
Impact of the Protein Environment on Two-Photon Absorption Cross-Sections of the GFP Chromophore Anion Resolved at the XMCQDPT2 Level of Theory
 , ,
, , 
Abstract
:1. Introduction
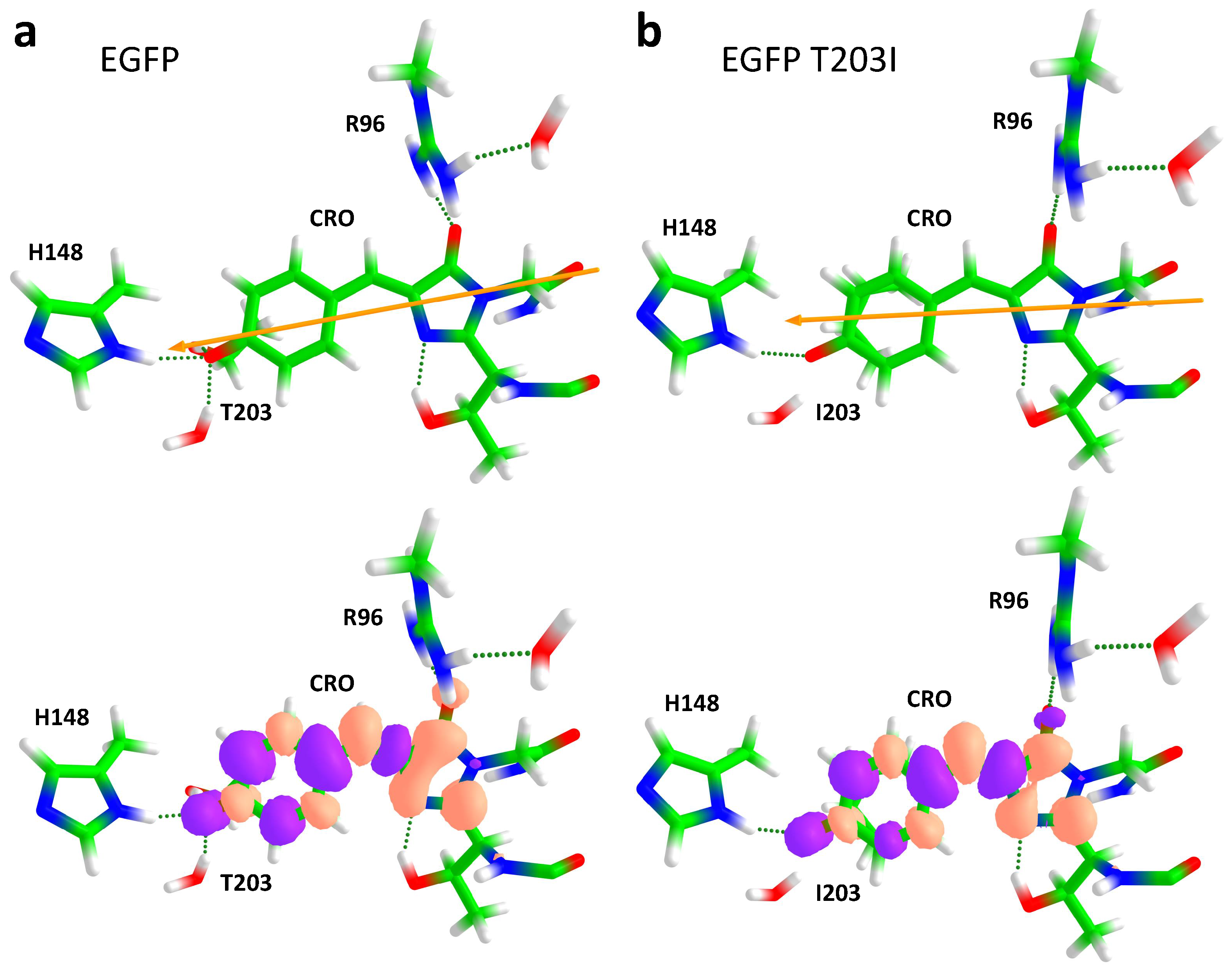
2. Results and Discussion
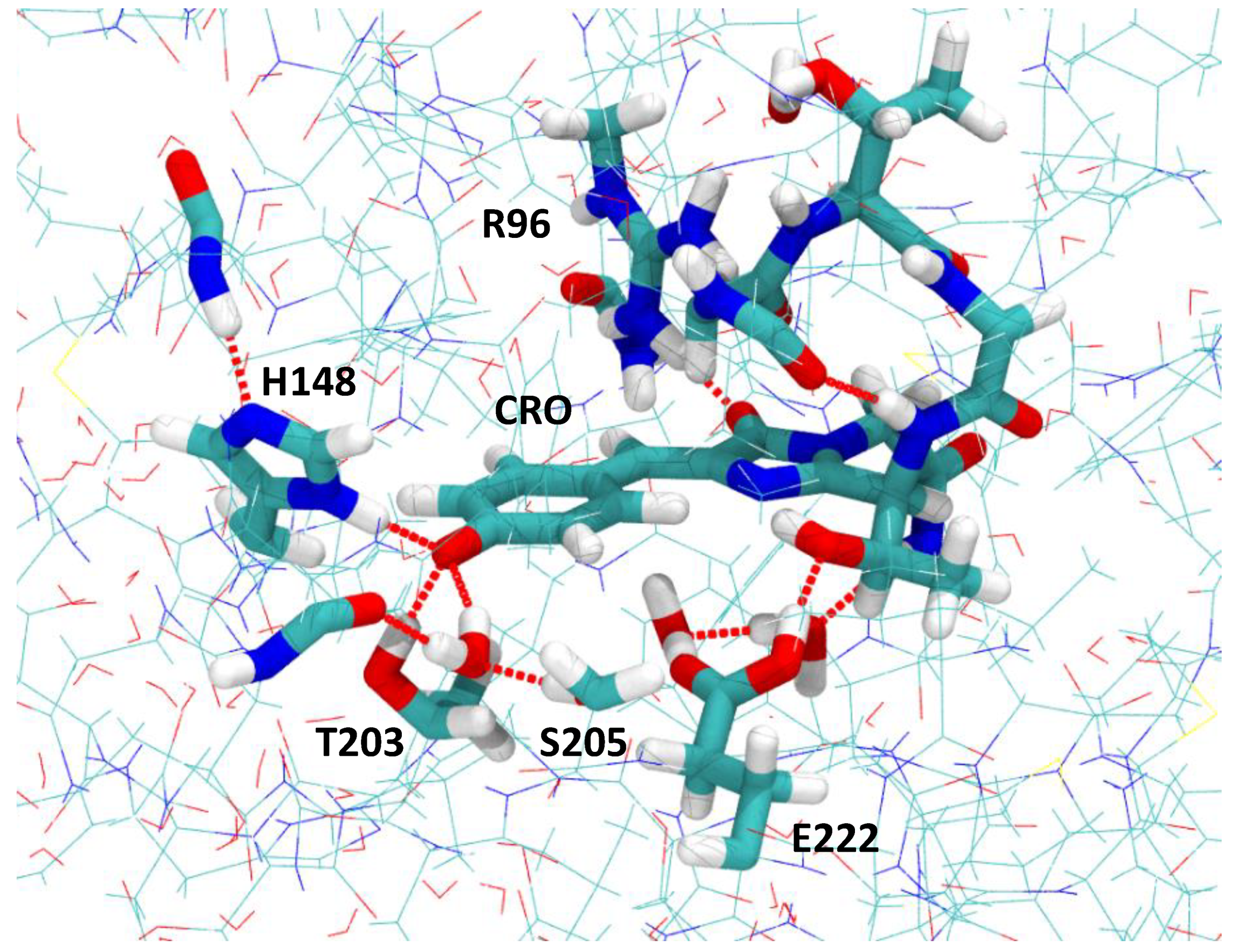
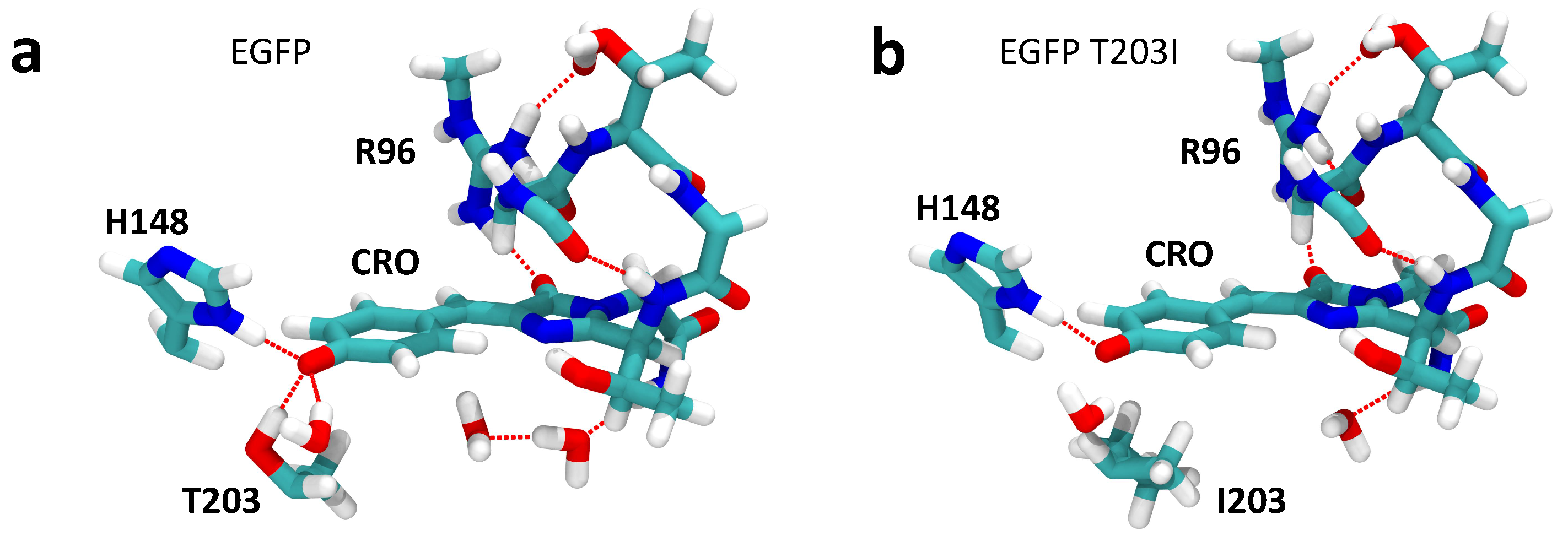
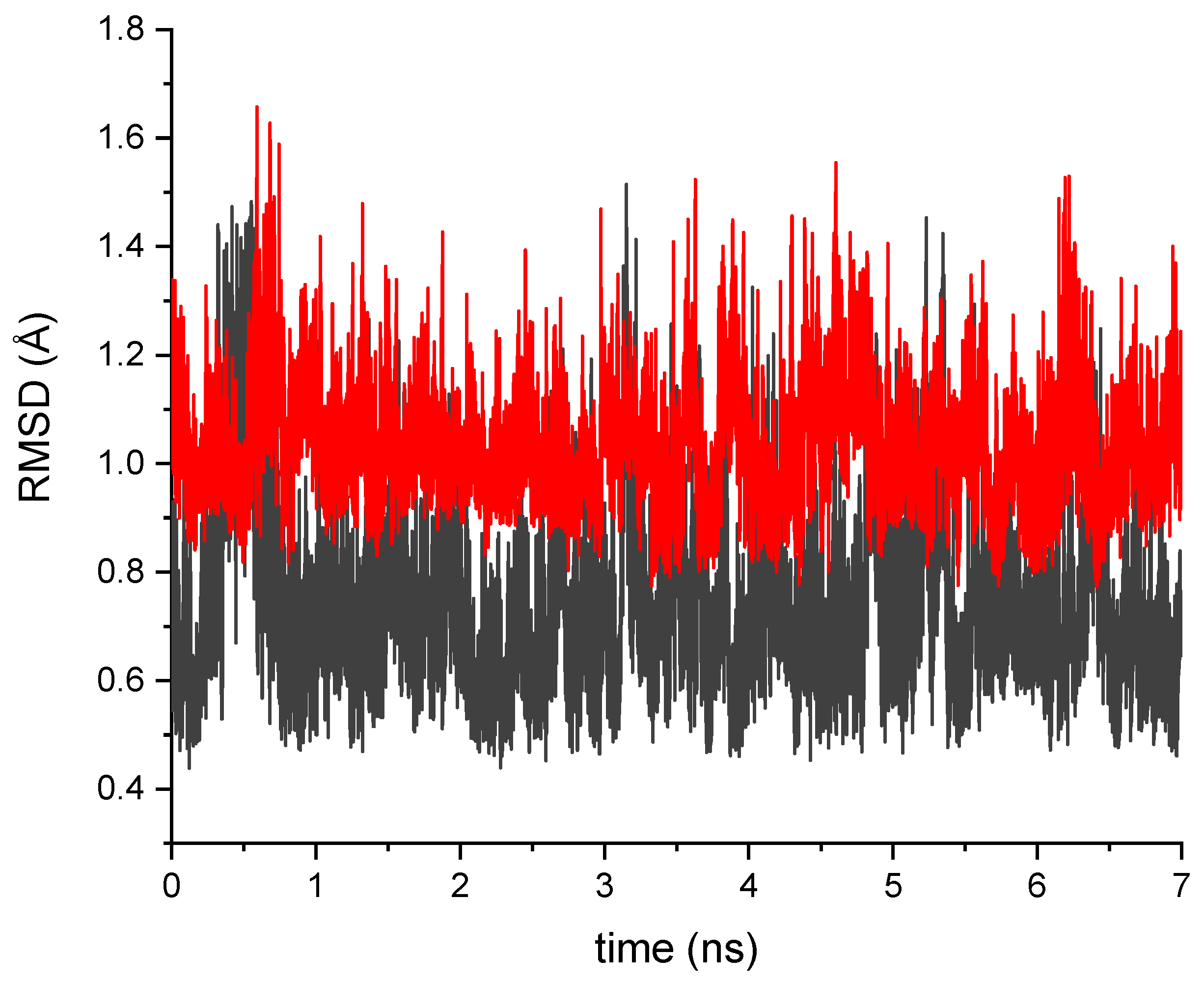
2.1. Impact of the T203I Mutation and Conformational Sampling
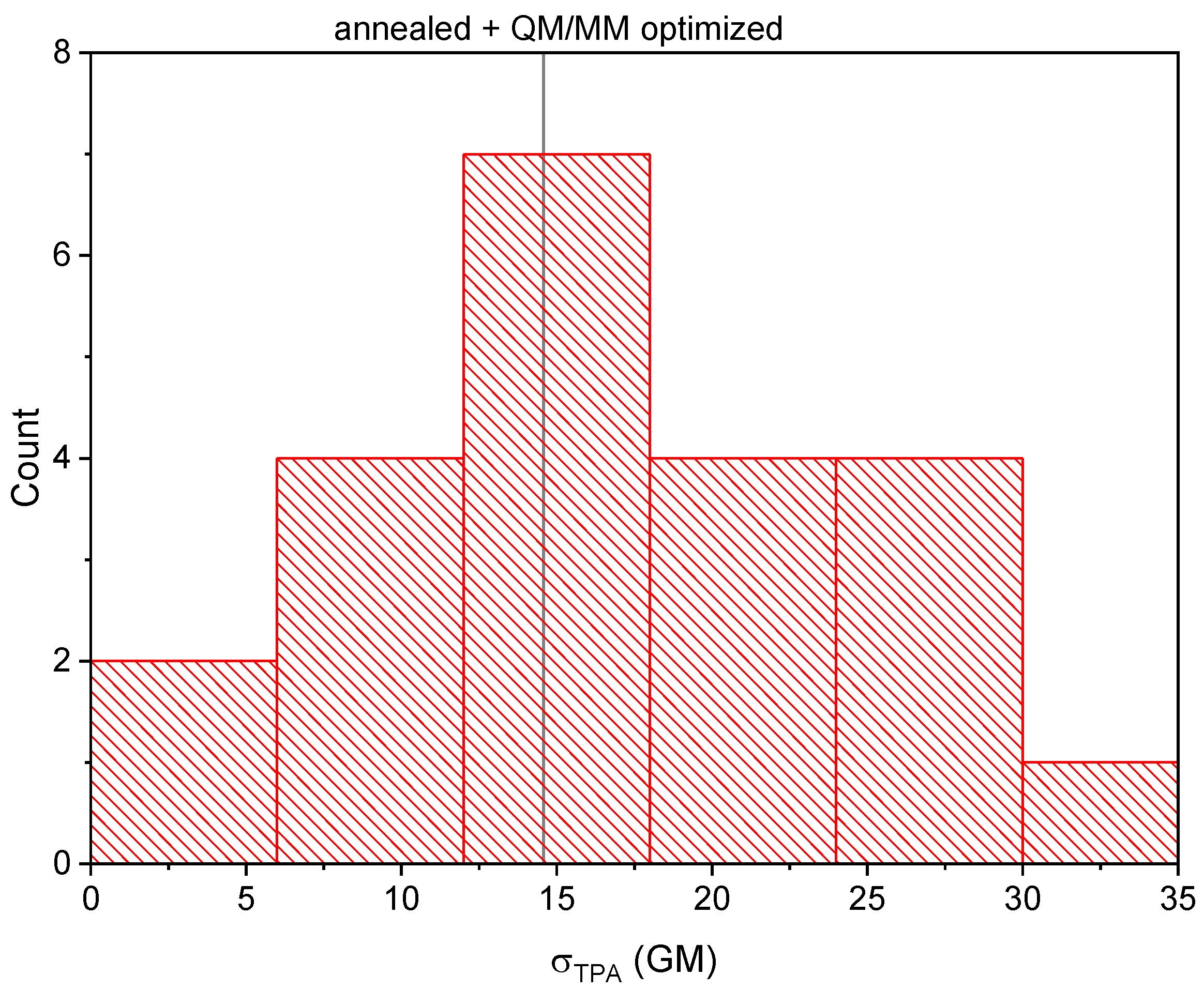
2.2. Analysis of the Calculated TPA Cross-Sections
3. Materials and Methods
3.1. Molecular Dynamics Simulations
3.2. Ground State QM/MM Optimization
3.3. Excited State QM/MM Calculations
3.4. Gas-Phase Calculations
3.5. Calculation of the TPA Cross-Section
Two-Level Model and N-Level Models
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Denk, W.; Strickler, J.H.; Webb, W.W. Two-Photon Laser Scanning Fluorescence Microscopy. Science 1990, 248, 73–76. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Zipfel, W.; Shear, J.B.; Williams, R.M.; Webb, W.W. Multiphoton fluorescence excitation: New spectral windows for biological nonlinear microscopy. Proc. Natl. Acad. Sci. USA 1996, 93, 10763–10768. [Google Scholar] [CrossRef]
- Zipfel, W.; Williams, R.; Webb, W. Nonlinear magic: Multiphoton microscopy in the biosciences. Nat. Biotechnol. 2003, 21, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Molina, R.S.; Tran, T.M.; Campbell, R.E.; Lambert, G.G.; Salih, A.; Shaner, N.C.; Hughes, T.E.; Drobizhev, M. Blue-Shifted Green Fluorescent Protein Homologues Are Brighter than Enhanced Green Fluorescent Protein under Two-Photon Excitation. J. Phys. Chem. Lett. 2017, 8, 2548–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.T.; Cheng, L.; Kain, S.R. Optimized Codon Usage and Chromophore Mutations Provide Enhanced Sensitivity with the Green Fluorescent Protein. Nucleic Acids Res. 1996, 24, 4592–4593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osamu, S.; Johnson, F.H.; Yo, S. Extraction, Purification and Properties of Aequorin, a Bioluminescent Protein from the Luminous Hydromedusan, Aequorea. J. Cell. Comp. Physiol. 1962, 59, 223–239. [Google Scholar] [CrossRef]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef]
- Heim, R.; Prasher, D.C.; Tsien, R.Y. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. USA 1994, 91, 12501–12504. [Google Scholar] [CrossRef]
- Arpino, J.A.J.; Rizkallah, P.J.; Jones, D.D. Crystal Structure of Enhanced Green Fluorescent Protein to 1.35 Å Resolution Reveals Alternative Conformations for Glu222. PLoS ONE 2012, 7, e47132. [Google Scholar] [CrossRef] [Green Version]
- Heim, R.; Cubitt, A.B.; Tsien, R.Y. Improved green fluorescence. Nature 1995, 373, 663–664. [Google Scholar] [CrossRef]
- Bochenkova, A.V.; Andersen, L.H. Ultrafast dual photoresponse of isolated biological chromophores: Link to the photoinduced mode-specific non-adiabatic dynamics in proteins. Faraday Discuss. 2013, 163, 297–319. [Google Scholar] [CrossRef]
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent Proteins and Their Applications in Imaging Living Cells and Tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef] [PubMed]
- Cubitt, A.B.; Heim, R.; Adams, S.R.; Boyd, A.E.; Gross, L.A.; Tsien, R.Y. Understanding, improving and using green fluorescent proteins. Trends Biochem. Sci. 1995, 20, 448–455. [Google Scholar] [CrossRef]
- Drobizhev, M.; Makarov, N.; Tillo, S.; Hughes, T.; Rebane, A. Two-photon absorption properties of fluorescent proteins. Nat. Methods 2011, 8, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoltzfus, C.; Barnett, L.; Drobizhev, M.; Wicks, G.; Mikhaylov, A.; Hughes, T.; Rebane, A. Two-photon directed evolution of green fluorescent proteins. Sci. Rep. 2015, 5, 11968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drobizhev, M.; Makarov, N.S.; Tillo, S.E.; Hughes, T.E.; Rebane, A. Describing Two-Photon Absorptivity of Fluorescent Proteins with a New Vibronic Coupling Mechanism. J. Phys. Chem. B. 2012, 116, 1736–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drobizhev, M.; Callis, P.; Nifosì, R.; Wicks, G.; Stoltzfus, C.; Barnett, L.; Hughes, T.; Sullivan, P.; Rebane, A. Long- and Short-Range Electrostatic Fields in GFP Mutants: Implications for Spectral Tuning. Sci. Rep. 2015, 5, 13223. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.; Jørgensen, P. Linear and nonlinear response function for an exact state and for an MCSCF state. J. Chem. Phys. 1985, 82, 3235–3264. [Google Scholar] [CrossRef] [Green Version]
- Nanda, K.D.; Krylov, A.I. Two-photon absorption cross sections within equation-of-motion coupled-cluster formalism using resolution-of-the-identity and Cholesky decomposition representations: Theory, implementation, and benchmarks. J. Chem. Phys. 2015, 142, 064118. [Google Scholar] [CrossRef] [Green Version]
- Dalgaard, E. Quadratic response functions within the time-dependent Hartree-Fock approximation. Phys. Rev. A 1982, 26, 42–52. [Google Scholar] [CrossRef]
- Parkinson, W.A.; Sengeløv, P.W.; Oddershede, J. Two-photon transition moments as determined from the quadratic response function. Int. J. Quantum Chem. 1990, 38, 487–499. [Google Scholar] [CrossRef]
- Hettema, H.; Jensen, H.J.A.; Jørgensen, P.; Olsen, J. Quadratic response functions for a multiconfigurational self-consistent field wave function. J. Chem. Phys. 1992, 97, 1174–1190. [Google Scholar] [CrossRef]
- Koch, H.; Jørgensen, P. Coupled cluster response functions. J. Chem. Phys. 1990, 93, 3333–3344. [Google Scholar] [CrossRef]
- Hättig, C.; Christiansen, O.; Koch, H.; Jørgensen, P. Frequency-dependent first hyperpolarizabilities using coupled cluster quadratic response theory. Chem. Phys. Lett. 1997, 269, 428–434. [Google Scholar] [CrossRef]
- Hättig, C.; Christiansen, O.; Jørgensen, P. Multiphoton transition moments and absorption cross sections in coupled cluster response theory employing variational transition moment functionals. J. Chem. Phys. 1998, 108, 8331–8354. [Google Scholar] [CrossRef]
- Hättig, C.; Christiansen, O.; Jørgensen, P. Coupled cluster response calculations of two-photon transition probability rate constants for helium, neon and argon. J. Chem. Phys. 1998, 108, 8355–8359. [Google Scholar] [CrossRef]
- Iwata, J.I.; Yabana, K.; Bertsch, G.F. Real-space computation of dynamic hyperpolarizabilities. J. Chem. Phys. 2001, 115, 8773–8783. [Google Scholar] [CrossRef] [Green Version]
- Hait Heinze, H.; Della Sala, F.; Görling, A. Efficient methods to calculate dynamic hyperpolarizability tensors by time-dependent density-functional theory. J. Chem. Phys. 2002, 116, 9624–9640. [Google Scholar] [CrossRef]
- Sałek, P.; Vahtras, O.; Helgaker, T.; Ågren, H. Density-functional theory of linear and nonlinear time-dependent molecular properties. J. Chem. Phys. 2002, 117, 9630–9645. [Google Scholar] [CrossRef]
- Wang, F.; Yam, C.Y.; Chen, G. Time-dependent density-functional theory/localized density matrix method for dynamic hyperpolarizability. J. Chem. Phys. 2007, 126, 244102. [Google Scholar] [CrossRef] [Green Version]
- Zahariev, F.; Gordon, M.S. Nonlinear response time-dependent density functional theory combined with the effective fragment potential method. J. Chem. Phys. 2014, 140, 18A523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knippenberg, S.; Rehn, D.R.; Wormit, M.; Starcke, J.H.; Rusakova, I.L.; Trofimov, A.B.; Dreuw, A. Calculations of nonlinear response properties using the intermediate state representation and the algebraic-diagrammatic construction polarization propagator approach: Two-photon absorption spectra. J. Chem. Phys. 2012, 136, 064107. [Google Scholar] [CrossRef] [PubMed]
- Nanda, K.D.; Gulania, S.; Krylov, A.I. Theory, implementation, and disappointing results for two-photon absorption cross sections within the doubly electron-attached equation-of-motion coupled-cluster framework. J. Chem. Phys. 2023, 158, 054102. [Google Scholar] [CrossRef] [PubMed]
- Beerepoot, M.T.P.; Friese, D.H.; List, N.H.; Kongsted, J.; Ruud, K. Benchmarking two-photon absorption cross sections: Performance of CC2 and CAM-B3LYP. Phys. Chem. Chem. Phys. 2015, 17, 19306–19314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steindal, A.H.; Olsen, J.M.H.; Ruud, K.; Frediani, L.; Kongsted, J. A combined quantum mechanics/molecular mechanics study of the one- and two-photon absorption in the green fluorescent protein. Phys. Chem. Chem. Phys. 2012, 14, 5440–5451. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Zhao, Y.; Liang, W. Assessment of mode-mixing and Herzberg-Teller effects on two-photon absorption and resonance hyper-Raman spectra from a time-dependent approach. J. Chem. Phys. 2014, 140, 094107. [Google Scholar] [CrossRef] [PubMed]
- de Wergifosse, M.; Beaujean, P.; Grimme, S. Ultrafast Evaluation of Two-Photon Absorption with Simplified Time-Dependent Density Functional Theory. J. Phys. Chem. A 2022, 126, 7534–7547. [Google Scholar] [CrossRef]
- Gholami, S.; Pedraza-González, L.; Yang, X.; Granovsky, A.; Ioffe, I.; Olivucci, M. Multi-State Multi-Configuration Quantum Chemical Computation of the Two-Photon Absorption Spectra of Bovine Rhodopsin. J. Phys. Chem. Lett. 2019, 2019, 6293–6300. [Google Scholar] [CrossRef]
- Dick, B.; Hohlneicher, G. Importance of initial and final states as intermediate states in two-photon spectroscopy of polar molecules. J. Chem. Phys. 1982, 76, 5755–5760. [Google Scholar] [CrossRef] [Green Version]
- Birge, R.R.; Zhang, C. Two-photon double resonance spectroscopy of bacteriorhodopsin. Assignment of the electronic and dipolar properties of the low-lying 1Ag*--like and 1Bu*+-like π, π* states. J. Chem. Phys. 1990, 92, 7178–7195. [Google Scholar] [CrossRef]
- Drobizhev, M.; Tillo, S.; Makarov, N.S.; Hughes, T.E.; Rebane, A. Color Hues in Red Fluorescent Proteins Are Due to Internal Quadratic Stark Effect. J. Phys. Chem. B 2009, 113, 12860–12864. [Google Scholar] [CrossRef] [Green Version]
- Grabarek, D.; Andruniów, T. Illuminating the origins of two-photon absorption properties in fluorescent protein chromophores. Int. J. Quantum Chem. 2019, 120, e26086. [Google Scholar] [CrossRef]
- Drobizhev, M.; Molina, R.S.; Callis, P.R.; Scott, J.N.; Lambert, G.G.; Salih, A.; Shaner, N.C.; Hughes, T.E. Local Electric Field Controls Fluorescence Quantum Yield of Red and Far-Red Fluorescent Proteins. Front. Mol. Biosci. 2021, 8, 633217. [Google Scholar] [CrossRef] [PubMed]
- Delysse, S.; Raimond, P.; Nunzi, J.M. Two-photon absorption in non-centrosymmetric dyes. Chem. Phys. 1997, 219, 341–351. [Google Scholar] [CrossRef]
- Rebane, A.; Makarov, N.S.; Drobizhev, M.; Spangler, B.; Tarter, E.S.; Reeves, B.D.; Spangler, C.W.; Meng, F.; Suo, Z. Quantitative Prediction of Two-Photon Absorption Cross Section Based on Linear Spectroscopic Properties. J. Phys. Chem. C 2008, 112, 7997–8004. [Google Scholar] [CrossRef]
- Rebane, A.; Drobizhev, M.; Makarov, N.S.; Beuerman, E.; Haley, J.E.; Krein, D.M.; Burke, A.R.; Flikkema, J.L.; Cooper, T.M. Relation between Two-Photon Absorption and Dipolar Properties in a Series of Fluorenyl-Based Chromophores with Electron Donating or Electron Withdrawing Substituents. J. Phys. Chem. A 2011, 115, 4255–4262. [Google Scholar] [CrossRef]
- Cronstrand, P.; Luo, Y.; Ågren, H. Generalized few-state models for two-photon absorption of conjugated molecules. Chem. Phys. Lett. 2002, 352, 262–269. [Google Scholar] [CrossRef]
- Cronstrand, P.; Luo, Y.; Ågren, H. Effects of dipole alignment and channel interference on two-photon absorption cross sections of two-dimensional charge-transfer systems. J. Chem. Phys. 2002, 117, 11102–11106. [Google Scholar] [CrossRef]
- Alam, M.M.; Chattopadhyaya, M.; Chakrabarti, S. Solvent induced channel interference in the two-photon absorption process—A theoretical study with a generalized few-state-model in three dimensions. Phys. Chem. Chem. Phys. 2012, 14, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Beerepoot, M.; Alam, M.; Bednarska, J.; Bartkowiak, W.; Ruud, K.; Zalesny, R. Benchmarking the Performance of Exchange-Correlation Functionals for Predicting Two-Photon Absorption Strengths. J. Chem. Theory Comput. 2018, 14, 3677–3685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ośmiałowski, B.; Petrusevich, E.F.; Antoniak, M.A.; Grela, I.; Bin Jassar, M.A.; Nyk, M.; Luis, J.M.; Jędrzejewska, B.; Zaleśny, R.; Jacquemin, D. Controlling Two-Photon Action Cross Section by Changing a Single Heteroatom Position in Fluorescent Dyes. J. Phys. Chem. Lett. 2020, 11, 5920–5925. [Google Scholar] [CrossRef] [PubMed]
- Ośmiałowski, B.; Petrusevich, E.F.; Nawrot, K.C.; Paszkiewicz, B.K.; Nyk, M.; Zielak, J.; Jedrzejewska, B.; Luis, J.M.; Jacquemin, D.; Zaleśny, R. Tailoring the nonlinear absorption of fluorescent dyes by substitution at a boron center. J. Mater. Chem. C 2021, 9, 6225–6233. [Google Scholar] [CrossRef]
- Petrusevich, E.F.; Ośmiałowski, B.; Zaleśny, R.; Alam, M.M. Two-Photon Absorption Activity of BOPHY Derivatives: Insights from Theory. J. Phys. Chem. A 2021, 125, 2581–2587. [Google Scholar] [CrossRef] [PubMed]
- Chołuj, M.; Behera, R.; Petrusevich, E.F.; Bartkowiak, W.; Alam, M.M.; Zaleśny, R. Much of a Muchness: On the Origins of Two- and Three-Photon Absorption Activity of Dipolar Y-Shaped Chromophores. J. Phys. Chem. A 2022, 126, 752–759. [Google Scholar] [CrossRef]
- Chołuj, M.; Alam, M.; Beerepoot, M.; Sitkiewicz, S.; Matito, E.; Ruud, K.; Zaleśny, R. Choosing Bad versus Worse: Predictions of Two-Photon-Absorption Strengths Based on Popular Density Functional Approximations. J. Chem. Theory Comput. 2022, 18, 1046–1060. [Google Scholar] [CrossRef]
- Takaba, K.; Tai, Y.; Eki, H.; Dao, H.A.; Hanazono, Y.; Hasegawa, K.; Miki, K.; Takeda, K. Subatomic resolution X-ray structures of green fluorescent protein. IUCrJ 2019, 6, 387–400. [Google Scholar] [CrossRef] [Green Version]
- Ehrig, T.; O’Kane, D.J.; Prendergast, F.G. Green-fluorescent protein mutants with altered fluorescence excitation spectra. FEBS Lett. 1995, 367, 163–166. [Google Scholar] [CrossRef] [Green Version]
- Kummer, A.D.; Wiehler, J.; Rehaber, H.; Kompa, C.; Steipe, B.; Michel-Beyerle, M.E. Effects of Threonine 203 Replacements on Excited-State Dynamics and Fluorescence Properties of the Green Fluorescent Protein (GFP). J. Phys. Chem. B 2000, 104, 4791–4798. [Google Scholar] [CrossRef]
- Ormö, M.; Cubitt, A.B.; Kallio, K.; Gross, L.A.; Tsien, R.Y.; Remington, S.J. Crystal Structure of the Aequorea victoria Green Fluorescent Protein. Science 1996, 273, 1392–1395. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regmi, C.K. Structural Flexibility and Oxygen Diffusion Pathways in Monomeric Fluorescent Proteins. Ph.D. Thesis, FIU Electronic Theses and Dissertations, Florida International University, Miami, FL, USA, 2014. Available online: https://digitalcommons.fiu.edu/etd/1122 (accessed on 3 July 2023).
- Granovsky, A.A. Firefly Version 8. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 3 July 2023).
- Schmidt, M.; Baldridge, K.; Boatz, J.; Elbert, S.; Gordon, M.; Jenson, J.; Koseki, S.; Matsunaga, N.; Nguyen, K.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comp. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Rackers, J.A.; Wang, Z.; Lu, C.; Laury, M.L.; Lagardère, L.; Schnieders, M.J.; Piquemal, J.P.; Ren, P.; Ponder, J.W. Tinker 8: Software Tools for Molecular Design. J. Chem. Theory Comput. 2018, 14, 5273–5289. [Google Scholar] [CrossRef] [PubMed]
- Day, P.N.; Jensen, J.H.; Gordon, M.S.; Webb, S.P.; Stevens, W.J.; Morris, K.; David, G.; Harold, B.; Drora, C. An effective fragment method for modeling solvent effects in quantum mechanical calculations. J. Chem. Phys. 1996, 105, 1968–1986. [Google Scholar] [CrossRef]
- Gordon, M.S.; Freitag, M.A.; Bandyopadhyay, P.; Jensen, J.H.; Kairys, V.; Stevens, W.J. The Effective Fragment Potential Method: A QM-Based MM Approach to Modeling Environmental Effects in Chemistry. J. Phys. Chem. A 2001, 105, 293–307. [Google Scholar] [CrossRef] [Green Version]
- Granovsky, A. Extended multi-configuration quasi-degenerate perturbation theory: The new approach to multi-state multi-reference perturbation theory. J. Chem. Phys. 2011, 134, 214113. [Google Scholar] [CrossRef] [Green Version]
- Macak, P.; Luo, Y.; Ågren, H. Simulations of vibronic profiles in two-photon absorption. Chem. Phys. Lett. 2000, 330, 447–456. [Google Scholar] [CrossRef]
- Monson, P.R.; McClain, W.M. Polarization Dependence of the Two-Photon Absorption of Tumbling Molecules with Application to Liquid 1-Chloronaphthalene and Benzene. J. Chem. Phys. 1970, 53, 29–37. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HBDI | EGFP T203I | EGFP | |
|---|---|---|---|
| , D | 10.5 | 10.9 | 10.5 |
| , D | 2.9 | 2.5 | 4.4 |
| VEE, eV | 2.62 | 2.43 | 2.52 |
| N | |||
| 2 | 19 (7448) | 15 (6953) | 45 (19,262) |
| 3 | 19 (7439) | 15 (6991) | 45 (19,333) |
| 4 | 18 (7340) | 15 (7011) | 45 (19,474) |
| 5 | 18 (7270) | 15 (6762) | 45 (19,213) |
| 6 | 18 (7312) | 15 (6817) | 45 (19,209) |
| 7 | 19 (7396) | 15 (6759) | 44 (19,184) |
| Experiment [15] | — | 19 | 42 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aslopovsky, V.R.; Scherbinin, A.V.; Kleshchina, N.N.; Bochenkova, A.V. Impact of the Protein Environment on Two-Photon Absorption Cross-Sections of the GFP Chromophore Anion Resolved at the XMCQDPT2 Level of Theory. Int. J. Mol. Sci. 2023, 24, 11266. https://doi.org/10.3390/ijms241411266
Aslopovsky VR, Scherbinin AV, Kleshchina NN, Bochenkova AV. Impact of the Protein Environment on Two-Photon Absorption Cross-Sections of the GFP Chromophore Anion Resolved at the XMCQDPT2 Level of Theory. International Journal of Molecular Sciences. 2023; 24(14):11266. https://doi.org/10.3390/ijms241411266
Chicago/Turabian StyleAslopovsky, Vladislav R., Andrei V. Scherbinin, Nadezhda N. Kleshchina, and Anastasia V. Bochenkova. 2023. "Impact of the Protein Environment on Two-Photon Absorption Cross-Sections of the GFP Chromophore Anion Resolved at the XMCQDPT2 Level of Theory" International Journal of Molecular Sciences 24, no. 14: 11266. https://doi.org/10.3390/ijms241411266