green Version]
- Paletta, J.T.; Pink, M.; Foley, B.; Rajca, S.; Rajca, A. Synthesis and Reduction Kinetics of Sterically Shielded Pyrrolidine Nitroxides. Org. Lett. 2012, 14, 5322–5325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sowiński, M.P.; Gahlawat, S.; Warnke, A.-L.; Lund, B.A.; Hopmann, K.H.; Lovett, J.E.; Haugland, M.M. Conformational tuning improves the stability of spirocyclic nitroxides with long paramagnetic relaxation times. Commun. Chem. 2023, 6, 111. [Google Scholar] [CrossRef]
- Kuzhelev, A.A.; Strizhakov, R.K.; Krumkacheva, O.A.; Polienko, Y.F.; Morozov, D.A.; Shevelev, G.Y.; Pyshnyi, D.V.; Kirilyuk, I.A.; Fedin, M.V.; Bagryanskaya, E.G. Room-temperature electron spin relaxation of nitroxides immobilized in trehalose: Effect of substituents adjacent to NO-group. J. Magn. Reson. 2016, 266, 1–7. [Google Scholar] [CrossRef]
- Morozov, D.A.; Kirilyuk, I.A.; Komarov, D.A.; Goti, A.; Bagryanskaya, I.Y.; Kuratieva, N.V.; Grigor’ev, I.A. Synthesis of a Chiral C2-Symmetric Sterically Hindered Pyrrolidine Nitroxide Radical via Combined Iterative Nucleophilic Additions and Intramolecular 1,3-Dipolar Cycloadditions to Cyclic Nitrones. J. Org. Chem. 2012, 77, 10688–10698. [Google Scholar] [CrossRef] [PubMed]
- Kirilyuk, I.A.; Polienko, Y.F.; Krumkacheva, O.A.; Strizhakov, R.K.; Gatilov, Y.V.; Grigor’ev, I.A.; Bagryanskaya, E.G. Synthesis of 2,5-Bis(spirocyclohexane)-Substituted Nitroxides of Pyrroline and Pyrrolidine Series, Including Thiol-Specific Spin Label: An Analogue of MTSSL with Long Relaxation Time. J. Org. Chem. 2012, 77, 8016–8027. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Paletta, J.T.; Berg, K.; Reinhart, E.; Rajca, S.; Rajca, A. Synthesis of unnatural amino acids functionalized with sterically shielded pyrroline nitroxides. Org. Lett. 2014, 16, 5298–5300. [Google Scholar] [CrossRef]
- Khoroshunova, Y.V.; Morozov, D.A.; Taratayko, A.I.; Gladkikh, P.D.; Glazachev, Y.I.; Kirilyuk, I.A. Synthesis of 1-azaspiro[4.4]nonan-1-oxyls via intramolecular 1,3-dipolar cycloaddition. Beilstein J. Org. Chem. 2019, 15, 2036–2042. [Google Scholar] [CrossRef] [Green Version]
- Cicchi, S.; Hold, I.; Brandi, A.J. New Synthesis of Five-Membered Cyclic Nitrones from Tartaric Acid. J. Org. Chem. 1993, 58, 5274–5275. [Google Scholar] [CrossRef]
- Saruengkhanphasit, R.; Collier, D.; Coldham, I. Synthesis of Spirocyclic Amines by Using Dipolar Cycloadditions of Nitrones. J. Org. Chem. 2017, 82, 6489–6496. [Google Scholar] [CrossRef]
- Grigg, R.; Markandu, J.; Surendrakumar, S.; Thornton-Pett, M.; Warnock, W.J. X = Y − ZH Systems as potential 1,3-dipoles. Part 37 generation of nitrones from oximes. Tandem intramolecular 1,3-azaprotio cyclotransfer—Intramolecular 1,3-dipolar cycloaddition reactions. Class 4 processes. Tetrahedron 1992, 48, 10399–10432. [Google Scholar] [CrossRef]
- Khoroshunova, Y.V.; Morozov, D.A.; Taratayko, A.I.; Dobrynin, S.A.; Eltsov, I.V.; Rybalova, T.V.; Sotnikova, Y.S.; Polovyanenko, D.N.; Asanbaeva, N.B.; Kirilyuk, I.A. The Reactions of 6-(Hydroxymethyl)-2,2-dimethyl-1-azaspiro[4.4]nonanes with Methanesulfonyl Chloride or PPh3-CBr4. Molecules 2021, 26, 6000. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Li, J.; Mano, E.; Song, Z.; Tschaen, D.M.; Grabowski, E.J.J.; Reider, P.J. Oxidation of Primary Alcohols to Carboxylic Acids with Sodium Chlorite Catalyzed by TEMPO and Bleach. J. Org. Chem. 1999, 64, 2564–2566. [Google Scholar] [CrossRef]
- Khramtsov, V.V.; Weiner, L.M.; Eremenko, S.I.; Belchenko, O.I.; Scastnev, P.V.; Grigir’ev, I.A.; Reznikov, V.A. Proton exchange in stable nitroxyl radicals of the imidazoline and imidazolidine series. J. Magn. Reson. 1985, 61, 397–408. [Google Scholar] [CrossRef]
- Hankovszky, O.H.; Hideg, K.; Lex, L.; Tigiy, J. Nitroxyls; IV. Synthesis of Spin-Labelled N-(4-Piperidinyloxycarbonyl)-imidazole and 4-Piperidinyloxycarbonyl Azides and Their Reaction with Amino Acid Derivatives. Synthesis 1979, 1979, 530–531. [Google Scholar] [CrossRef]
- Trofimov, D.G.; Glazachev, Y.I.; Gorodetsky, A.A.; Komarov, D.A.; Rybalova, T.V.; Kirilyuk, I.A. 4-Dialkylamino-2,5-dihydroimidazol-1-oxyls with Functional Groups at the Position 2 and at the Exocyclic Nitrogen: The pH-Sensitive Spin Labels. Gels 2022, 8, 11. [Google Scholar] [CrossRef]
- Widder, P.; Schuck, J.; Summerer, D.; Drescher, M. Combining site-directed spin labeling in vivo and in-cell EPR distance determination. Phys. Chem. Chem. Phys. 2020, 22, 4875–4879. [Google Scholar] [CrossRef] [Green Version]
- Rajaram, H.; Palanivelu, M.K.; Arumugam, T.V.; Rao, V.M.; Shaw, P.N.; McGeary, R.P.; Ross, B.P. ‘Click’ assembly of glycoclusters and discovery of a trehalose analogue that retards Aβ40 aggregation and inhibits Aβ40-induced neurotoxicity. Bioorg. Med. Chem. Lett. 2014, 24, 4523–4528. [Google Scholar] [CrossRef] [Green Version]
- Shukla, A.K.; Shrivash, M.K.; Pandey, A.; Pandey, J. Synthesis, in vitro and computational studies of novel glycosyl-1, 2, 3-1H-triazolyl methyl benzamide derivatives as potential α-glucosidase inhibitory activity. Bioorganic Chem. 2021, 109, 104687. [Google Scholar] [CrossRef]
- El-Sayed, W.A.; Alminderej, F.M.; Mounier, M.M.; Nossier, E.S.; Saleh, S.M.; Kassem, A.F. Novel 1,2,3-Triazole-Coumarin Hybrid Glycosides and Their Tetrazolyl Analogues: Design, Anticancer Evaluation and Molecular Docking Targeting EGFR, VEGFR-2 and CDK-2. Molecules 2022, 27, 2047. [Google Scholar] [CrossRef]
- Okazaki, S.; Mannan, M.A.; Sawai, K.; Masumizu, T.; Miura, Y.; Takeshita, K. Enzymatic reduction-resistant nitroxyl spin probes with spirocyclohexyl rings. Free. Radic. Res. 2007, 41, 1069–1077. [Google Scholar] [CrossRef]
- Soetbeer, J.; Hülsmann, M.; Godt, A.; Polyhach, Y.; Jeschke, G. Dynamical decoupling of nitroxides in o-terphenyl: A study of temperature, deuteration and concentration effects. Phys. Chem. Chem. Phys. 2018, 20, 1615–1628. [Google Scholar] [CrossRef] [PubMed]
- Soetbeer, J.; Millen, M.; Zouboulis, K.; Hülsmann, M.; Godt, A.; Polyhach, Y.; Jeschke, G. Dynamical decoupling in water–glycerol glasses: A comparison of nitroxides, trityl radicals and gadolinium complexes. Phys. Chem. Chem. Phys. 2021, 23, 5352–5369. [Google Scholar] [CrossRef] [PubMed]
- Kirilina, E.P.; Dzuba, S.A.; Maryasov, A.G.; Tsvetkov, Y.D. Librational dynamics of nitroxide molecules in a molecular glass studied by echo-detected EPR. Appl. Magn. Reson. 2001, 21, 203–221. [Google Scholar] [CrossRef]
- Dzuba, S.A.; Kirilina, E.P.; Salnikov, E.S. On the possible manifestation of harmonic-anharmonic dynamical transition in glassy media in electron paramagnetic resonance of nitroxide spin probes. J. Chem. Phys. 2006, 125, 054502. [Google Scholar] [CrossRef]
- Bobko, A.A.; Kirilyuk, I.A.; Grigor’ev, I.A.; Zweier, J.L.; Khramtsov, V.V. Reversible reduction of nitroxides to hydroxylamines: Roles for ascorbate and glutathione. Free Radic. Biol. Med. 2007, 42, 404–412. [Google Scholar] [CrossRef] [Green Version]
- Kirilyuk, I.A.; Bobko, A.A.; Semenov, S.V.; Komarov, D.A.; Irtegova, I.G.; Grigor’ev, I.A.; Bagryanskaya, E. Effect of Sterical Shielding on the Redox Properties of Imidazoline and Imidazolidine Nitroxides. J. Org. Chem. 2015, 80, 9118–9125. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS, v. 2008/1; Bruker AXS, Inc.: Madison, WI, USA, 2008. [Google Scholar]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C 2015, C71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Isaev, N.P.; Melnikov, A.R.; Lomanovich, K.A.; Dugin, M.V.; Ivanov, M.Y.; Polovyanenko, D.N.; Veber, S.L.; Bowman, M.K.; Bagryanskaya, E.G. A broadband pulse EPR spectrometer for high-throughput measurements in the X-band. J. Magn. Reson. Open 2023, 14–15, 100092. [Google Scholar] [CrossRef]
- Hideg, K.; Hankovszky, H.O.; Lex, L.; Kulcsár, G. Nitroxyls; VI1. Synthesis and Reactions of 3-Hydroxymethyl-2,2,5,5-tetramethyl-2,5-dihydropyrrole-1-oxyl and 3-Formyl Derivatives. Synthesis 1980, 1980, 911–914. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nitroxide | ωg (mT) (±0.02) | ωl (mT) (±0.002) | aN (H2O) (mT), (±0.05) | aN (Toluene) (mT), (±0.05) |
|---|---|---|---|---|
 | 0.23 | 0.016 | 1.52 | 1.44 |
 | 0.23 | 0.019 | 1.52 | |
 | 0.10 | 0.005 | 1.62 | 1.42 |
 | 0.21 | 0.076 | 1.61 |
| Nitroxide | 80 K | 120 K | ||||
|---|---|---|---|---|---|---|
| T1, ms (±0.01) | Tm, µs (±0.1) | n (±0.03) | T1, ms (±0.01) | Tm, µs (±0.1) | n (±0.03) | |
| 5 | 0.94 | 3.0 | 1.70 | 0.34 | 2.8 | 1.67 |
| 33 | 1.01 | 3.9 | 1.97 | 0.34 | 3.9 | 2.1 |
| 34 | 0.51 | 3.1 | 1.70 | 0.20 | 0.9 | 1.0 |
| 35 | 0.53 | 3.4 | 1.97 | 0.22 | 1.1 | 1.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khoroshunova, Y.V.; Morozov, D.A.; Kuznetsov, D.A.; Rybalova, T.V.; Glazachev, Y.I.; Bagryanskaya, E.G.; Kirilyuk, I.A. Synthesis and Properties of (1R(S),5R(S),7R(S),8R(S))-1,8-Bis(hydroxymethyl)-6-azadispiro[4.1.4.2]tridecane-6-oxyl: Reduction-Resistant Spin Labels with High Spin Relaxation Times. Int. J. Mol. Sci. 2023, 24, 11498. https://doi.org/10.3390/ijms241411498
Khoroshunova YV, Morozov DA, Kuznetsov DA, Rybalova TV, Glazachev YI, Bagryanskaya EG, Kirilyuk IA. Synthesis and Properties of (1R(S),5R(S),7R(S),8R(S))-1,8-Bis(hydroxymethyl)-6-azadispiro[4.1.4.2]tridecane-6-oxyl: Reduction-Resistant Spin Labels with High Spin Relaxation Times. International Journal of Molecular Sciences. 2023; 24(14):11498. https://doi.org/10.3390/ijms241411498
Chicago/Turabian StyleKhoroshunova, Yulia V., Denis A. Morozov, Danil A. Kuznetsov, Tatyana V. Rybalova, Yurii I. Glazachev, Elena G. Bagryanskaya, and Igor A. Kirilyuk. 2023. "Synthesis and Properties of (1R(S),5R(S),7R(S),8R(S))-1,8-Bis(hydroxymethyl)-6-azadispiro[4.1.4.2]tridecane-6-oxyl: Reduction-Resistant Spin Labels with High Spin Relaxation Times" International Journal of Molecular Sciences 24, no. 14: 11498. https://doi.org/10.3390/ijms241411498