

Lamivudine (3TC), a Nucleoside Reverse Transcriptase Inhibitor, Prevents the Neuropathological Alterations Present in Mutant Tau Transgenic Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
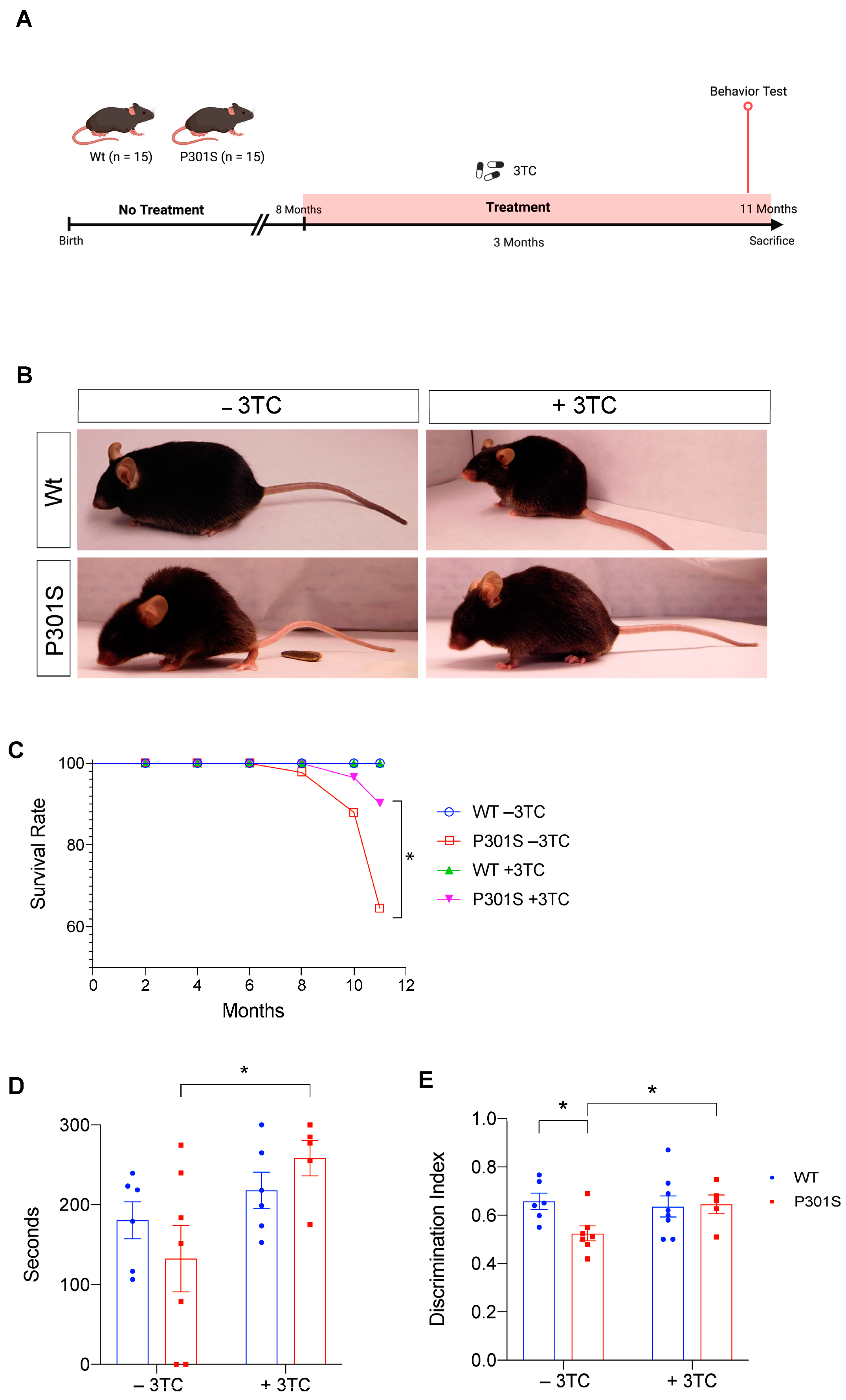
2.1. P301S Mice Show an Increase in Survival and an Improvement in Learning after 3TC Treatment
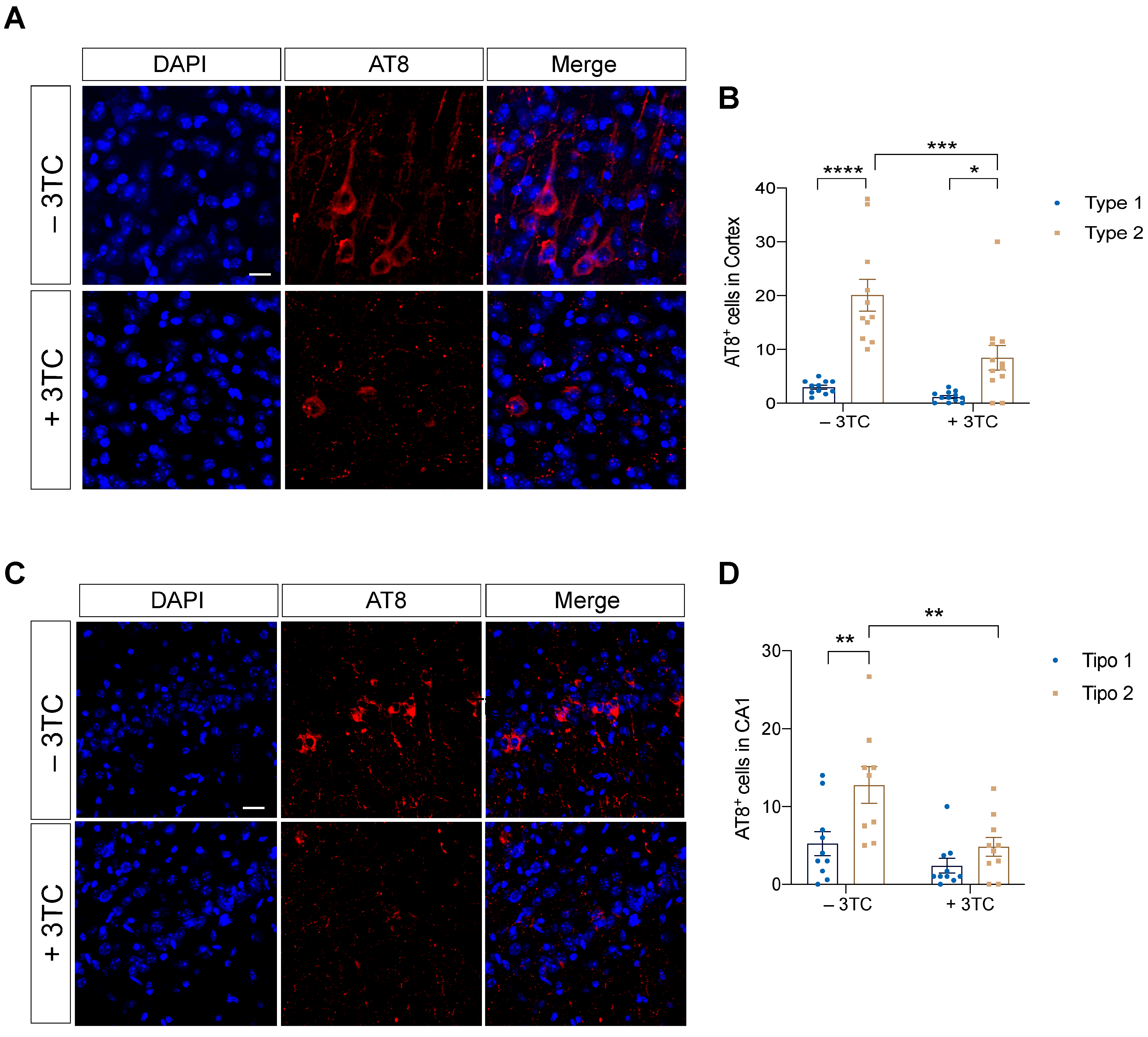
2.2. Pharmacological 3TC Treatment Modulates P301S-Dependent Tau Pathology In Vivo
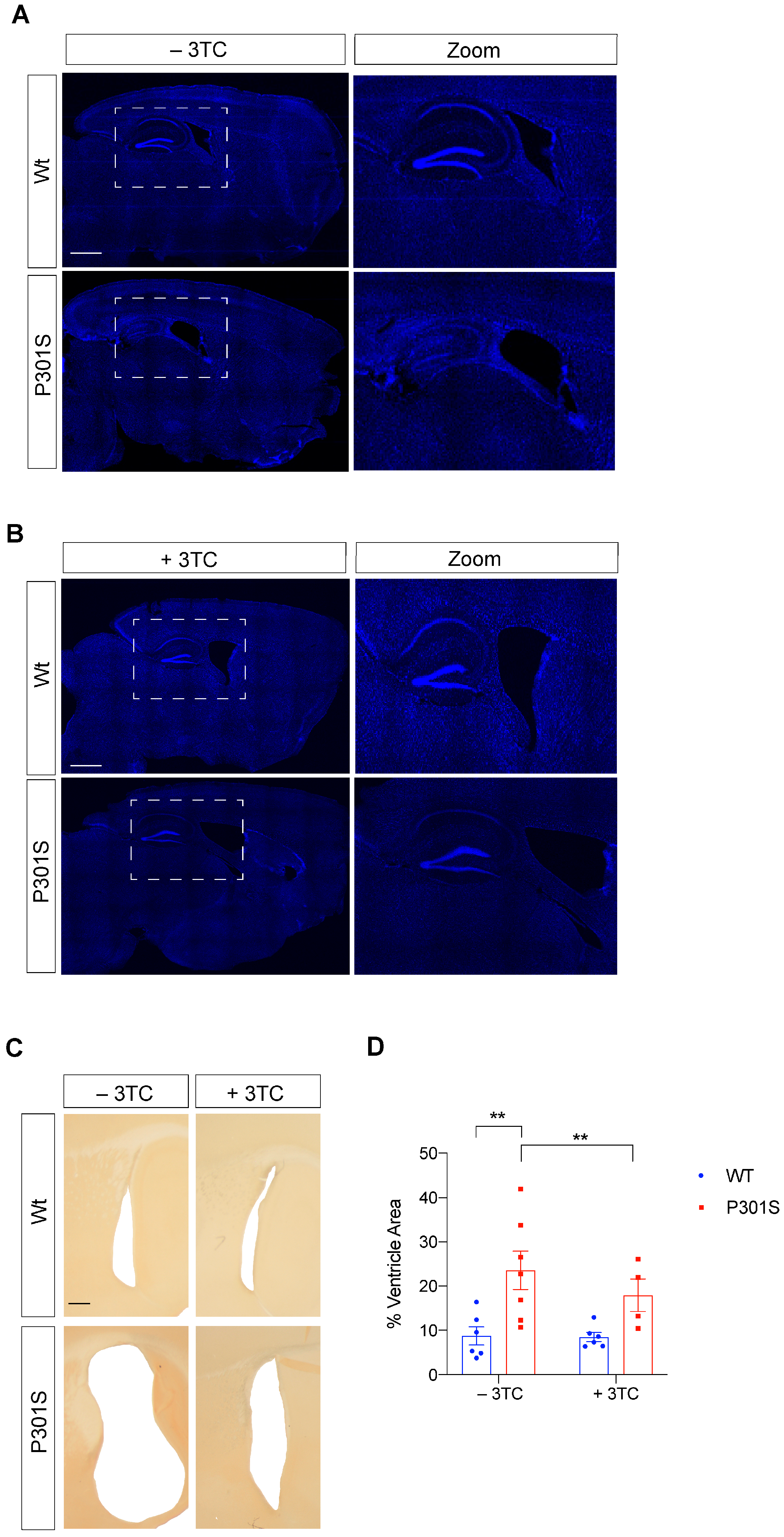
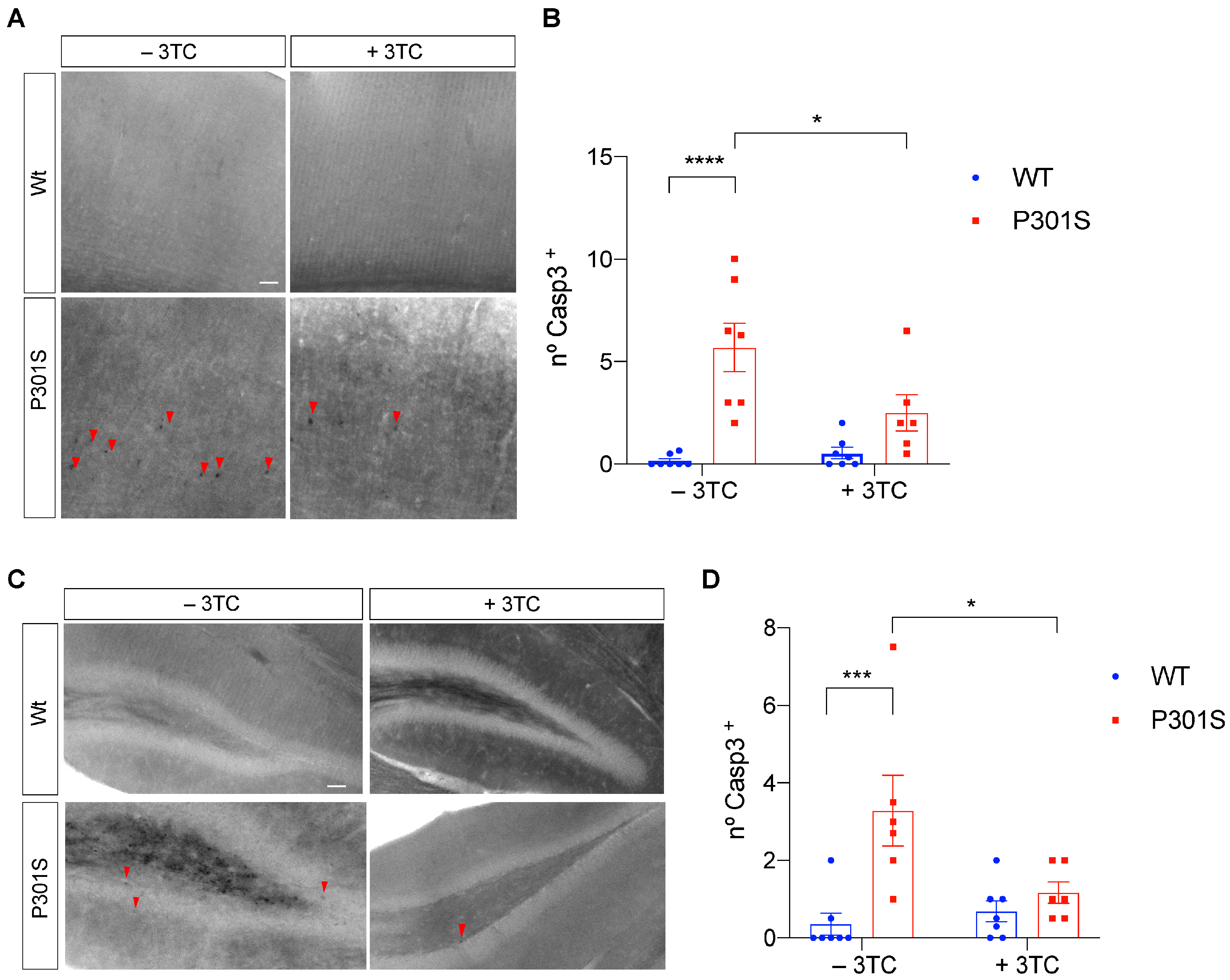
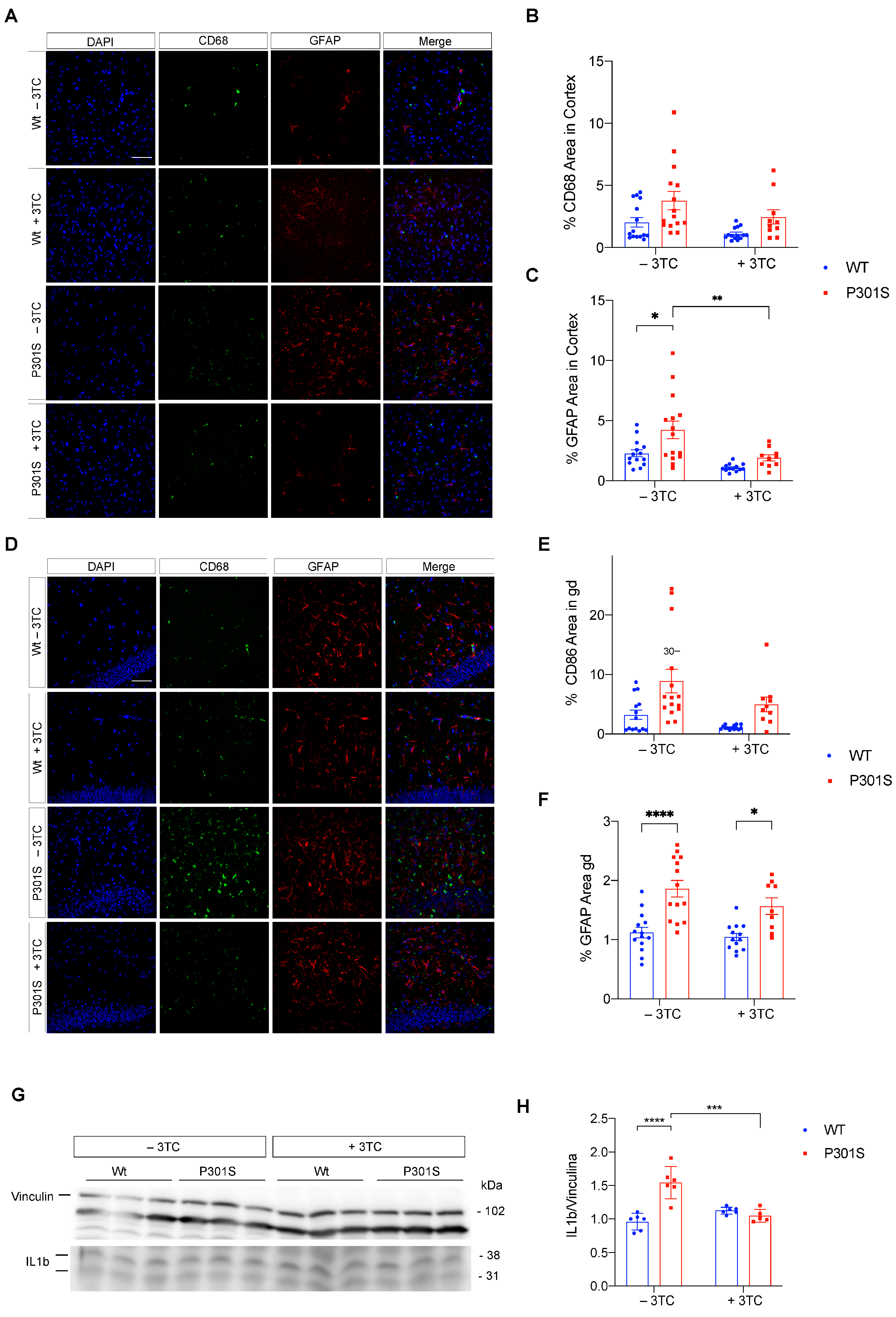
2.3. Partial Reversal of Atrophy, Neuronal Death, and Gliosis in 3TC-Treated P301S Mice
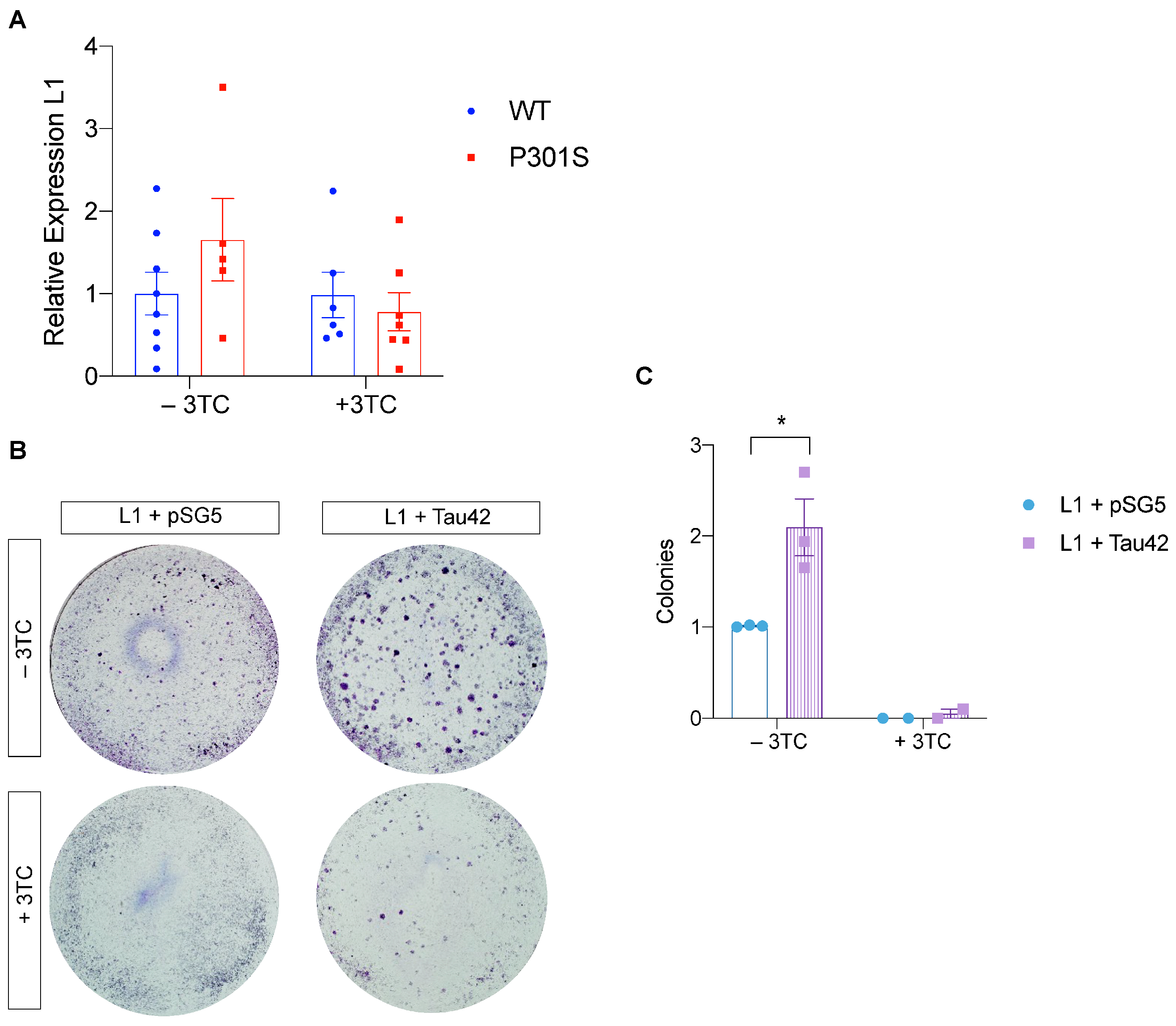
2.4. Histone-3 Trimethylation Levels and LINE-1 Insertion in Genomic DNA
2.5. Tau Promotes LINE-1 Insertion In Vitro
3. Discussion
4. Material and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Driver, C.J.; McKechnie, S.W. Transposable elements as a factor in the aging of Drosophila melanogaster. Ann. N. Y. Acad. Sci. 1992, 673, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.G.; Helfand, S.L. Chromatin structure and transposable elements in organismal aging. Front. Genet. 2013, 4, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Prazak, L.; Chatterjee, N.; Gruninger, S.; Krug, L.; Theodorou, D.; Dubnau, J. Activation of transposable elements during aging and neuronal decline in Drosophila. Nat. Neurosci. 2013, 16, 529–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [Green Version]
- Baillie, J.K.; Barnett, M.W.; Upton, K.R.; Gerhardt, D.J.; Richmond, T.A.; De Sapio, F.; Brennan, P.M.; Rizzu, P.; Smith, S.; Fell, M.; et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature 2011, 479, 534–537. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Moran, J.V.; Kazazian, H.H., Jr.; Boeke, J.D. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 1996, 87, 905–916. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.L.; Cruceanu, M.; Branciforte, D.; Wai-Lun Li, P.; Kwok, S.C.; Hodges, R.S.; Williams, M.C. LINE-1 retrotransposition requires the nucleic acid chaperone activity of the ORF1 protein. J. Mol. Biol. 2005, 348, 549–561. [Google Scholar] [CrossRef]
- Hancks, D.C.; Kazazian, H.H., Jr. Active human retrotransposons: Variation and disease. Curr. Opin. Genet. Dev. 2012, 22, 191–203. [Google Scholar] [CrossRef] [Green Version]
- De Cecco, M.; Criscione, S.W.; Peckham, E.J.; Hillenmeyer, S.; Hamm, E.A.; Manivannan, J.; Peterson, A.L.; Kreiling, J.A.; Neretti, N.; Sedivy, J.M. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 2013, 12, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Wood, J.G.; Jones, B.C.; Jiang, N.; Chang, C.; Hosier, S.; Wickremesinghe, P.; Garcia, M.; Hartnett, D.A.; Burhenn, L.; Neretti, N.; et al. Chromatin-modifying genetic interventions suppress age-associated transposable element activation and extend life span in Drosophila. Proc. Natl. Acad. Sci. USA 2016, 113, 11277–11282. [Google Scholar] [CrossRef]
- Ravel-Godreuil, C.; Znaidi, R.; Bonnifet, T.; Joshi, R.L.; Fuchs, J. Transposable elements as new players in neurodegenerative diseases. FEBS Lett. 2021, 595, 2733–2755. [Google Scholar] [CrossRef] [PubMed]
- Murray, V. Are transposons a cause of ageing? Mutat. Res. 1990, 237, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Jeong, H.H.; Hsieh, Y.C.; Klein, H.U.; Bennett, D.A.; De Jager, P.L.; Liu, Z.; Shulman, J.M. Tau Activates Transposable Elements in Alzheimer’s Disease. Cell Rep. 2018, 23, 2874–2880. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Samimi, H.; Gamez, M.; Zare, H.; Frost, B. Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat. Neurosci. 2018, 21, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, P.; Zuniga, G.; Sun, W.; Beckmann, A.; Ochoa, E.; DeVos, S.L.; Hyman, B.; Chiu, G.; Roy, E.R.; Cao, W.; et al. Pathogenic tau accelerates aging-associated activation of transposable elements in the mouse central nervous system. Prog. Neurobiol. 2022, 208, 102181. [Google Scholar] [CrossRef]
- Grundman, J.; Spencer, B.; Sarsoza, F.; Rissman, R.A. Transcriptome analyses reveal tau isoform-driven changes in transposable element and gene expression. PLoS ONE 2021, 16, e0251611. [Google Scholar] [CrossRef]
- Misiak, B.; Ricceri, L.; Sasiadek, M.M. Transposable Elements and Their Epigenetic Regulation in Mental Disorders: Current Evidence in the Field. Front. Genet. 2019, 10, 580. [Google Scholar] [CrossRef] [Green Version]
- Terry, D.M.; Devine, S.E. Aberrantly High Levels of Somatic LINE-1 Expression and Retrotransposition in Human Neurological Disorders. Front. Genet. 2019, 10, 1244. [Google Scholar] [CrossRef] [Green Version]
- Siangphoe, U.; Archer, K.J.; Nguyen, C.; Lee, K.R. Associations of antiretroviral therapy and comorbidities with neurocognitive outcomes in HIV-1-infected patients. AIDS 2020, 34, 893–902. [Google Scholar] [CrossRef]
- Hache, C.; Villeneuve, J.P. Lamivudine treatment in patients with chronic hepatitis B and cirrhosis. Expert Opin. Pharmacother. 2006, 7, 1835–1843. [Google Scholar] [CrossRef]
- Quercia, R.; Perno, C.F.; Koteff, J.; Moore, K.; McCoig, C.; St Clair, M.; Kuritzkes, D. Twenty-Five Years of Lamivudine: Current and Future Use for the Treatment of HIV-1 Infection. J. Acquir. Immune Defic. Syndr. 2018, 78, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Martinez de Lagran, M.; Elizalde-Torrent, A.; Paredes, R.; Clotet, B.; Dierssen, M. Lamivudine, a reverse transcriptase inhibitor, rescues cognitive deficits in a mouse model of down syndrome. J. Cell. Mol. Med. 2022, 26, 4210–4215. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, J.; Tang, Q.; Zhang, Q.; Wang, Y.; Zhang, J.; Hao, Y.; Bai, X.; Lu, Z. Lamivudine improves cognitive decline in SAMP8 mice: Integrating in vivo pharmacological evaluation and network pharmacology. J. Cell. Mol. Med. 2021, 25, 8490–8503. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Merchan-Rubira, J.; Sebastian-Serrano, A.; Diaz-Hernandez, M.; Avila, J.; Hernandez, F. Peripheral nervous system effects in the PS19 tau transgenic mouse model of tauopathy. Neurosci. Lett. 2019, 698, 204–208. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Vanmechelen, E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci. Lett. 1995, 189, 167–169. [Google Scholar] [CrossRef]
- Engel, T.; Goni-Oliver, P.; Lucas, J.J.; Avila, J.; Hernandez, F. Chronic lithium administration to FTDP-17 tau and GSK-3beta overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J. Neurochem. 2006, 99, 1445–1455. [Google Scholar] [CrossRef]
- Hashem, J.; Hu, M.; Zhang, J.; Gao, F.; Chen, C. Inhibition of 2-Arachidonoylglycerol Metabolism Alleviates Neuropathology and Improves Cognitive Function in a Tau Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 4122–4133. [Google Scholar] [CrossRef]
- Sarkar, T.J.; Quarta, M.; Mukherjee, S.; Colville, A.; Paine, P.; Doan, L.; Tran, C.M.; Chu, C.R.; Horvath, S.; Qi, L.S.; et al. Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat. Commun. 2020, 11, 1545. [Google Scholar] [CrossRef] [Green Version]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 2016, 167, 1719–1733.E12. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Matellan, A.; Alcazar, N.; Hernandez, F.; Serrano, M.; Avila, J. In Vivo Reprogramming Ameliorates Aging Features in Dentate Gyrus Cells and Improves Memory in Mice. Stem Cell Rep. 2020, 15, 1056–1066. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, B.J.; Kroutter, E.N.; Derbes, R.S.; Belancio, V.P.; Roy-Engel, A.M. Molecular reconstruction of extinct LINE-1 elements and their interaction with nonautonomous elements. Mol. Biol. Evol. 2013, 30, 88–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montejo de Garcini, E.; de la Luna, S.; Dominguez, J.E.; Avila, J. Overexpression of tau protein in COS-1 cells results in the stabilization of centrosome-independent microtubules and extension of cytoplasmic processes. Mol. Cell Biochem. 1994, 130, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Frost, B. Alzheimer’s disease: An acquired neurodegenerative laminopathy. Nucleus 2016, 7, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Galas, M.C.; Bonnefoy, E.; Buee, L.; Lefebvre, B. Emerging Connections Between Tau and Nucleic Acids. Adv. Exp. Med. Biol. 2019, 1184, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Zekanowski, C.; Wojda, U. Aneuploidy, chromosomal missegregation, and cell cycle reentry in Alzheimer’s disease. Acta Neurobiol. Exp. 2009, 69, 232–253. [Google Scholar]
- Hancks, D.C.; Kazazian, H.H., Jr. Roles for retrotransposon insertions in human disease. Mob. DNA 2016, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef] [Green Version]
- Benhelli-Mokrani, H.; Mansuroglu, Z.; Chauderlier, A.; Albaud, B.; Gentien, D.; Sommer, S.; Schirmer, C.; Laqueuvre, L.; Josse, T.; Buee, L.; et al. Genome-wide identification of genic and intergenic neuronal DNA regions bound by Tau protein under physiological and stress conditions. Nucleic Acids Res. 2018, 46, 11405–11422. [Google Scholar] [CrossRef] [Green Version]
- Gil, L.; Nino, S.A.; Guerrero, C.; Jimenez-Capdeville, M.E. Phospho-Tau and Chromatin Landscapes in Early and Late Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 283. [Google Scholar] [CrossRef]
- Pegoraro, G.; Misteli, T. The central role of chromatin maintenance in aging. Aging 2009, 1, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S.; et al. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron 2018, 99, 925–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, L.; Federico, C.; Pinedo, F.; Bruno, F.; Rebolledo, A.B.; Montoya, J.J.; Olazabal, I.M.; Ferrer, I.; Saccone, S. Aging dependent effect of nuclear tau. Brain Res. 2017, 1677, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Monroy-Ramirez, H.C.; Basurto-Islas, G.; Mena, R.; Cisneros, B.; Binder, L.I.; Avila, J.; Garcia-Sierra, F. Alterations in the nuclear architecture produced by the overexpression of tau protein in neuroblastoma cells. J. Alzheimer’s Dis. 2013, 36, 503–520. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Nogales, M.; Cabrera, J.R.; Santos-Galindo, M.; Hoozemans, J.J.; Ferrer, I.; Rozemuller, A.J.; Hernandez, F.; Avila, J.; Lucas, J.J. Huntington’s disease is a four-repeat tauopathy with tau nuclear rods. Nat. Med. 2014, 20, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Nogales, M.; Santos-Galindo, M.; Merchan-Rubira, J.; Hoozemans, J.J.M.; Rabano, A.; Ferrer, I.; Avila, J.; Hernandez, F.; Lucas, J.J. Tau-positive nuclear indentations in P301S tauopathy mice. Brain Pathol. 2017, 27, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Paonessa, F.; Evans, L.D.; Solanki, R.; Larrieu, D.; Wray, S.; Hardy, J.; Jackson, S.P.; Livesey, F.J. Microtubules Deform the Nuclear Membrane and Disrupt Nucleocytoplasmic Transport in Tau-Mediated Frontotemporal Dementia. Cell Rep. 2019, 26, 582–593. [Google Scholar] [CrossRef] [Green Version]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Paxinos, G.; Franklin, K.B.J. The Mouse Brain; Academic Press: Cambridge, MA, USA, 2001. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallés-Saiz, L.; Ávila, J.; Hernández, F. Lamivudine (3TC), a Nucleoside Reverse Transcriptase Inhibitor, Prevents the Neuropathological Alterations Present in Mutant Tau Transgenic Mice. Int. J. Mol. Sci. 2023, 24, 11144. https://doi.org/10.3390/ijms241311144
Vallés-Saiz L, Ávila J, Hernández F. Lamivudine (3TC), a Nucleoside Reverse Transcriptase Inhibitor, Prevents the Neuropathological Alterations Present in Mutant Tau Transgenic Mice. International Journal of Molecular Sciences. 2023; 24(13):11144. https://doi.org/10.3390/ijms241311144
Chicago/Turabian StyleVallés-Saiz, Laura, Jesús Ávila, and Félix Hernández. 2023. "Lamivudine (3TC), a Nucleoside Reverse Transcriptase Inhibitor, Prevents the Neuropathological Alterations Present in Mutant Tau Transgenic Mice" International Journal of Molecular Sciences 24, no. 13: 11144. https://doi.org/10.3390/ijms241311144