

A C57BL/6J Fancg-KO Mouse Model Generated by CRISPR/Cas9 Partially Captures the Human Phenotype

 and
and
Abstract
:1. Introduction
2. Results
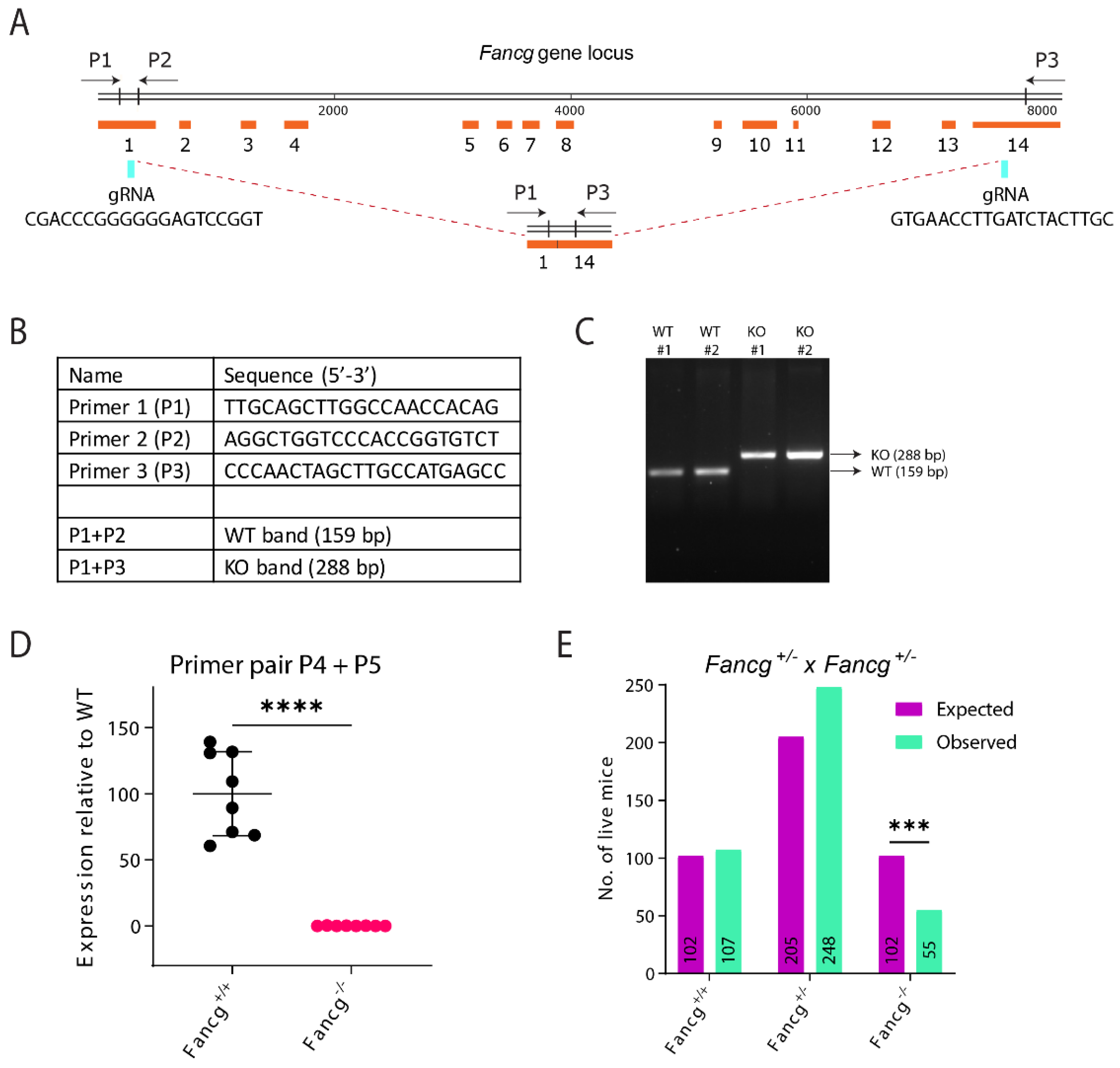
2.1. Establishing a Genetically and Immunologically Defined C57BL/6J Fancg-KO Mouse Model
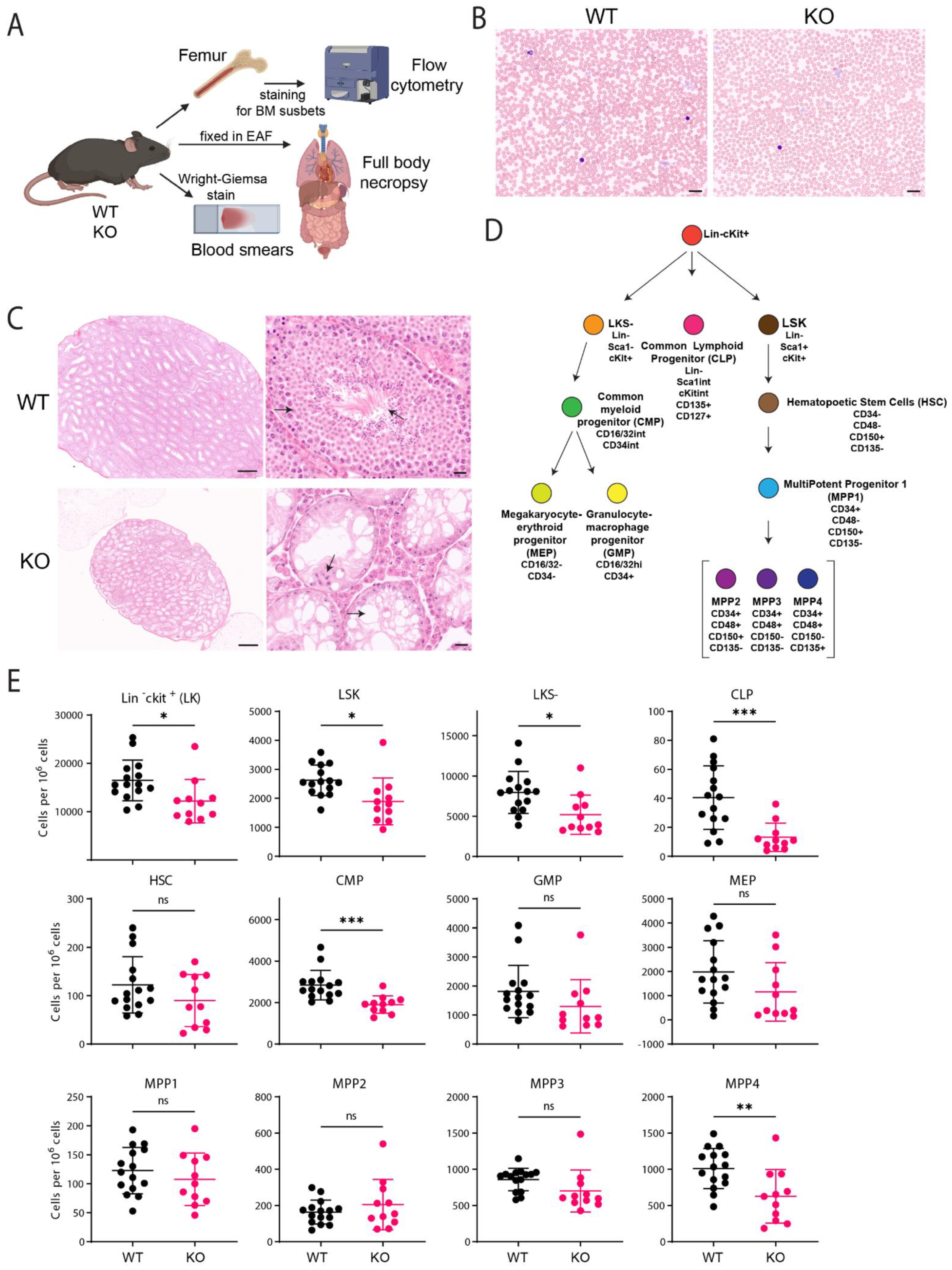
2.2. C57BL/6J Fancg-KO Mice Display Stem Cell Defects
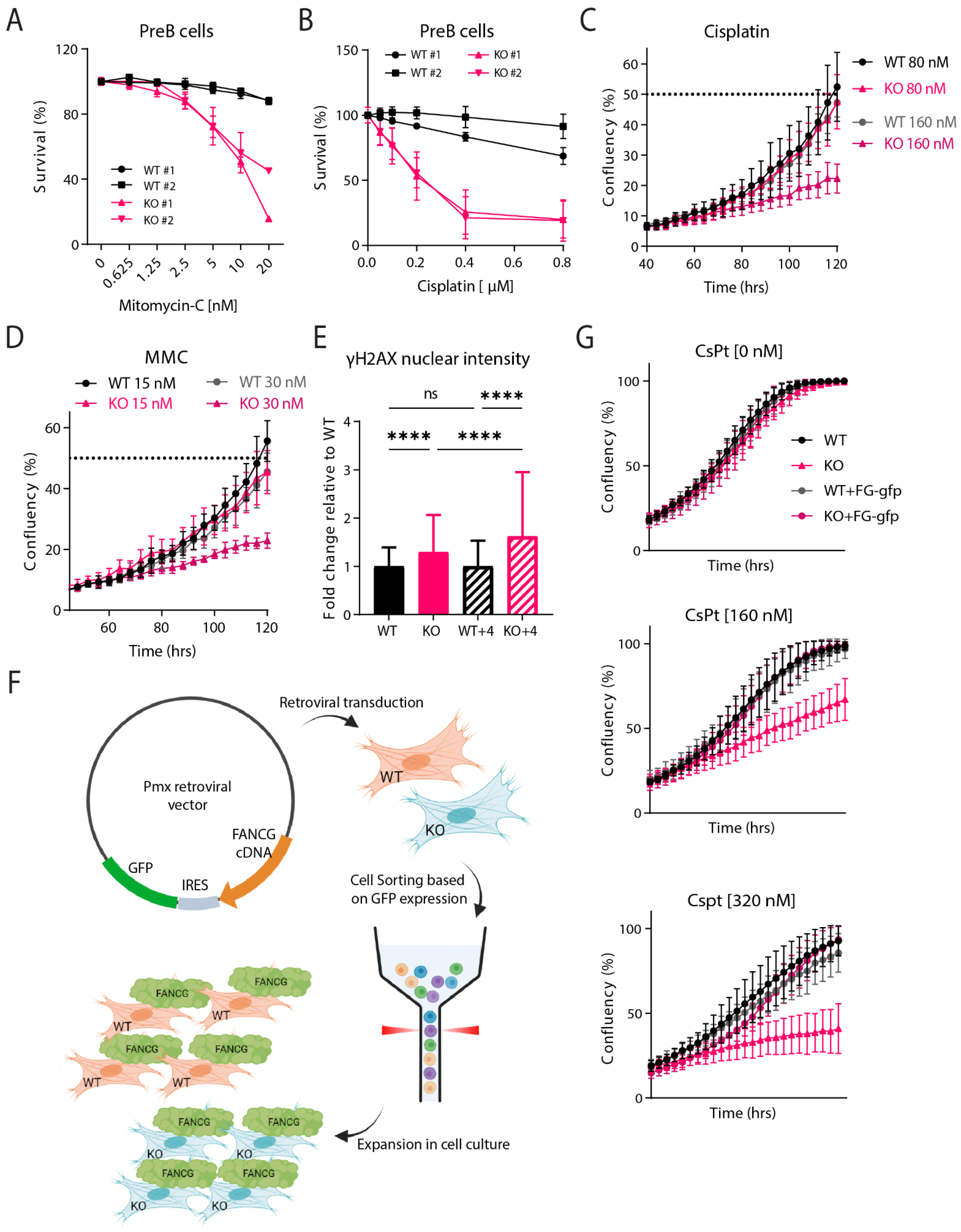
2.3. Fancg-KO Cells Are Hypersensitive to Crosslinking Agents
3. Discussion
4. Materials and Methods
4.1. Generation of Fancg−/− Mouse
4.2. Generation of Primary Cell Lines and Cell Culture
4.3. PCR Genotyping
4.4. qRT-PCR
4.5. Immunofluorescence of Bone Marrow (BM) Cells
4.6. Histopathology
4.7. PreB Survival Assay
4.8. IncuCyte Proliferation Assay
4.9. γ-H2AX Immunofluorescence
4.10. Fancg-Flag Reconstitution
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamashita, T.; Nakahata, T. Current Knowledge on the Pathophysiology of Fanconi Anemia: From Genes to Phenotypes. Int. J. Hematol. 2001, 74, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Guitton-Sert, L.; Gao, Y.; Masson, J.-Y. Animal models of Fanconi anemia: A developmental and therapeutic perspective on a multifaceted disease. Semin. Cell. Dev. Biol. 2021, 113, 113–131. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell. Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Errazquin, R.; Page, A.; Suñol, A.; Segrelles, C.; Carrasco, E.; Peral, J.; Garrido-Aranda, A.; Del Marro, S.; Ortiz, J.; Lorz, C.; et al. Development of a mouse model for spontaneous oral squamous cell carcinoma in Fanconi anemia. Oral Oncol. 2022, 134, 106184. [Google Scholar] [CrossRef]
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef]
- Che, R.; Zhang, J.; Nepal, M.; Han, B.; Fei, P. Multifaceted Fanconi Anemia Signaling. Trends Genet. 2018, 34, 171–183. [Google Scholar] [CrossRef]
- Alpi, A.F.; Patel, K.J. Monoubiquitylation in the Fanconi anemia DNA damage response pathway. DNA Repair. 2009, 8, 430–435. [Google Scholar] [CrossRef]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef] [Green Version]
- De Winter, J.P.; Waisfisz, Q.; Rooimans, M.A.; van Berkel, C.G.; Bosnoyan-Collins, L.; Alon, N.; Carreau, M.; Bender, O.; Demuth, I.; Schindler, D.; et al. The Fanconi anaemia group G gene FANCG is identical with XRCC9. Nat. Genet. 1998, 20, 281–283. [Google Scholar] [CrossRef]
- Liu, N.; Lamerdin, J.E.; Tucker, J.D.; Zhou, Z.-Q.; Walter, C.A.; Albala, J.S.; Busch, D.B.; Thompson, L.H. The human XRCC9 gene corrects chromosomal instability and mutagen sensitivities in CHO UV40 cells. Proc. Natl. Acad. Sci. USA 1997, 94, 9232–9237. [Google Scholar] [CrossRef]
- Saar, K.; Schindler, D.; Wegner, R.-D.; Reis, A.; Wienker, T.F.; Hoehn, H.; Joenje, H.; Sperling, K.; Digweed, M. Localisation of a Fanconi anaemia gene to chromosome 9p. Eur. J. Hum. Genet. 1998, 6, 501–508. [Google Scholar] [CrossRef] [Green Version]
- Van De Vrugt, H.J.; Koomen, M.; Berns, M.A.D.; De Vries, Y.; Rooimans, M.A.; Van Der Weel, L.; Blom, E.; De Groot, J.; Schepers, R.J.; Stone, S.; et al. Characterization, expression and complex formation of the murine Fanconi anaemia gene product Fancg. Genes Cells 2002, 7, 333–342. [Google Scholar] [CrossRef]
- Demuth, I.; Wlodarski, M.; Tipping, A.J.; Morgan, N.V.; de Winter, J.P.; Thiel, M.; Gräsl, S.; Schindler, D.; D’Andrea, A.D.; Altay, C.; et al. Spectrum of mutations in the Fanconi anaemia group G gene, FANCG/XRCC9. Eur. J. Hum. Genet. 2000, 8, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Higuera, I.; Kuang, Y.; Näf, D.; Wasik, J.; D’Andrea, A.D. Fanconi anemia proteins FANCA, FANCC, and FANCG/XRCC9 interact in a functional nuclear complex. Mol. Cell. Biol. 1999, 19, 4866–4873. [Google Scholar] [CrossRef] [Green Version]
- Waisfisz, Q.; de Winter, J.P.; Kruyt, F.A.; de Groot, J.; van der Weel, L.; Dijkmans, L.M.; Zhi, Y.; Arwert, F.; Scheper, R.J.; Youssoufian, H.; et al. A physical complex of the Fanconi anemia proteins FANCG/XRCC9 and FANCA. Proc. Natl. Acad. Sci. USA 1999, 96, 10320–10325. [Google Scholar] [CrossRef]
- Garcia-Higuera, I.; Kuang, Y.; Denham, J.; D’Andrea, A.D. The fanconi anemia proteins FANCA and FANCG stabilize each other and promote the nuclear accumulation of the Fanconi anemia complex. Blood 2000, 96, 3224–3230. [Google Scholar] [CrossRef]
- De Winter, J.P.; van der Weel, L.; de Groot, J.; Stone, S.; Waisfisz, Q.; Arwert, F.; Scheper, R.J.; Kruyt, F.A.; Hoatlin, M.E.; Joenje, H. The Fanconi anemia protein FANCF forms a nuclear complex with FANCA, FANCC and FANCG. Hum. Mol. Genet. 2000, 9, 2665–2674. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Ishiai, M.; Matsushita, N.; Arakawa, H.; Lamerdin, J.E.; Buerstedde, J.-M.; Tanimoto, M.; Harada, M.; Thompson, L.H.; Takata, M. Fanconi anemia FANCG protein in mitigating radiation- and enzyme-induced DNA double-strand breaks by homologous recombination in vertebrate cells. Mol. Cell. Biol. 2003, 23, 5421–5430. [Google Scholar] [CrossRef] [Green Version]
- Kersten, K.; de Visser, K.E.; van Miltenburg, M.H.; Jonkers, J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol. Med. 2017, 9, 137–153. [Google Scholar] [CrossRef]
- Hill, W.; Caswell, D.R.; Swanton, C. Capturing cancer evolution using genetically engineered mouse models (GEMMs). Trends Cell Biol. 2021, 31, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Bakker, S.T.; de Winter, J.P.; te Riele, H. Learning from a paradox: Recent insights into Fanconi anaemia through studying mouse models. Dis. Model. Mech. 2013, 6, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, E.L.; Guitton-Sert, L.; Béliveau, M.; Parmar, K.; Chagraoui, J.; Vignard, J.; Pauty, J.; Caron, M.-C.; Coulombe, Y.; Buisson, R.; et al. A Fanci knockout mouse model reveals common and distinct functions for FANCI and FANCD2. Nucleic Acids Res. 2019, 47, 7532–7547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, N.C.; van de Vrugt, H.J.; van der Valk, M.A.; Oostra, A.B.; Krimpenfort, P.; de Vries, Y.; Joenje, H.; Berns, A.; Arwert, F. Mice with a targeted disruption of the Fanconi anemia homolog Fanca. Hum. Mol. Genet. 2000, 9, 1805–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wen, Y.; Guo, X. CRISPR/Cas9 for genome editing: Progress, implications and challenges. Hum. Mol. Genet. 2014, 23, R40–R46. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Zhang, J.; Wu, H.; Wang, J.; Ma, K.; Li, Z.; Zhang, X.; Zhang, P.; Huang, X. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res. 2013, 23, 720–723. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [Green Version]
- Koomen, M.; Cheng, N.C.; van de Vrugt, H.J.; Godthelp, B.C.; van der Valk, M.A.; Oostra, A.B.; Zdzienicka, M.Z.; Joenje, H.; Arwert, F. Reduced fertility and hypersensitivity to mitomycin C characterize Fancg/Xrcc9 null mice. Hum. Mol. Genet. 2002, 11, 273–281. [Google Scholar] [CrossRef]
- Yang, Y.; Kuang, Y.; De Oca, R.M.; Hays, T.; Moreau, L.; Lu, N.; Seed, B.; D’Andrea, A.D. Targeted disruption of the murine Fanconi anemia gene, Fancg/Xrcc9. Blood 2001, 98, 3435–3440. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.M.; Greenaway, S.; White, J.K.; Fuchs, H.; Gailus-Durner, V.; Wells, S.; Sorg, T.; Wong, K.; Bedu, E.; Cartwright, E.J.; et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 2013, 14, R82. [Google Scholar] [CrossRef]
- Kutler, D.I.; Singh, B.; Satagopan, J.; Batish, S.D.; Berwick, M.; Giampietro, P.F.; Hanenberg, H.; Auerbach, A.D. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood 2003, 101, 1249–1256. [Google Scholar] [CrossRef] [Green Version]
- Mathew, C.G. Fanconi anaemia genes and susceptibility to cancer. Oncogene 2006, 25, 5875–5884. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Domenech, C.; Maillard, L.; Rousseau, A.; Guidez, F.; Petit, L.; Pla, M.; Clay, D.; Guimiot, F.; Sanfilippo, S.; Jacques, S.; et al. Studies in an Early Development Window Unveils a Severe HSC Defect in both Murine and Human Fanconi Anemia. Stem Cell Rep. 2018, 11, 1075–1091. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.; Laurenti, E.; Oser, G.; van der Wath, R.C.; Blanco-Bose, W.; Jaworski, M.; Offner, S.; Dunant, C.F.; Eshkind, L.; Bockamp, E.; et al. Hematopoietic Stem Cells Reversibly Switch from Dormancy to Self-Renewal during Homeostasis and Repair. Cell 2008, 135, 1118–1129. [Google Scholar] [CrossRef] [Green Version]
- Pulliam-Leath, A.C.; Ciccone, S.L.; Nalepa, G.; Li, X.; Si, Y.; Miravalle, L.; Smith, D.; Yuan, J.; Li, J.; Anur, P.; et al. Genetic disruption of both Fancc and Fancg in mice recapitulates the hematopoietic manifestations of Fanconi anemia. Blood 2010, 116, 2915–2920. [Google Scholar] [CrossRef] [Green Version]
- Pietras, E.M.; Reynaud, D.; Kang, Y.-A.; Carlin, D.; Calero-Nieto, F.J.; Leavitt, A.D.; Stuart, J.M.; Göttgens, B.; Passegué, E. Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell Stem Cell 2015, 17, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Tomkins, D.J.; Auerbach, W.; McKerlie, C.; Youssoufian, H.; Liu, L.; Gan, O.; Carreau, M.; Auerbach, A.; Groves, T.; et al. Inactivation of Fac in mice produces inducible chromosomal instability and reduced fertility reminiscent of Fanconi anaemia. Nat. Genet. 1996, 12, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018, 553, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Dingler, F.A.; Patel, K.J. Genotoxic aldehydes in the hematopoietic system. Blood 2022, 139, 2119–2129. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, I.M.; Chen, J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarysta, A.; Riou, L.; Firlej, V.; Lapoujade, C.; Kortulewski, T.; Barroca, V.; Gille, A.-S.; Dumont, F.; Jacques, S.; Letourneur, F.; et al. Abnormal migration behavior linked to Rac1 signaling contributes to primordial germ cell exhaustion in Fanconi anemia pathway-deficient Fancg−/− embryos. Hum. Mol. Genet. 2021, 31, 97–110. [Google Scholar] [CrossRef]
- Barroca, V.; Mouthon, M.A.; Lewandowski, D.; Brunet de la Grange, P.; Gauthier, L.R.; Pflumio, F.; Boussin, F.D.; Arwert, F.; Riou, L.; Allemand, I.; et al. Impaired functionality and homing of Fancg-deficient hematopoietic stem cells. Hum. Mol. Genet. 2012, 21, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, S.; Yuan, J.; Yang, Y.; Li, J.; Ma, J.; Wu, X.; Freund, M.; Pollok, K.; Hanenberg, H.; et al. Mesenchymal stem/progenitor cells promote the reconstitution of exogenous hematopoietic stem cells in Fancg−/− mice in vivo. Blood 2009, 113, 2342–2351. [Google Scholar] [CrossRef] [Green Version]
- Whitney, M.A.; Royle, G.; Low, M.J.; Kelly, M.A.; Axthelm, M.K.; Reifsteck, C.; Olson, S.; Braun, R.E.; Heinrich, M.C.; Rathbun, R.K.; et al. Germ cell defects and hematopoietic hypersensitivity to gamma-interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood 1996, 88, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Whitney, M.A.; Jakobs, P.; Kaback, M.; Moses, R.E.; Grompe, M. The Ashkenazi Jewish Fanconi anemia mutation: Incidence among patients and carrier frequency in the at-risk population. Hum. Mutat. 1994, 3, 339–341. [Google Scholar] [CrossRef]
- Zhou, Y.; He, Y.; Xing, W.; Zhang, P.; Shi, H.; Chen, S.; Shi, J.; Bai, J.; Rhodes, S.D.; Zhang, F.; et al. An abnormal bone marrow microenvironment contributes to hematopoietic dysfunction in Fanconi anemia. Haematologica 2017, 102, 1017–1027. [Google Scholar] [CrossRef] [Green Version]
- Parmar, K.; Kim, J.; Sykes, S.M.; Shimamura, A.; Stuckert, P.; Zhu, K.; Hamilton, A.; Deloach, M.K.; Kutok, J.L.; Akashi, K.; et al. Hematopoietic stem cell defects in mice with deficiency of Fancd2 or Usp1. Stem Cells 2010, 28, 1186–1195. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; Parmar, K.; Huang, M.; Weinstock, D.M.; Ruit, C.A.; Kutok, J.L.; D’Andrea, A.D. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev. Cell 2009, 16, 314–320. [Google Scholar] [CrossRef] [Green Version]
- Veillette, A. SLAM-Family Receptors: Immune Regulators with or without SAP-Family Adaptors. Cold Spring Harb. Perspect. Biol. 2010, 2, a002469. [Google Scholar] [CrossRef]
- Pritchard, C.E.J.; Kroese, L.J.; Huijbers, I.J. Direct Generation of Conditional Alleles Using CRISPR/Cas9 in Mouse Zygotes. In Site-Specific Recombinases: Methods and Protocols; Methods in Molecular Biology; Eroshenko, N., Ed.; Springer: New York, NY, USA, 2017; pp. 21–35. ISBN 978-1-4939-7169-5. [Google Scholar]
- Wit, N.; Buoninfante, O.A.; van den Berk, P.C.M.; Jansen, J.G.; Hogenbirk, M.A.; de Wind, N.; Jacobs, H. Roles of PCNA ubiquitination and TLS polymerases κ and η in the bypass of methyl methanesulfonate-induced DNA damage. Nucleic Acids Res. 2015, 43, 282–294. [Google Scholar] [CrossRef] [Green Version]
- Dirac, A.M.G.; Bernards, R. Reversal of Senescence in Mouse Fibroblasts through Lentiviral Suppression of p53. J. Biol. Chem. 2003, 278, 11731–11734. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Spandidos, A.; Wang, H.; Seed, B. PrimerBank: A PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 2012, 40, D1144–D1149. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′-3′) |
|---|---|
| pmxFG_Internal primer A fwd | GAGGGATGTCCTTCTGACTGC |
| pmxFG_Internal primer A rev | GAGAACCTTGTCTCTGAGCCACCC |
| pmxFG_Internal primer B fwd | CTGCCGTGTTGCCCAGTCTGGGTC |
| pmxFG_Internal primer B rev | GGCTTCGCGGTTAGGGGGATGGAT |
| pmxFG_Internal primer C fwd | TCTTCCACTGTATTTAGAAACCTG |
| pmxFG_Internal primer C rev | TTATACACGTGGCTTTTGGCCGCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, R.; van den Berk, P.C.M.; Pritchard, C.E.J.; Song, J.-Y.; Kreft, M.; Pilzecker, B.; Jacobs, H. A C57BL/6J Fancg-KO Mouse Model Generated by CRISPR/Cas9 Partially Captures the Human Phenotype. Int. J. Mol. Sci. 2023, 24, 11129. https://doi.org/10.3390/ijms241311129
Shah R, van den Berk PCM, Pritchard CEJ, Song J-Y, Kreft M, Pilzecker B, Jacobs H. A C57BL/6J Fancg-KO Mouse Model Generated by CRISPR/Cas9 Partially Captures the Human Phenotype. International Journal of Molecular Sciences. 2023; 24(13):11129. https://doi.org/10.3390/ijms241311129
Chicago/Turabian StyleShah, Ronak, Paul C. M. van den Berk, Colin E. J. Pritchard, Ji-Ying Song, Maaike Kreft, Bas Pilzecker, and Heinz Jacobs. 2023. "A C57BL/6J Fancg-KO Mouse Model Generated by CRISPR/Cas9 Partially Captures the Human Phenotype" International Journal of Molecular Sciences 24, no. 13: 11129. https://doi.org/10.3390/ijms241311129