

The Role of Activating Transcription Factor 3 in Metformin’s Alleviation of Gastrointestinal Injury Induced by Restraint Stress in Mice

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
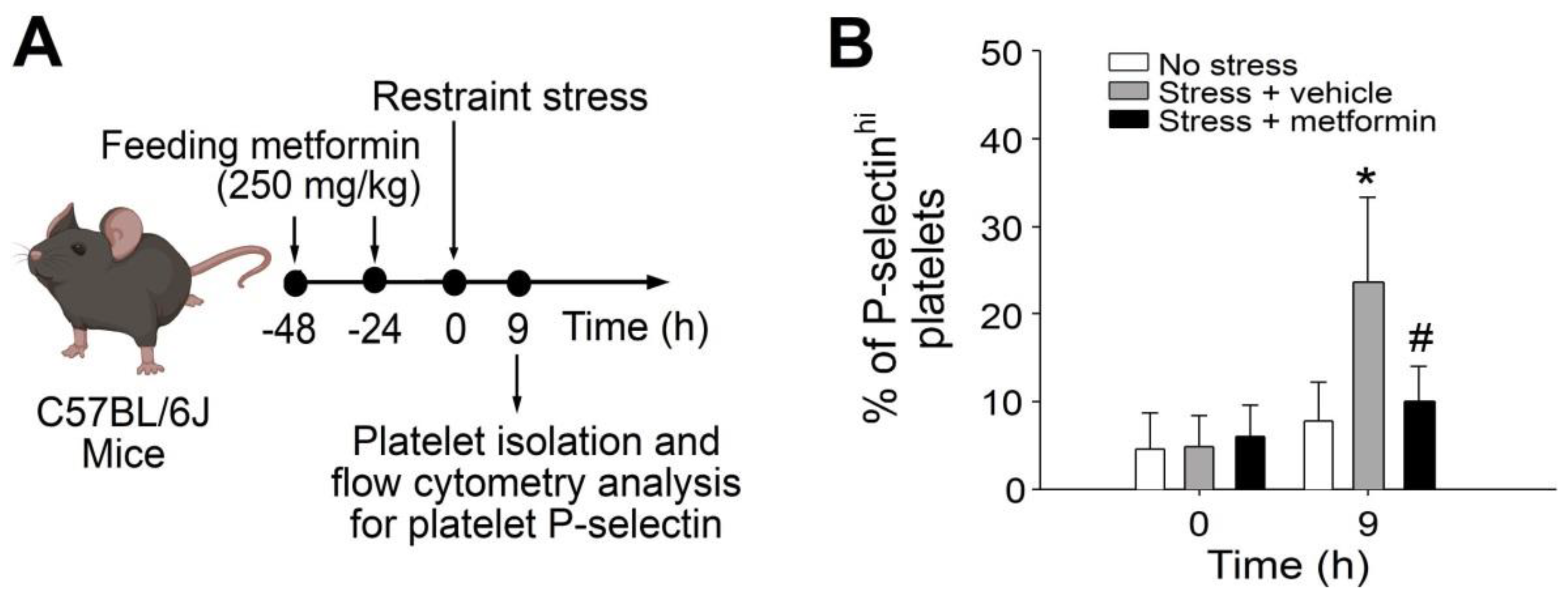
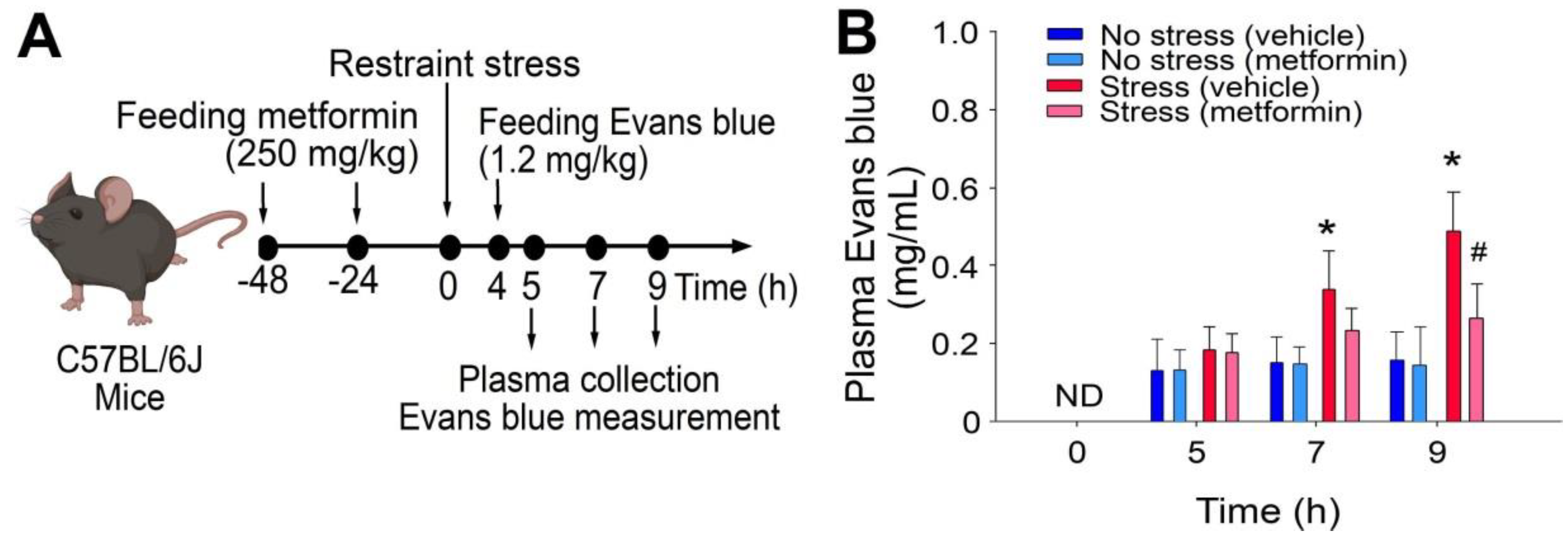
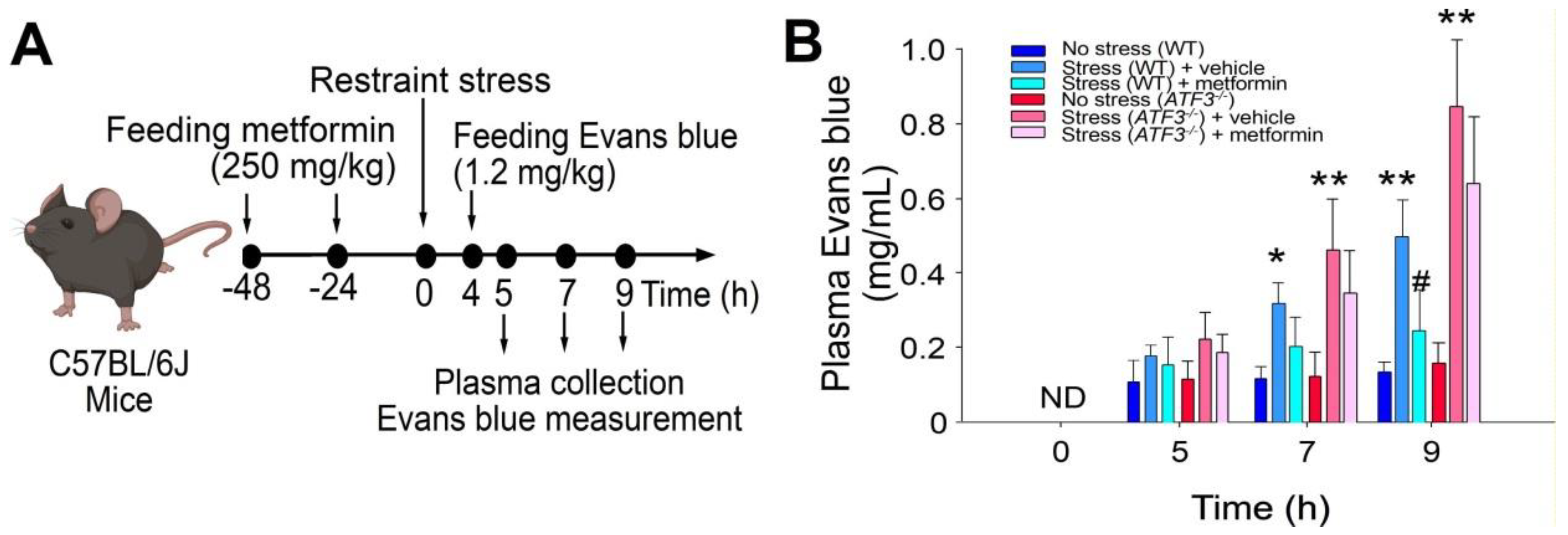
2.1. Treatments of Metformin-Rescued Restraint-Stress-Induced Platelet Activation and GI Injury
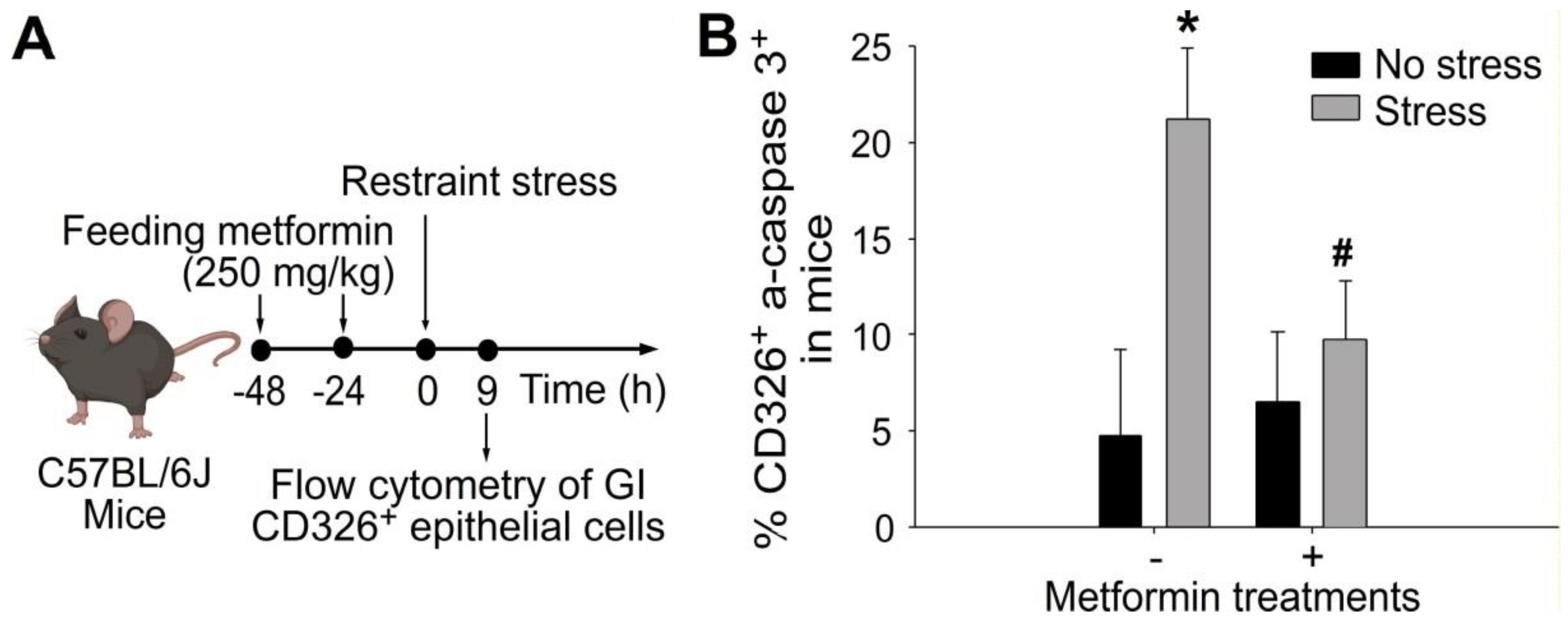
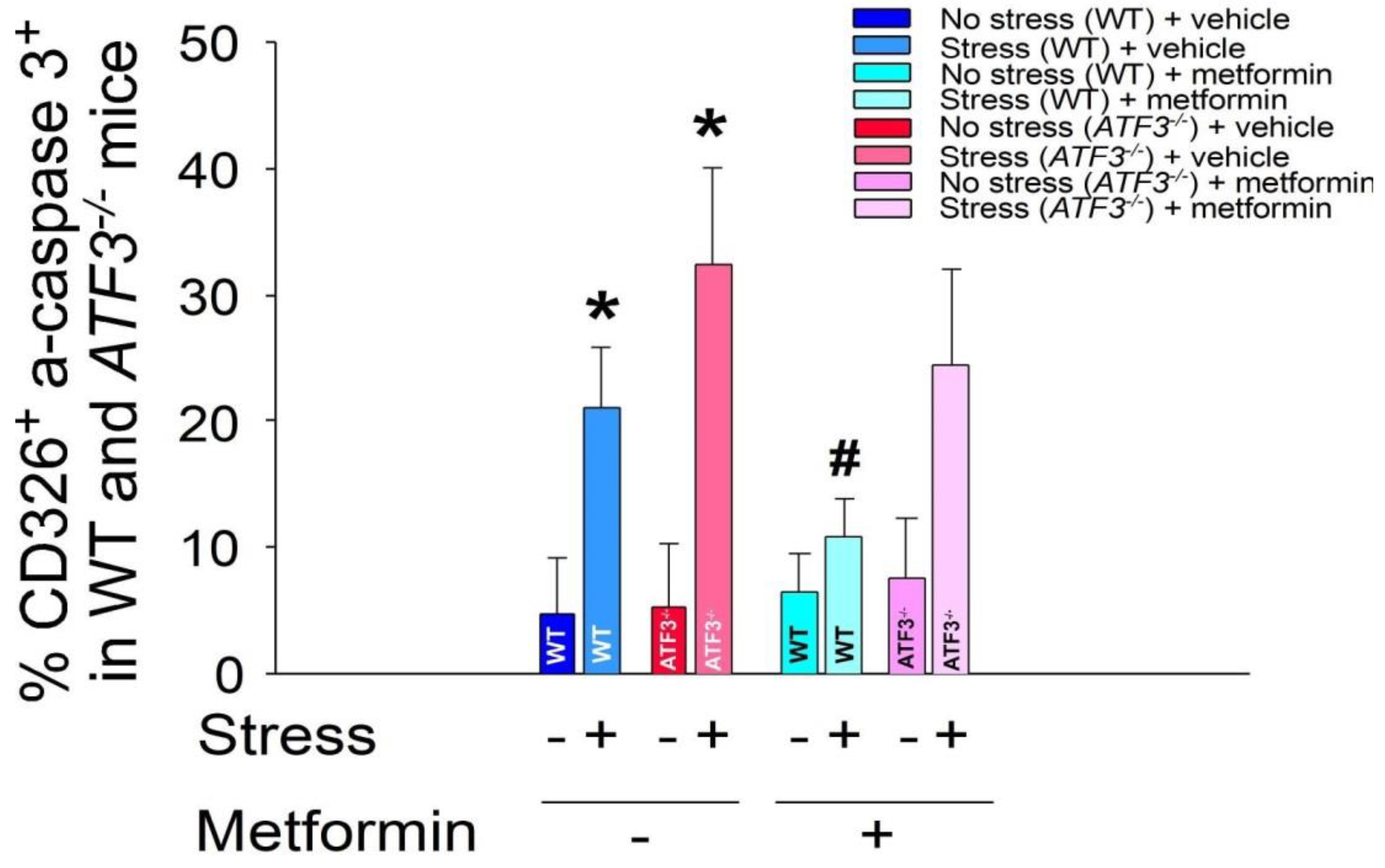
2.2. Treatments of Metformin-Rescued Restraint-Stress-Induced GI Epithelial Cell Apoptosis
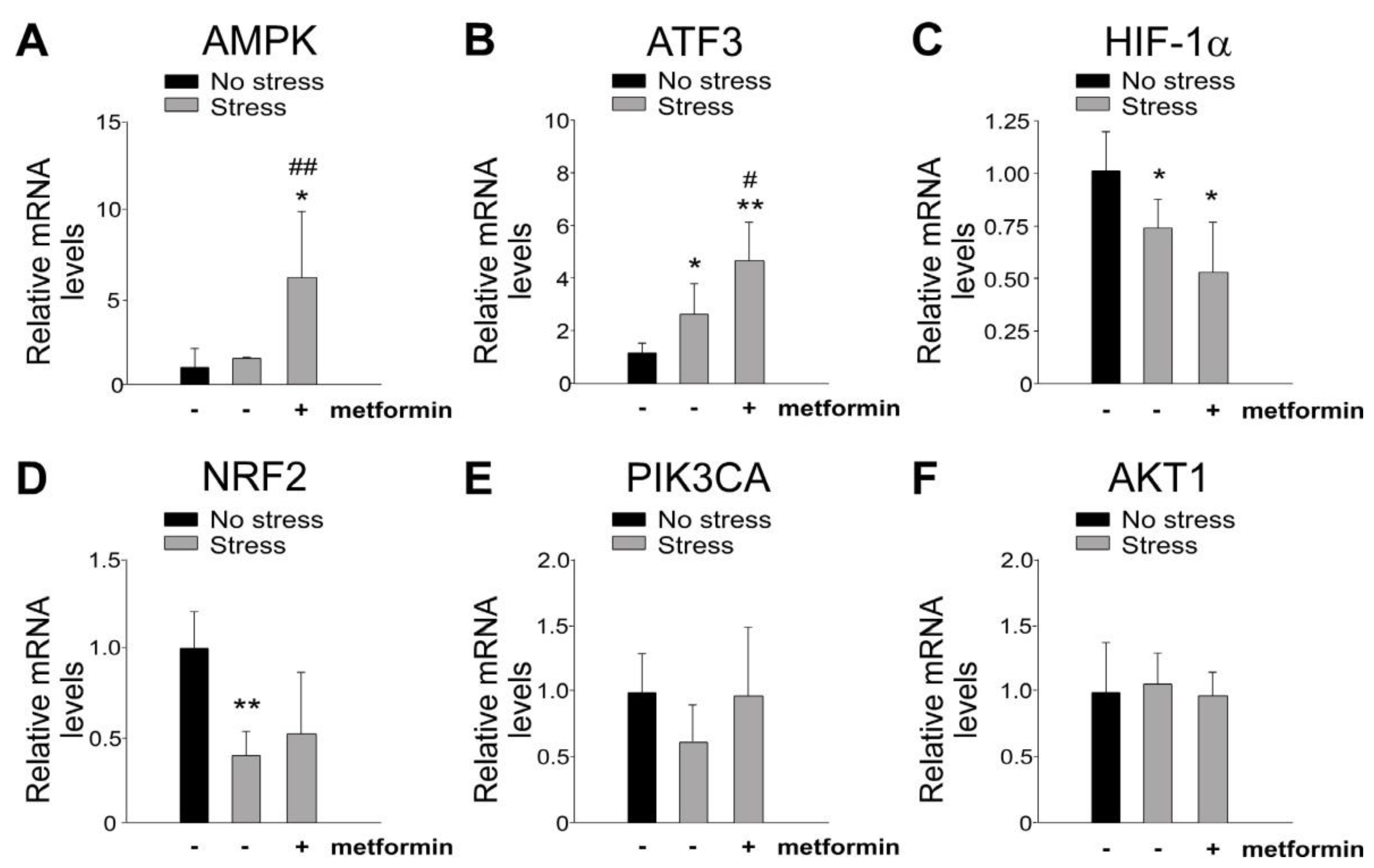
2.3. Metformin-Mediated Protection Is Associated with the Induction of GI AMPK and ATF3 mRNA Expression
2.4. ATF3 Deficiency Reduced Metformin-Mediated Rescue in Restraint-Stress-Induced GI Leakage and the Rescue of Stress-Induced GI Epithelial Cell Apoptosis
3. Discussion
4. Materials and Methods
4.1. Laboratory Mice
4.2. Induction, Reversal and Measurement of Stress-Induced GI Leakage
4.3. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.3.1. RNA and cDNA Preparation
4.3.2. qRT-PCR Analyses
4.4. Flow Cytometry Analysis
4.5. Confocal Microscopy for Immunohistochemistry Samples
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Y.-W.; He, S.-J.; Feng, X.; Cheng, J.; Luo, Y.-T.; Tian, L.; Huang, Q. Metformin: A review of its potential indications. Drug Des. Dev. Ther. 2017, 11, 2421–2429. [Google Scholar] [CrossRef] [Green Version]
- Foretz, M.; Guigas, B.; Viollet, B. Metformin: Update on mechanisms of action and repurposing potential. Nat. Rev. Endocrinol. 2023, 1–17. [Google Scholar] [CrossRef]
- Bai, B.; Chen, H. Metformin: A Novel Weapon Against Inflammation. Front. Pharmacol. 2021, 12, 622262. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, I.; Hollenberg, M.D.; Ding, H.; Triggle, C.R. A Critical Review of the Evidence That Metformin Is a Putative Anti-Aging Drug That Enhances Healthspan and Extends Lifespan. Front. Endocrinol. 2021, 12, 718942. [Google Scholar] [CrossRef] [PubMed]
- Sapolsky, R.M. Why Zebras Don’t Get Ulcers, 3rd ed.; W. H. Freeman: New York, NY, USA, 2004. [Google Scholar]
- Oligschlaeger, Y.; Yadati, T.; Houben, T.; Oliván, C.M.C.; Shiri-Sverdlov, R. Inflammatory Bowel Disease: A Stressed “Gut/Feeling”. Cells 2019, 8, 659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, M.; Gershon, M.D. The bowel and beyond: The enteric nervous system in neurological disorders. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Disease, G.B.D.; Injury, I.; Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [Green Version]
- Wasilewska, J.; Klukowski, M. Gastrointestinal symptoms and autism spectrum disorder: Links and risks–a possible new overlap syndrome. Pediatr. Health Med. Ther. 2015, 6, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Karling, P.; Maripuu, M.; Wikgren, M.; Adolfsson, R.; Norrback, K.-F. Association between gastrointestinal symptoms and affectivity in patients with bipolar disorder. World J. Gastroenterol. 2016, 22, 8540–8548. [Google Scholar] [CrossRef] [Green Version]
- Severance, E.G.; Prandovszky, E.; Castiglione, J.; Yolken, R.H. Gastroenterology Issues in Schizophrenia: Why the Gut Matters. Curr. Psychiatry Rep. 2015, 17, 27. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Wang, H.E.; Bai, Y.-M.; Tsai, S.-J.; Su, T.-P.; Chen, T.-J.; Wang, Y.-P.; Chen, M.-H. Inflammatory bowel disease is associated with higher dementia risk: A nationwide longitudinal study. Gut 2021, 70, 85–91. [Google Scholar] [CrossRef]
- Alkasir, R.; Li, J.; Li, X.; Jin, M.; Zhu, B. Human gut microbiota: The links with dementia development. Protein Cell 2017, 8, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Long-Smith, C.; O’Riordan, K.J.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Microbiota-Gut-Brain Axis: New Therapeutic Opportunities. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 477–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef]
- Campos, A.C.; Fogaca, M.V.; Aguiar, D.C.; Guimaraes, F.S. Animal models of anxiety disorders and stress. Braz. J. Psychiatry 2013, 35 (Suppl. 2), S101–S111. [Google Scholar] [CrossRef] [Green Version]
- Paré, W.P.; Glavin, G.B. Restraint stress in biomedical research: A review. Neurosci. Biobehav. Rev. 1986, 10, 339–370. [Google Scholar] [CrossRef] [PubMed]
- Glavin, G.B.; Paré, W.P.; Sandbak, T.; Bakke, H.-K.; Murison, R. Restraint stress in biomedical research: An update. Neurosci. Biobehav. Rev. 1994, 18, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.S.; Lien, T.S.; Chang, H.H. Restraint stress-associated gastrointestinal injury and implications from the Evans-blue fed restraint stress mouse model. Tzu Chi Med. J. 2023, 35. in press. [Google Scholar]
- Chuang, D.-J.; Pethaperumal, S.; Siwakoti, B.; Chien, H.-J.; Cheng, C.-F.; Hung, S.-C.; Lien, T.-S.; Sun, D.-S.; Chang, H.-H. Activating Transcription Factor 3 Protects against Restraint Stress-Induced Gastrointestinal Injury in Mice. Cells 2021, 10, 3530. [Google Scholar] [CrossRef]
- Pethaperumal, S.; Hung, S.-C.; Lien, T.-S.; Sun, D.-S.; Chang, H.-H. P-Selectin is a Critical Factor for Platelet-Mediated Protection on Restraint Stress-Induced Gastrointestinal Injury in Mice. Int. J. Mol. Sci. 2022, 23, 11909. [Google Scholar] [CrossRef]
- Eisinger, F.; Patzelt, J.; Langer, H.F. The Platelet Response to Tissue Injury. Front. Med. 2018, 5, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.-S.; Chang, H.-H. Platelets in Inflammation and Immune Modulations: Functions Beyond Hemostasis. Arch. Immunol. Et Ther. Exp. 2012, 60, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zeng, H.; Lei, L.; Tong, X.; Yang, L.; Yang, Y.; Li, S.; Zhou, Y.; Luo, L.; Huang, J.; et al. Tight junctions and their regulation by non-coding RNAs. Int. J. Biol. Sci. 2021, 17, 712–727. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef]
- Deng, J.; Zeng, L.; Lai, X.; Li, J.; Liu, L.; Lin, Q.; Chen, Y. Metformin protects against intestinal barrier dysfunction via AMPKα1-dependent inhibition of JNK signalling activation. J. Cell. Mol. Med. 2018, 22, 546–557. [Google Scholar] [CrossRef] [Green Version]
- Induri, S.N.R.; Kansara, P.; Thomas, S.C.; Xu, F.; Saxena, D.; Li, X. The Gut Microbiome, Metformin, and Aging. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 85–108. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.; Razazan, A.; Nagpal, R.; Jain, S.; Wang, B.; Mishra, S.P.; Wang, S.; Justice, J.; Ding, J.; McClain, D.A.; et al. Metformin Reduces Aging-Related Leaky Gut and Improves Cognitive Function by Beneficially Modulating Gut Microbiome/Goblet Cell/Mucin Axis. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, e9–e21. [Google Scholar] [CrossRef]
- Ke, H.; Li, F.; Deng, W.; Li, Z.; Wang, S.; Lv, P.; Chen, Y. Metformin Exerts Anti-inflammatory and Mucus Barrier Protective Effects by Enriching Akkermansia muciniphila in Mice With Ulcerative Colitis. Front. Pharmacol. 2021, 12, 726707. [Google Scholar] [CrossRef]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, A.; Hernández-Arriaga, A.; Kehm, R.; Sánchez, V.; Jin, C.J.; Nier, A.; Baumann, A.; Camarinha-Silva, A.; Bergheim, I. Metformin attenuates the onset of non-alcoholic fatty liver disease and affects intestinal microbiota and barrier in small intestine. Sci. Rep. 2019, 9, 6668. [Google Scholar] [CrossRef] [Green Version]
- Ku, H.-C.; Cheng, C.-F. Master Regulator Activating Transcription Factor 3 (ATF3) in Metabolic Homeostasis and Cancer. Front. Endocrinol. 2020, 11, 556. [Google Scholar] [CrossRef]
- Sun, D.-S.; Chang, H.-H. Emerging role of the itaconate-mediated rescue of cellular metabolic stress. Tzu Chi Med. J. 2021, 33, 134–138. [Google Scholar] [CrossRef]
- Thompson, M.R.; Xu, D.; Williams, B.R.G. ATF3 transcription factor and its emerging roles in immunity and cancer. J. Mol. Med. 2009, 87, 1053–1060. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Recarte, D.; Barroso, E.; Zhang, M.; Rada, P.; Pizarro-Delgado, J.; Peña, L.; Palomer, X.; Valverde, A.M.; Wahli, W.; Vázquez-Carrera, M. A positive feedback loop between AMPK and GDF15 promotes metformin antidiabetic effects. Pharmacol. Res. 2023, 187, 106578. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Meng, D.; Zou, F.-F.; Fan, L.; Zhang, P.; Yu, Y.; Fang, J. Activating transcription factor 3 regulates survivability and migration of vascular smooth muscle cells. IUBMB Life 2011, 63, 62–69. [Google Scholar] [CrossRef]
- Nakagomi, S.; Suzuki, Y.; Namikawa, K.; Kiryu-Seo, S.; Kiyama, H. Expression of the Activating Transcription Factor 3 Prevents c-Jun N-Terminal Kinase-Induced Neuronal Death by Promoting Heat Shock Protein 27 Expression and Akt Activation. J. Neurosci. 2003, 23, 5187–5196. [Google Scholar] [CrossRef] [Green Version]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Mandal, J.P.; Shiue, C.-N.; Chen, Y.-C.; Lee, M.-C.; Yang, H.-H.; Chang, H.-H.; Hu, C.-T.; Liao, P.-C.; Hui, L.-C.; You, R.-I.; et al. PKCdelta mediates mitochondrial ROS generation and oxidation of HSP60 to relieve RKIP inhibition on MAPK pathway for HCC progression. Free. Radic. Biol. Med. 2020, 163, 69–87. [Google Scholar] [CrossRef]
- Demine, S.; Schiavo, A.A.; Marín-Cañas, S.; Marchetti, P.; Cnop, M.; Eizirik, D.L. Pro-inflammatory cytokines induce cell death, inflammatory responses, and endoplasmic reticulum stress in human iPSC-derived beta cells. Stem Cell Res. Ther. 2020, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murthy, V.; Oshi, M.; Tokumaru, Y.; Endo, I.; Takabe, K. Increased apoptosis is associated with robust immune cell infiltration and cytolytic activity in breast cancer. Am. J. Cancer Res. 2021, 11, 3674–3687. [Google Scholar] [PubMed]
- Anderton, H.; Wicks, I.P.; Silke, J. Cell death in chronic inflammation: Breaking the cycle to treat rheumatic disease. Nat. Rev. Rheumatol. 2020, 16, 496–513. [Google Scholar] [CrossRef]
- Lien, T.-S.; Sun, D.-S.; Wu, C.-Y.; Chang, H.-H. Exposure to Dengue Envelope Protein Domain III Induces Nlrp3 Inflammasome-Dependent Endothelial Dysfunction and Hemorrhage in Mice. Front. Immunol. 2021, 12, 617251. [Google Scholar] [CrossRef] [PubMed]
- Lien, T.-S.; Sun, D.-S.; Hung, S.-C.; Wu, W.-S.; Chang, H.-H. Dengue Virus Envelope Protein Domain III Induces Nlrp3 Inflammasome-Dependent NETosis-Mediated Inflammation in Mice. Front. Immunol. 2021, 12, 618577. [Google Scholar] [CrossRef]
- Lien, T.-S.; Chan, H.; Sun, D.-S.; Wu, J.-C.; Lin, Y.-Y.; Lin, G.-L.; Chang, H.-H. Exposure of Platelets to Dengue Virus and Envelope Protein Domain III Induces Nlrp3 Inflammasome-Dependent Platelet Cell Death and Thrombocytopenia in Mice. Front. Immunol. 2021, 12, 616394. [Google Scholar] [CrossRef]
- Hung, S.-C.; Ke, L.-C.; Lien, T.-S.; Huang, H.-S.; Sun, D.-S.; Cheng, C.-L.; Chang, H.-H. Nanodiamond-Induced Thrombocytopenia in Mice Involve P-Selectin-Dependent Nlrp3 Inflammasome-Mediated Platelet Aggregation, Pyroptosis and Apoptosis. Front. Immunol. 2022, 13, 806686. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Zhang, L.; Fu, L.; Liu, B. A small-molecule activator induces ULK1-modulating autophagy-associated cell death in triple negative breast cancer. Autophagy 2017, 13, 777–778. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Fu, L.; Zhang, S.; Zhang, J.; Zhao, Y.; Zheng, Y.; He, G.; Yang, S.; Ouyang, L.; Liu, B. Discovery of a small molecule targeting ULK1-modulated cell death of triple negative breast cancer in vitro and in vivo. Chem. Sci. 2017, 8, 2687–2701. [Google Scholar] [CrossRef] [Green Version]
- Das, C.K.; Banerjee, I.; Mandal, M. Pro-survival autophagy: An emerging candidate of tumor progression through maintaining hallmarks of cancer. Semin. Cancer Biol. 2020, 66, 59–74. [Google Scholar] [CrossRef]
- Das, C.K.; Mandal, M.; Kögel, D. Pro-survival autophagy and cancer cell resistance to therapy. Cancer Metastasis Rev. 2018, 37, 749–766. [Google Scholar] [CrossRef]
- Deretic, V. Autophagy in inflammation, infection, and immunometabolism. Immunity 2021, 54, 437–453. [Google Scholar] [CrossRef] [PubMed]
- Noor, H.B.; Mou, N.A.; Salem, L.; Shimul, M.F.A.; Biswas, S.; Akther, R.; Khan, S.; Raihan, S.; Mohib, M.M.; Sagor, M.A.T. Anti-inflammatory Property of AMP-activated Protein Kinase. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2020, 19, 2–41. [Google Scholar] [CrossRef]
- Keerthana, C.K.; Rayginia, T.P.; Shifana, S.C.; Anto, N.P.; Kalimuthu, K.; Isakov, N.; Anto, R.J. The role of AMPK in cancer metabolism and its impact on the immunomodulation of the tumor microenvironment. Front. Immunol. 2023, 14, 1114582. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.-A.; Cheon, H.G. Activating transcription factor-3 induction is involved in the anti-inflammatory action of berberine in RAW264.7 murine macrophages. Korean J. Physiol. Pharmacol. 2016, 20, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kwak, H.J.; Cha, J.-Y.; Jeong, Y.-S.; Rhee, S.D.; Kim, K.R.; Cheon, H.G. Metformin Suppresses Lipopolysaccharide (LPS)-induced Inflammatory Response in Murine Macrophages via Activating Transcription Factor-3 (ATF-3) Induction. J. Biol. Chem. 2014, 289, 23246–23255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, D.M.; Herway, S.T.; Fillmore, N.; Kim, H.; Brown, J.D.; Barrow, J.R.; Winder, W.W. AMP-activated protein kinase phosphorylates transcription factors of the CREB family. J. Appl. Physiol. 2008, 104, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.-J.; Buchthal, B.; Lau, D.; Hayer, S.; Dick, O.; Schwaninger, M.; Veltkamp, R.; Zou, M.; Weiss, U.; Bading, H. A Signaling Cascade of Nuclear Calcium-CREB-ATF3 Activated by Synaptic NMDA Receptors Defines a Gene Repression Module That Protects against Extrasynaptic NMDA Receptor-Induced Neuronal Cell Death and Ischemic Brain Damage. J. Neurosci. 2011, 31, 4978–4990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, A.; Hall, M.N.; Lin, S.-C.; Hardie, D.G. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef]
- Zhu, Q.; Wang, H.; Jiang, B.; Ni, X.; Jiang, L.; Li, C.; Wang, X.; Zhang, F.; Ke, B.; Lu, L. Loss of ATF3 exacerbates liver damage through the activation of mTOR/p70S6K/ HIF-1α signaling pathway in liver inflammatory injury. Cell Death Dis. 2018, 9, 910. [Google Scholar] [CrossRef] [Green Version]
- Pernice, H.F.; Schieweck, R.; Kiebler, M.A.; Popper, B. mTOR and MAPK: From localized translation control to epilepsy. BMC Neurosci. 2016, 17, 73. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Chen, J.; Hai, T. The regulation of ATF3 gene expression by mitogen-activated protein kinases. Biochem. J. 2007, 401, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.-Y.; Sun, D.-S.; Chang, H.-H. Silver Nanoparticles Protect Skin from Ultraviolet B-Induced Damage in Mice. Int. J. Mol. Sci. 2020, 21, 7082. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-L.; Sun, D.-S.; Su, M.-T.; Lien, T.-S.; Chen, Y.-H.; Lin, C.-Y.; Huang, C.-H.; King, C.-C.; Li, C.-R.; Chen, T.-H.; et al. Suppressed humoral immunity is associated with dengue nonstructural protein NS1-elicited anti-death receptor antibody fractions in mice. Sci. Rep. 2020, 10, 6294. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-Y.; Hu, C.-T.; Sun, D.-S.; Lien, T.-S.; Chang, H.-H. Thioacetamide-induced liver damage and thrombocytopenia is associated with induction of antiplatelet autoantibody in mice. Sci. Rep. 2019, 9, 17497. [Google Scholar] [CrossRef] [Green Version]
- Perevedentseva, E.; Krivokharchenko, A.; Karmenyan, A.V.; Chang, H.-H.; Cheng, C.-L. Raman spectroscopy on live mouse early embryo while it continues to develop into blastocyst in vitro. Sci. Rep. 2019, 9, 6636. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.; Huang, H.-S.; Sun, D.-S.; Lee, C.-J.; Lien, T.-S.; Chang, H.-H. TRPM8 and RAAS-mediated hypertension is critical for cold-induced immunosuppression in mice. Oncotarget 2018, 9, 12781–12795. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-Y.; Yu, W.-S.; Liu, G.-C.; Hung, S.-C.; Chang, J.-H.; Chang, J.C.; Cheng, C.-L.; Sun, D.-S.; Lin, M.-D.; Lin, W.-Y.; et al. Opportunistic gill infection is associated with TiO2 nanoparticle-induced mortality in zebrafish. PLoS ONE 2021, 16, e0247859. [Google Scholar] [CrossRef]
- Lien, T.-S.; Sun, D.-S.; Wu, W.-S.; Chang, H.-H. Simulation of Hemorrhage Pathogenesis in Mice through Dual Stimulation with Dengue Envelope Protein Domain III-Coated Nanoparticles and Antiplatelet Antibody. Int. J. Mol. Sci. 2023, 24, 9270. [Google Scholar] [CrossRef]
- Hartman, M.G.; Lu, D.; Kim, M.-L.; Kociba, G.J.; Shukri, T.; Buteau, J.; Wang, X.; Frankel, W.L.; Guttridge, D.; Prentki, M.; et al. Role for Activating Transcription Factor 3 in Stress-Induced β-Cell Apoptosis. Mol. Cell. Biol. 2004, 24, 5721–5732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.-F.; Ku, H.-C.; Cheng, J.-J.; Chao, S.-W.; Li, H.-F.; Lai, P.-F.; Chang, C.-C.; Don, M.-J.; Chen, H.-H.; Lin, H. Adipocyte browning and resistance to obesity in mice is induced by expression of ATF3. Commun. Biol. 2019, 2, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimprich, A.; Garrett, L.; Deussing, J.M.; Wotjak, C.T.; Fuchs, H.; Gailus-Durner, V.; de Angelis, M.H.; Wurst, W.; Holter, S.M. A robust and reliable non-invasive test for stress responsivity in mice. Front. Behav. Neurosci. 2014, 8, 125. [Google Scholar] [CrossRef] [Green Version]
- Chu, X.; Zhou, Y.; Hu, Z.; Lou, J.; Song, W.; Li, J.; Liang, X.; Chen, C.; Wang, S.; Yang, B.; et al. 24-hour-restraint stress induces long-term depressive-like phenotypes in mice. Sci. Rep. 2016, 6, 32935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Groschwitz, K.R.; Hogan, S.P. Intestinal barrier function: Molecular regulation and disease pathogenesis. J. Allergy Clin. Immunol. 2009, 124, 3–20. [Google Scholar] [CrossRef] [Green Version]
- Rath, E.; Moschetta, A.; Haller, D. Mitochondrial function—Gatekeeper of intestinal epithelial cell homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 497–516. [Google Scholar] [CrossRef]
- Sheng, H.; Shao, J.; Townsend, C.M., Jr.; Evers, B.M. Phosphatidylinositol 3-kinase mediates proliferative signals in intestinal epithelial cells. Gut 2003, 52, 1472–1478. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.-S.; Ho, P.-H.; Chang, H.-H. Soluble P-selectin rescues viper venom–induced mortality through anti-inflammatory properties and PSGL-1 pathway-mediated correction of hemostasis. Sci. Rep. 2016, 6, 35868. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.-S.; Chang, Y.-W.; Kau, J.-H.; Huang, H.-H.; Ho, P.-H.; Tzeng, Y.-J.; Chang, H.-H. Soluble P-selectin rescues mice from anthrax lethal toxin-induced mortality through PSGL-1 pathway-mediated correction of hemostasis. Virulence 2017, 8, 1216–1228. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.-S.; Sun, D.-S.; Lien, T.-S.; Chang, H.-H. Dendritic cells modulate platelet activity in IVIg-mediated amelioration of ITP in mice. Blood 2010, 116, 5002–5009. [Google Scholar] [CrossRef] [Green Version]
- Ouchi, T.; Morimura, S.; Dow, L.E.; Miyoshi, H.; Udey, M.C. EpCAM (CD326) Regulates Intestinal Epithelial Integrity and Stem Cells via Rho-Associated Kinase. Cells 2021, 10, 256. [Google Scholar] [CrossRef]
- Balfe, A.; Lennon, G.; Lavelle, A.; Docherty, N.G.; Coffey, J.C.; Sheahan, K.; Winter, D.C.; O’Connell, P.R. Isolation and gene expression profiling of intestinal epithelial cells: Crypt isolation by calcium chelation from in vivo samples. Clin. Exp. Gastroenterol. 2018, 11, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bialkowska, A.B.; Ghaleb, A.M.; Nandan, M.O.; Yang, V.W. Improved Swiss-rolling Technique for Intestinal Tissue Preparation for Immunohistochemical and Immunofluorescent Analyses. J. Vis. Exp. JoVE 2016, 113, e54161. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siwakoti, B.; Lien, T.-S.; Lin, Y.-Y.; Pethaperumal, S.; Hung, S.-C.; Sun, D.-S.; Cheng, C.-F.; Chang, H.-H. The Role of Activating Transcription Factor 3 in Metformin’s Alleviation of Gastrointestinal Injury Induced by Restraint Stress in Mice. Int. J. Mol. Sci. 2023, 24, 10995. https://doi.org/10.3390/ijms241310995
Siwakoti B, Lien T-S, Lin Y-Y, Pethaperumal S, Hung S-C, Sun D-S, Cheng C-F, Chang H-H. The Role of Activating Transcription Factor 3 in Metformin’s Alleviation of Gastrointestinal Injury Induced by Restraint Stress in Mice. International Journal of Molecular Sciences. 2023; 24(13):10995. https://doi.org/10.3390/ijms241310995
Chicago/Turabian StyleSiwakoti, Bijaya, Te-Sheng Lien, You-Yen Lin, Subhashree Pethaperumal, Shih-Che Hung, Der-Shan Sun, Ching-Feng Cheng, and Hsin-Hou Chang. 2023. "The Role of Activating Transcription Factor 3 in Metformin’s Alleviation of Gastrointestinal Injury Induced by Restraint Stress in Mice" International Journal of Molecular Sciences 24, no. 13: 10995. https://doi.org/10.3390/ijms241310995