
Methylammonium Tetrel Halide Perovskite Ion Pairs and Their Dimers: The Interplay between the Hydrogen-, Pnictogen- and Tetrel-Bonding Interactions



Abstract
:1. Introduction
2. Results
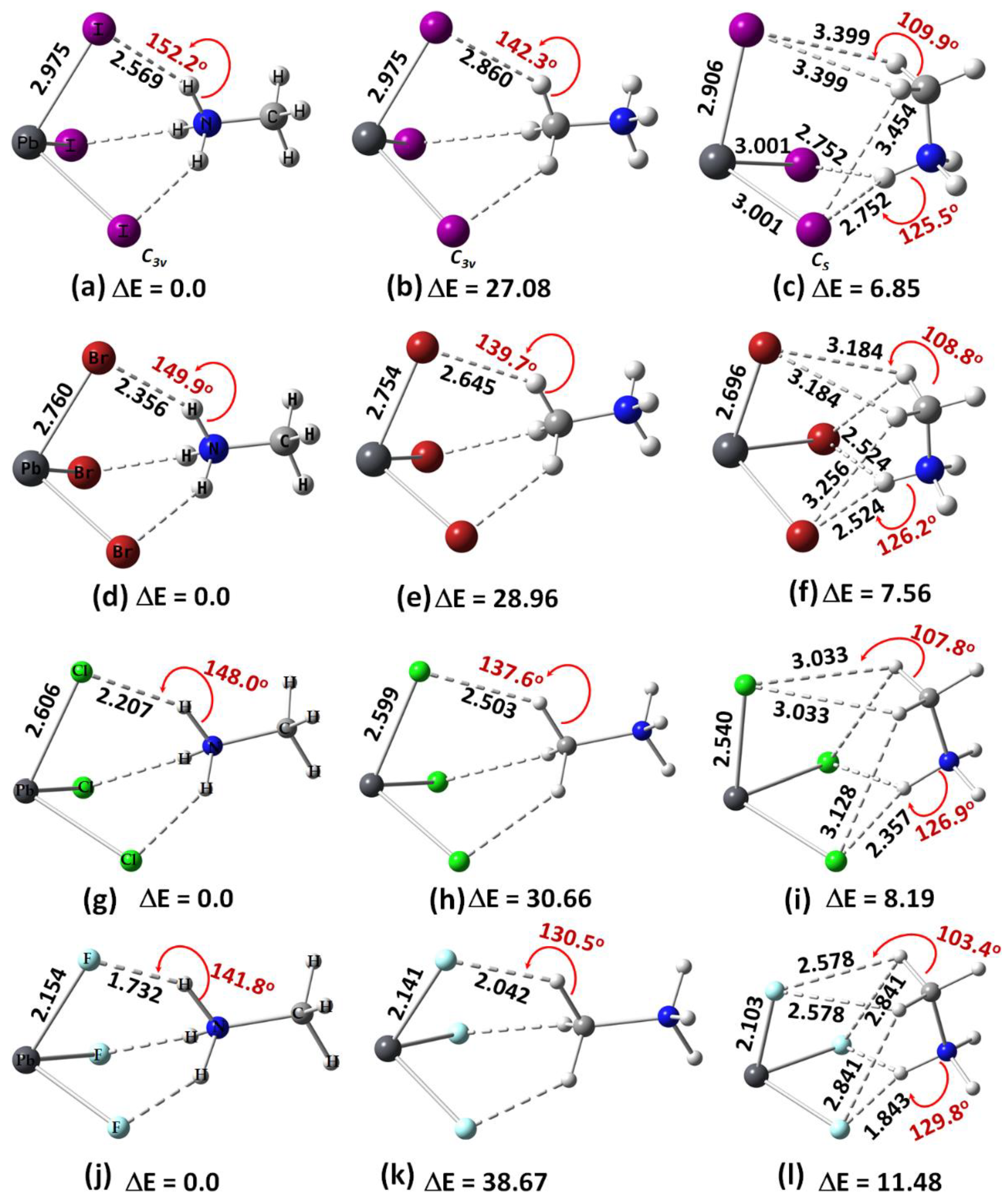
2.1. Methylammonium Tetrel Halide Perovskite Ion Pairs
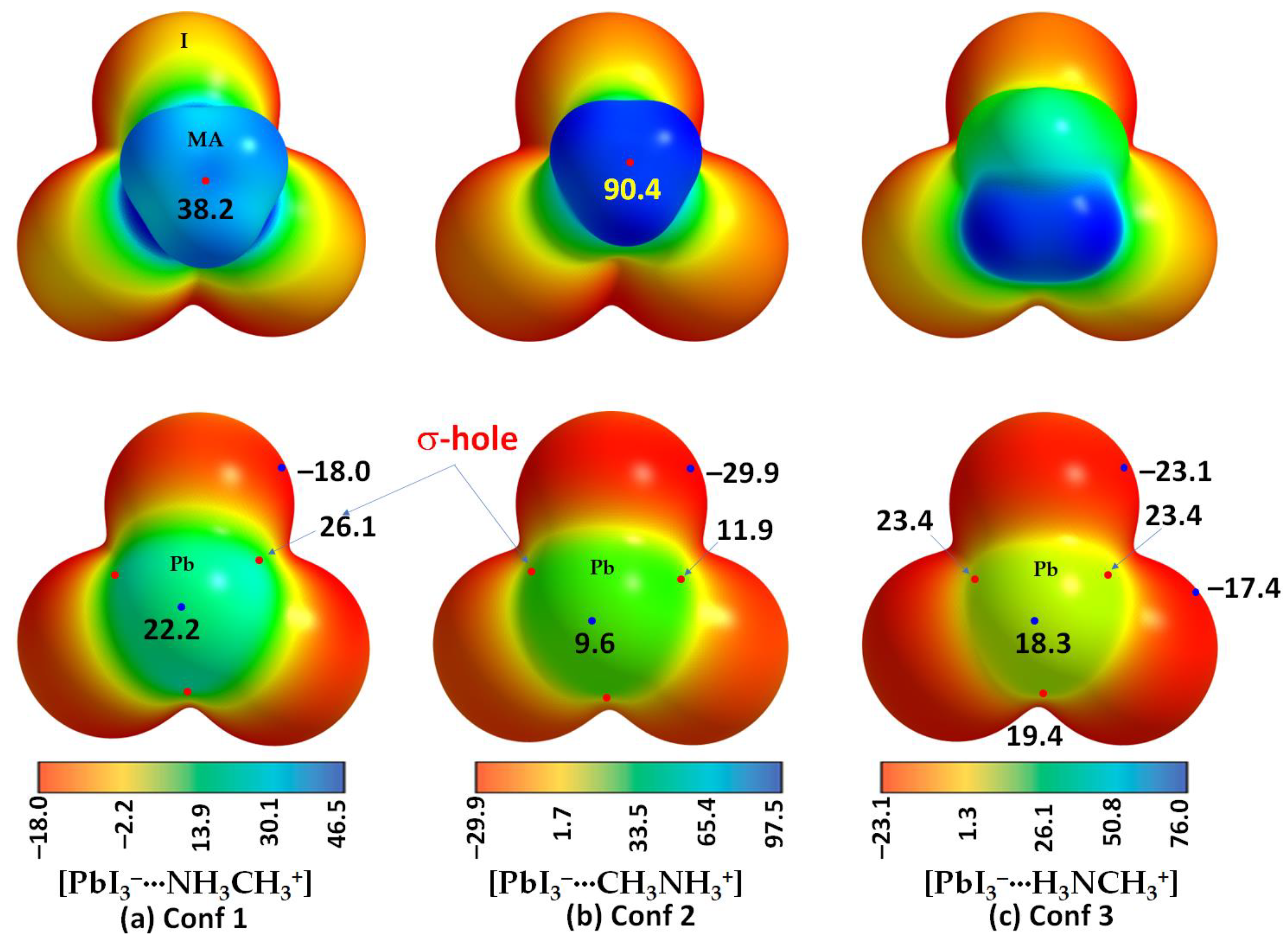
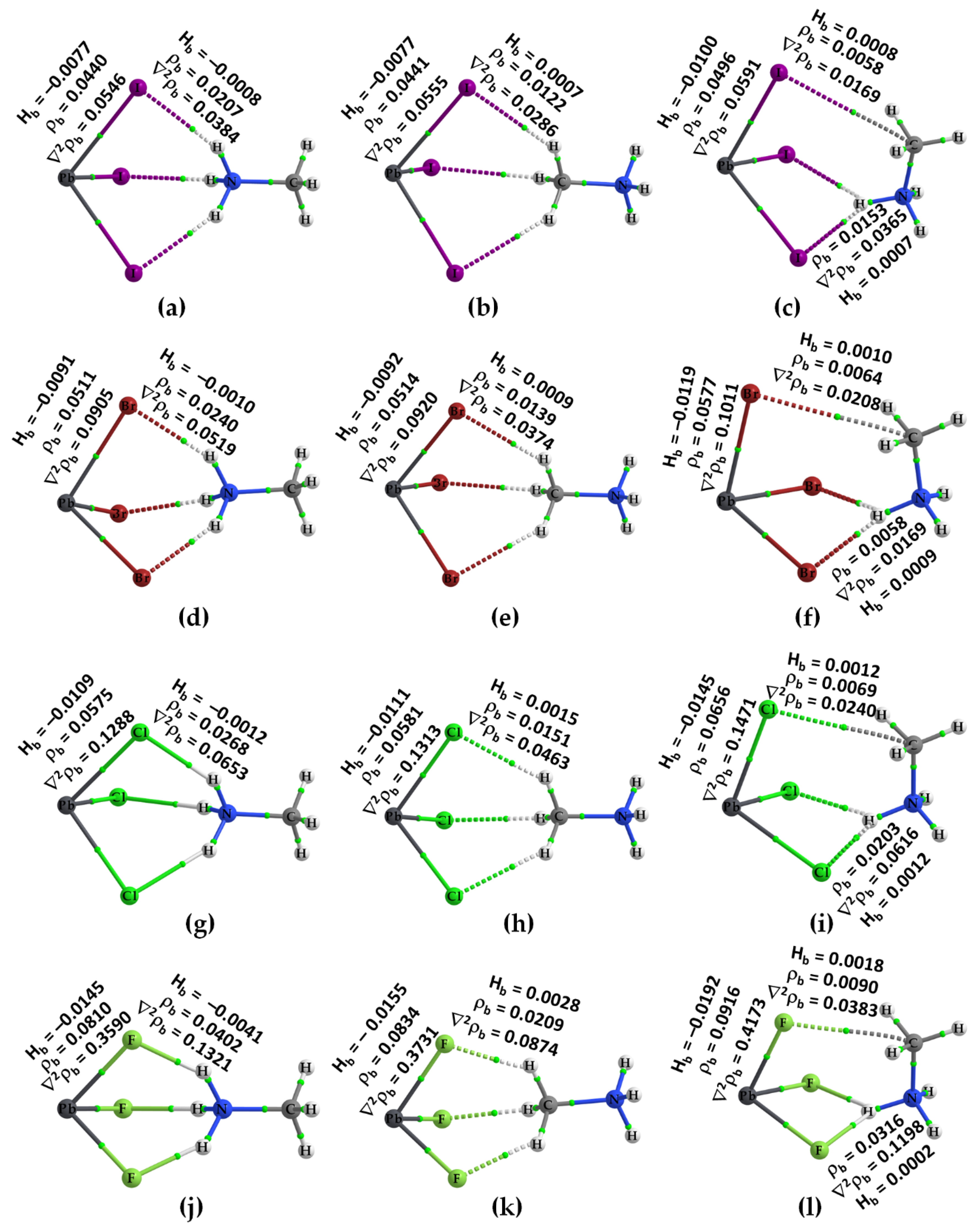
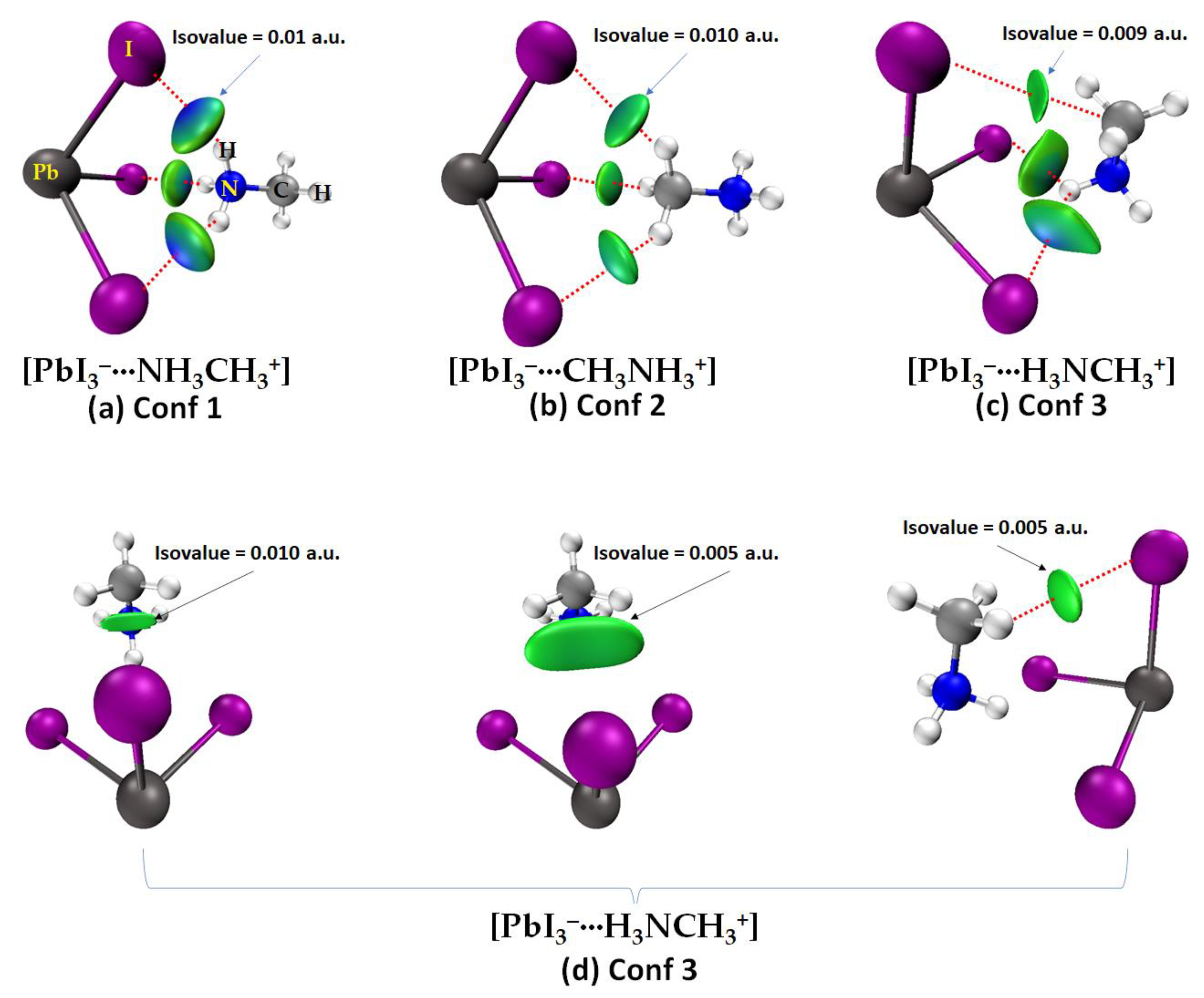
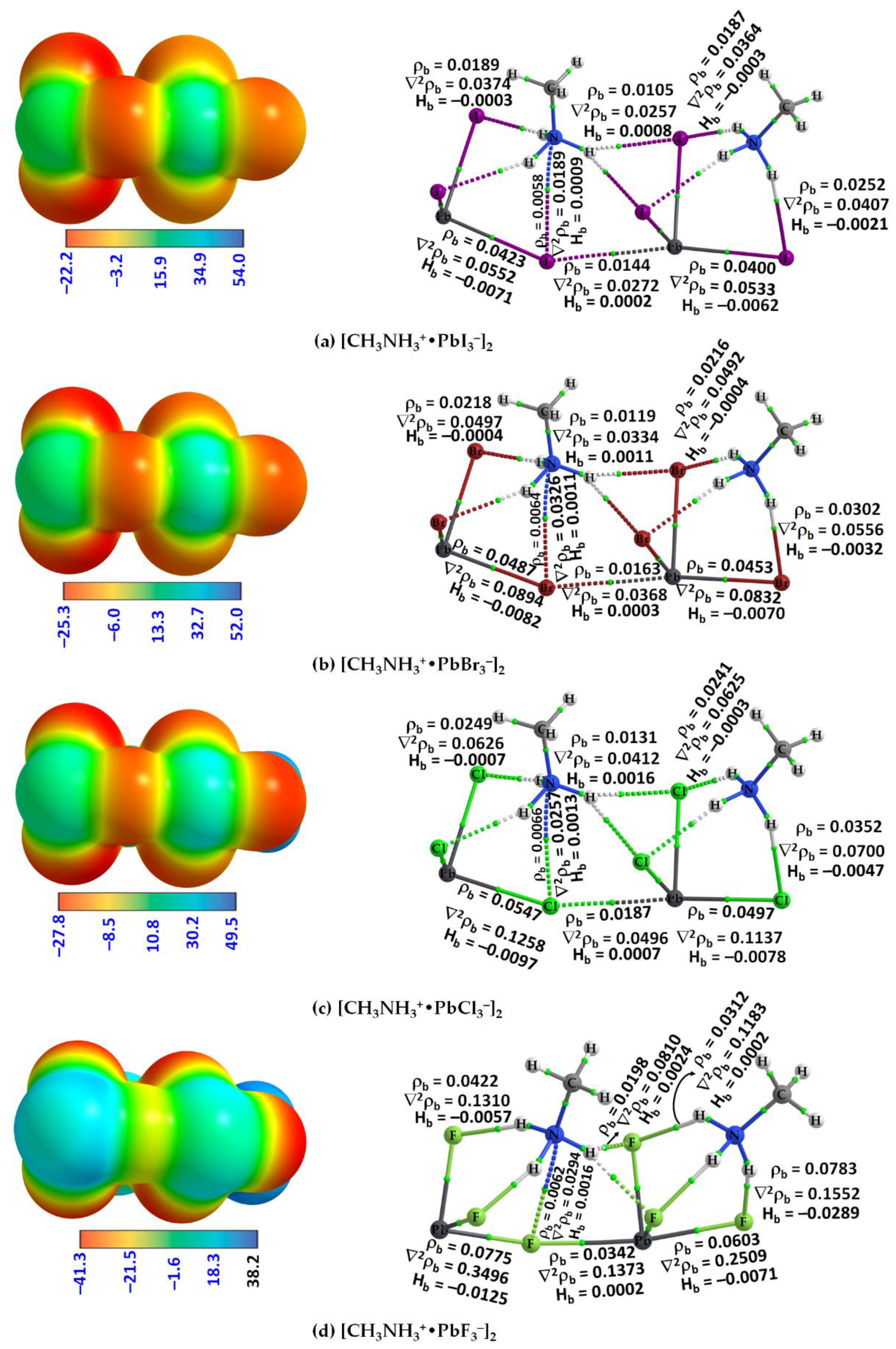
2.2. QTAIM, IGM-δginter and NBO Analysis of Ion Pairs
2.3. Energy Stability of the Ion Pairs
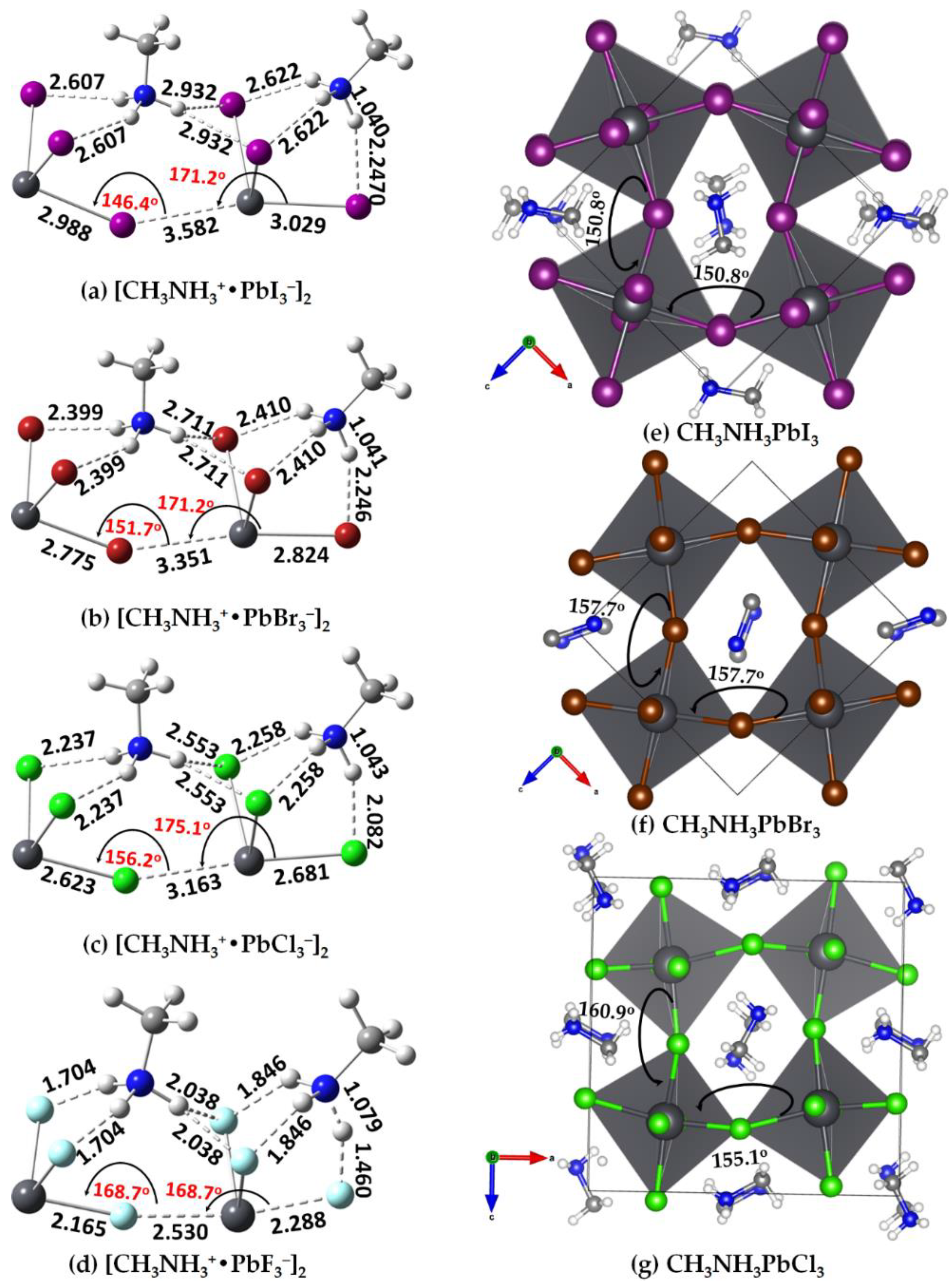
2.4. [CH3NH3+•TtX3−]2 (Tt = Si, Ge, Sn, Pb; X = F, Cl, Br, I) Dimers
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, W.; Eperon, G.E.; Snaith, H.J. Metal halide perovskites for energy applications. Nat. Energy 2016, 1, 16048. [Google Scholar] [CrossRef]
- Zhou, Y.; Saliba, M. Zooming In on Metal Halide Perovskites: New Energy Frontiers Emerge. ACS Energy Lett. 2021, 6, 2750–2754. [Google Scholar] [CrossRef]
- Alaei, A.; Circelli, A.; Yuan, Y.; Yang, Y.; Lee, S.S. Polymorphism in metal halide perovskites. Mater. Adv. 2021, 2, 47–63. [Google Scholar] [CrossRef]
- Chen, Q.; De Marco, N.; Yang, Y.; Song, T.-B.; Chen, C.-C.; Zhao, H.; Hong, Z.; Zhou, H.; Yang, Y. Under the spotlight: The organic–inorganic hybrid halide perovskite for optoelectronic applications. Nanotoday 2015, 10, 355–396. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhu, K. Organic–inorganic hybrid lead halide perovskites for optoelectronic and electronic applications. Chem. Soc. Rev. 2016, 45, 655–689. [Google Scholar] [CrossRef] [PubMed]
- Selivanov, N.I.; Samsonova, A.Y.; Kevorkyants, R.; Krauklis, I.V.; Chizhov, Y.V.; Stroganov, B.V.; Triantafyllou-Rundell, M.E.; Bahnemann, D.W.; Stoumpos, C.C.; Emeline, A.V.; et al. Hybrid Organic–Inorganic Halide Post-Perovskite 3-Cyanopyridinium Lead Tribromide for Optoelectronic Applications. Adv. Funct. Mater. 2021, 31, 2102338. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, Y.-J.; Hou, C.; Liang, T.-X. Ground-State Surface of All-Inorganic Halide Perovskites. J. Phys. Chem. C 2022, 126, 21155–21161. [Google Scholar] [CrossRef]
- Xie, H.; Hao, S.; Bao, J.; Slade, T.J.; Snyder, G.J.; Wolverton, C.; Kanatzidis, M.G. All-Inorganic Halide Perovskites as Potential Thermoelectric Materials: Dynamic Cation off-Centering Induces Ultralow Thermal Conductivity. J. Am. Chem. Soc. 2020, 142, 9553–9563. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. The Tetrel Bond and Tetrel Halide Perovskite Semiconductors. Int. J. Mol. Sci. 2023, 24, 6659. [Google Scholar] [CrossRef]
- Kojima, K.; Teshima, Y.; Shirai, Y.; Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 2009, 131, 6050–6051. [Google Scholar] [CrossRef]
- De Wolf, S.; Holovsky, J.; Moon, S.-J.; Löper, P.; Niese, B.; Ledinsky, M.; Haug, F.-J.; Yum, J.-H.; Ballif, C. Organometallic Halide Perovskites: Sharp Optical Absorption Edge and Its Relation to Photovoltaic Performance. J. Phys. Chem. Lett. 2014, 5, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.V. Organometal halide perovskites for transformative photovoltaics. J. Am. Chem. Soc. 2014, 136, 3713–3714. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, F.; Kamat, P.V. Riding the New Wave of Perovskites. ACS Energy Lett. 2017, 2, 922–923. [Google Scholar] [CrossRef]
- Varadwaj, P.R. Methylammonium Lead Trihalide Perovskite Solar Cell Semiconductors Are Not Organometallic: A Perspective. Helv. Chim. Acta 2017, 100, e1700090. [Google Scholar] [CrossRef]
- Wang, X.; Li, M.; Zhang, B.; Wang, H.; Zhao, Y.; Wang, B. Recent progress in organometal halide perovskite photodetectors. Org. Electron. 2018, 52, 172–183. [Google Scholar] [CrossRef]
- Liang, M.; Ali, A.; Belaidi, A.; Hossain, M.I.; Ronan, O.; Downing, C.; Tabet, N.; Sanvito, S.; Ei-Mellouhi, F.; Nicolosi, V. Improving stability of organometallic-halide perovskite solar cells using exfoliation two-dimensional molybdenum chalcogenides. npj 2D Mater. Appl. 2020, 4, 40. [Google Scholar] [CrossRef]
- Almushaikeh, A.M.; Wang, H.; Gutiérrez-Arzaluz, L.; Yin, J.; Huang, R.-W.; Bakr, O.M.; Mohammed, O.F. Zero-dimensional Cu(I)-based organometallic halide with green cluster-centred emission for high resolution X-ray imaging screens. Chem. Commun. 2023, 59, 4447–4450. [Google Scholar] [CrossRef]
- Yerezhep, D.; Omarova, Z.; Aldiyarov, A.; Shinbayeva, A.; Tokmoldin, N. IR Spectroscopic Degradation Study of Thin Organometal Halide Perovskite Films. Molecules 2023, 28, 1288. [Google Scholar] [CrossRef]
- Handayani, Y.S.; Indari, E.D.; Hidayat, R.; Othsubo, Y.; Kimura, S.-i. Understanding the role of organic cations on the electronic structure of lead iodide perovskite from their UV photoemission spectra and their electronic structures calculated by DFT method. Mater. Res. Express 2019, 6, 084009. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Yamashita, K. Revealing the Chemistry between Band Gap and Binding Energy for Lead-/Tin-Based Trihalide Perovskite Solar Cell Semiconductors. ChemSusChem 2018, 11, 449–463. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Yamashita, K. Revealing the Cooperative Chemistry of the Organic Cation in the Methylammonium Lead Triiodide Perovskite Semiconductor System. ChemistrySelect 2018, 3, 7269–7282. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. Significance of hydrogen bonding and other noncovalent interactions in determining octahedral tilting in the CH3NH3PbI3 hybrid organic-inorganic halide perovskite solar cell semiconductor. Sci. Rep. 2019, 9, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. Halogen in materials design: Revealing the nature of hydrogen bonding and other non-covalent interactions in the polymorphic transformations of methylammonium lead tribromide perovskite. Mater. Today Chem. 2018, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, A.; Varadwaj, P.R.; Yamashita, K. Hybrid organic–inorganic CH3NH3PbI3 perovskite building blocks: Revealing ultra-strong hydrogen bonding and mulliken inner complexes and their implications in materials design. J. Comp. Chem. 2017, 38, 2802–2818. [Google Scholar] [CrossRef]
- Motta, C.; El-Mellouhi, F.; Kais, S.; Tabet, N.; Alharbi, F.; Sanvito, S. Revealing the role of organic cations in hybrid halide perovskite CH3NH3PbI3. Nat. Commun. 2015, 6, 7026. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, G.; Yamashita, K. Zero-Dimensional Hybrid Organic–Inorganic Halide Perovskite Modeling: Insights from First Principles. J. Phys. Chem. Lett. 2016, 7, 888–899. [Google Scholar] [CrossRef]
- Lin, H.; Zhou, C.; Tian, Y.; Siegrist; Ma, B. T. Low-Dimensional Organometal Halide Perovskites. ACS Energy Lett. 2018, 3, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Cao, F.; Zhang, P.; Li, L. Multidimensional perovskite solar cells. Fund. Res. 2022, 2, 237–253. [Google Scholar] [CrossRef]
- Cheng, X.; Han, Y.; Cui, B.-B. Hetero-perovskite engineering for stable and efficient perovskite solar cells. Sustain. Energy Fuels 2022, 6, 3304–3323. [Google Scholar] [CrossRef]
- Duan, D.; Ge, C.; Rahaman, M.Z.; Lin, C.-H.; Shi, Y.; Lin, H.; Hu, H.; Wu, T. Recent progress with one-dimensional metal halide perovskites: From rational synthesis to optoelectronic applications. NPG Asia Mater. 2023, 15, 8. [Google Scholar] [CrossRef]
- Lei, Y.; Chen, Y.; Xu, S. Single-crystal halide perovskites: Opportunities and challenges. Matter 2021, 4, 2266–2308. [Google Scholar] [CrossRef]
- Chen, Y.; Shen, J.; Yang, G.; Zhou, W.; Shao, Z. A single-/double-perovskite composite with an overwhelming single-perovskite phase for the oxygen reduction reaction at intermediate temperatures. J. Mater. Chem. A 2017, 5, 24842–24849. [Google Scholar] [CrossRef]
- Chu, Y.; Hu, Y.; Xiao, Z. First-Principles Insights into the Stability Difference between ABX3 Halide Perovskites and Their A2BX6 Variants. J. Phys. Chem. C 2021, 125, 9688–9694. [Google Scholar] [CrossRef]
- Ghosh, S.; Shankar, H.; Kar, P. Recent developments of lead-free halide double perovskites: A new superstar in the optoelectronic field. Mater. Adv. 2022, 3, 3742–3765. [Google Scholar] [CrossRef]
- Wolf, N.R.; Connor, B.A.; Slavney, A.H.; Karunadasa, H.I. Doubling the Stakes: The Promise of Halide Double Perovskites. Angew. Chem. Int. Ed. 2021, 60, 16264–16278. [Google Scholar] [CrossRef]
- Bibi, A.; Lee, I.; Nah, Y.; Allam, O.; Kim, H.; Quan, L.N.; Tang, J.; Walsh, A.; Jang, S.S.; Sargent, E.H.; et al. Lead-free halide double perovskites: Toward stable and sustainable optoelectronic devices. Mater. Today 2021, 49, 123–144. [Google Scholar] [CrossRef]
- Kangsabanik, J.; Sugathan, V.; Yadav, A.; Yella, A.; Alam, A. Double perovskites overtaking the single perovskites: A set of new solar harvesting materials with much higher stability and efficiency. Phys. Rev. Mater. 2018, 2, 055401. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R.; Marques, H.M. Physical and optoelectronic features of lead-free A2AgRhBr6 (A = Cs, Rb, K, Na, Li) with halide double perovskite composition. J. Mat. Chem. C 2020, 8, 12968–12983. [Google Scholar] [CrossRef]
- Varadwaj, P.R. A2AgCrCl6 (A = Li, Na, K, Rb, Cs) halide double perovskites: A transition metal-based semiconducting material series with appreciable optical characteristics. Phys. Chem. Chem. Phys. 2020, 22, 24337–24350. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Marques, H.M. The Cs2AgRhCl6 Halide Double Perovskite: A Dynamically Stable Lead-Free Transition-Metal Driven Semiconducting Material for Optoelectronics. Front. Chem. 2020, 8, 796. [Google Scholar] [CrossRef]
- Vargas, B.; Rodríguez-López, G.; Solis-Ibarra, D. The Emergence of Halide Layered Double Perovskites. ACS Energy Lett. 2020, 5, 3591–3608. [Google Scholar] [CrossRef]
- Yi, C.; Luo, J.; Meloni, S.; Boziki, A.; Ashari-Astani, N.; Grätzel, C.; Zakeeruddin, S.M.; Röthlisberger, U.; Grätzel, M. Entropic stabilization of mixed A-cation ABX3 metal halide perovskites for high performance perovskite solar cells. Energy Environ. Sci. 2016, 9, 656–662. [Google Scholar] [CrossRef]
- Stoumpos, C.C.; Kanatzidis, M.G. The Renaissance of Halide Perovskites and Their Evolution as Emerging Semiconductors. Acc. Chem. Res. 2015, 48, 2791–2802. [Google Scholar] [CrossRef] [PubMed]
- Unger, E.L. The PV-Researcher’s Siren: Hybrid metal halide perovskites. Curr. Opin. Green Sustain. Chem. 2017, 4, 72–76. [Google Scholar] [CrossRef]
- Chen, J.-K.; Zhao, Q.; Shirahata, N.; Yin, J.; Bakr, O.M.; Mohammed, O.F.; Sun, H.-T. Shining Light on the Structure of Lead Halide Perovskite Nanocrystals. ACS Mater. Lett. 2021, 3, 845–861. [Google Scholar] [CrossRef]
- Pachori, S.; Agarwal, R.; Prakash, B.; Kumari, S.; Verma, A.S. Fundamental Physical Properties of Nontoxic Tin-Based Formamidinium FASnX3 (X = I, Br, Cl) Hybrid Halide Perovskites: Future Opportunities in Photovoltaic Applications. Energy Technol. 2022, 10, 2100709. [Google Scholar] [CrossRef]
- Monika; Pachori, S.; Agrawal, R.; Choudhary, B.L.; Verma, A.S. An efficient and stable lead-free organic–inorganic tin iodide perovskite for photovoltaic device: Progress and challenges. Energy Rep. 2022, 8, 5753–5763. [Google Scholar] [CrossRef]
- Montoya De Los Santos, I.; Cortina-Marrero, H.J.; Ruíz-Sánchez, M.A.; Hechavarría-Difur, L.; Sánchez-Rodríguez, F.J.; Courel, M.; Hu, H. Optimization of CH3NH3PbI3 perovskite solar cells: A theoretical and experimental study. Solar Energy 2020, 199, 198–205. [Google Scholar] [CrossRef]
- López, C.A.; Abia, C.; Gainza, J.; Kayser, P.; Nemes, N.N.; Dura, O.J.; Martínez, J.L.; Fernández-Díaz, M.T.; Álvarez-Galván, C.; Alonso, J.A. Structural evolution, optical gap and thermoelectric properties of CH3NH3SnBr3 hybrid perovskite, prepared by mechanochemistry. Mater. Adv. 2021, 2, 3620–3628. [Google Scholar] [CrossRef]
- Dimesso, L.; Dimamay, M.; Hamburger, M.; Jaegermann, W. Properties of CH3NH3PbX3 (X = I, Br, Cl) Powders as Precursors for Organic/Inorganic Solar Cells. Chem. Mater. 2014, 26, 6762–6770. [Google Scholar] [CrossRef]
- Loryuenyong, V.; Khiaokaeo, N.; Koomsin, W.; Thongchu, S.; Buasri, A. Crystallisation of CH3NH3PbX3 (X = I, Br, and Cl) trihalide perovskite using PbI2 and PbCl2 precursors. Micro Nano Lett. 2018, 13, 486–489. [Google Scholar] [CrossRef]
- Hao, F.; Stoumpos, C.C.; Guo, P.; Zhou, N.; Marks, T.J.; Chang, R.P.H.; Kanatzidis, M.G. Solvent-Mediated Crystallization of CH3NH3SnI3 Films for Heterojunction Depleted Perovskite Solar Cells. J. Am. Chem. Soc. 2015, 137, 11445–11452. [Google Scholar] [CrossRef]
- Kim, H.D.; Miyamoto, Y.; Kubota, H.; Yamanari, T.; Ohkita, H. Open-circuit Voltage Loss in CH3NH3SnI3 Perovskite Solar Cells. Chem. Lett. 2017, 46, 253–256. [Google Scholar] [CrossRef] [Green Version]
- Protesescu, L.; Yakunin, S.; Bodnarchuk, M.I.; Krieg, F.; Caputo, R.; Hendon, C.H.; Yang, R.X.; Walsh, A.; Kovalenko, M.V. Nanocrystals of Cesium Lead Halide Perovskites (CsPbX3, X = Cl, Br, and I): Novel Optoelectronic Materials Showing Bright Emission with Wide Color Gamut. Nano Lett. 2015, 15, 3692–3696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kshirsagar, B.; Jaykhedkar, N.; Jain, K.; Kishor, S.; Shah, V.; Ramaniah, L.M.; Tiwari, S. Green CsSnX3 (X = Cl, Br, I)-Derived Quantum Dots for Photovoltaic Applications: First-Principles Investigations. J. Phys. Chem. C 2021, 125, 2592–2606. [Google Scholar] [CrossRef]
- Rahman, M.H.; Jubair, M.; Rahaman, M.Z.; Ahasan, M.S.; Ostrikov, K.; Roknuzzaman, M. RbSnX3 (X = Cl, Br, I): Promising lead-free metal halide perovskites for photovoltaics and optoelectronics. RSC Adv. 2022, 12, 7497–7505. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Hu, Y.; Dong, H.; Sun, L.; Zhao, B.; Jiang, X.; Zhang, L. An All-Inorganic Perovskite-Phase Rubidium Lead Bromide Nanolaser. Angew. Chem. Int. Ed. 2019, 58, 16134–16140. [Google Scholar] [CrossRef]
- Amgar, D.; Wierzbowska, M.; Uvarov, V.; Gutkin, V.; Etgar, L. Novel rubidium lead chloride nanocrystals: Synthesis and characterization. Nano Futures 2017, 1, 021002. [Google Scholar] [CrossRef] [Green Version]
- McGovern, L.; Futscher, M.H.; Muscarella, L.A.; Ehrler, B. Understanding the Stability of MAPbBr3 versus MAPbI3: Suppression of Methylammonium Migration and Reduction of Halide Migration. J. Phys. Chem. Lett. 2020, 11, 7127–7132. [Google Scholar] [CrossRef]
- Ahmad, Z.; Aziz, F.; Abdullah, H.Y. Study on the stability of the mixed (MAPbI3 and MAPbBr3) perovskite solar cells using dopant-free HTL. Org. Electron. 2020, 76, 105453. [Google Scholar] [CrossRef]
- Su, L.; Méndez, M.; Jiménez-López, J.; Zhu, M.; Xiao, Y.; Gil, E.J.P. Analysis of the Oxygen Passivation Effects on MAPbI3 and MAPbBr3 in Fresh and Aged Solar Cells by the Transient Photovoltage Technique. ChemPlusChem 2021, 86, 1316–1321. [Google Scholar] [CrossRef]
- Filip, M.R.; Eperon, G.E.; Snaith, H.J.; Giustino, F. Steric engineering of metal-halide perovskites with tunable optical band gaps. Nat. Commun. 2014, 5, 5757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakharia, V.; Castelli, I.E.; Bhavsar, K.; Solanki, A. Bandgap prediction of metal halide perovskites using regression machine learning models. Phys. Lett. A 2022, 422, 127800. [Google Scholar] [CrossRef]
- Ou, Q.; Bao, X.; Zhang, Y.; Shao, H.; Xing, G.; Li, X.; Shao, L.; Bao, Q. Band structure engineering in metal halide perovskite nanostructures for optoelectronic applications. Nano Mater. Sci. 2019, 1, 268–287. [Google Scholar] [CrossRef]
- Liu, S.; Wang, J.; Hu, Z.; Duan, Z.; Zhang, H.; Zhang, W.; Guo, R.; Xie, F. Role of organic cation orientation in formamidine based perovskite materials. Sci. Rep. 2021, 11, 20433. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Song, N.; Feng, L.; Deng, X. Effects of Organic Cation Additives on the Fast Growth of Perovskite Thin Films for Efficient Planar Heterojunction Solar Cells. ACS Appl. Mater. Interfaces 2016, 8, 24703–24711. [Google Scholar] [CrossRef] [PubMed]
- Alwarappan, G.; Alam, M.R.; Hassan, W.M.I.; Shibl, M.F.; Alfalah, S.; Patil, S.; Nekovei, R.; Verma, A. Role of organic cation in modern lead-based perovskites. Solar Energy 2019, 189, 86–93. [Google Scholar] [CrossRef]
- Tremblay, M.-H.; Boyington, A.; Rigin, S.; Jiang, J.; Bacsa, J.; Al Kurdi, K.; Khrustalev, V.N.; Pachter, R.; Timofeeva, T.V.; Jui, N.; et al. Hybrid Organic Lead Iodides: Role of Organic Cation Structure in Obtaining 1D Chains of Face-Sharing Octahedra vs 2D Perovskites. Chem. Mater. 2022, 34, 935–946. [Google Scholar] [CrossRef]
- Zhou, Z.; Lian, H.J.; Xie, J.; Qiao, W.C.; Wu, X.F.; Shi, Y.; Wang, X.L.; Dai, S.; Yuan, H.; Hou, Y.; et al. Non-selective adsorption of organic cations enables conformal surface capping of perovskite grains for stabilized photovoltaic operation. Cell Rep. Phys. Sci. 2022, 3, 100760. [Google Scholar] [CrossRef]
- Senno, M.; Tinte, S. Mixed formamidinium–methylammonium lead iodide perovskite from first-principles: Hydrogen-bonding impact on the electronic properties. Phys. Chem. Chem. Phys. 2021, 23, 7376–7385. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. The Pnictogen Bond, Together with Other Non-Covalent Interactions, in the Rational Design of One-, Two- and Three-Dimensional Organic-Inorganic Hybrid Metal Halide Perovskite Semiconducting Materials, and Beyond. Int. J. Mol. Sci. 2022, 23, 8816. [Google Scholar] [CrossRef] [PubMed]
- Svane, K.L.; Forse, A.C.; Grey, C.P.; Kieslich, G.; Cheetham, A.K.; Walsh, A.; Butler, K.T. How Strong Is the Hydrogen Bond in Hybrid Perovskites? J. Phys. Chem. Lett. 2017, 8, 6154–6159. [Google Scholar] [CrossRef] [Green Version]
- Wilks, R.G.; Erbing, A.; Sadoughi, G.; Starr, D.E.; Handick, E.; Meyer, F.; Benkert, A.; Iannuzzi, M.; Hauschild, D.; Yang, W.; et al. Dynamic Effects and Hydrogen Bonding in Mixed-Halide Perovskite Solar Cell Absorbers. J. Phys. Chem. Lett. 2021, 12, 3885–3890. [Google Scholar] [CrossRef]
- Ibaceta-Jaña, J.; Chugh, M.; Novikov, A.S.; Mirhosseini, H.; Kühne, T.D.; Szyszka, B.; Wagner, M.R.; Muydinov, R. Do Lead Halide Hybrid Perovskites Have Hydrogen Bonds? J. Phys. Chem. C 2022, 126, 16215–16226. [Google Scholar] [CrossRef]
- Lee, J.H.; Bristowe, P.D.; Lee, J.H.; Lee, S.H.; Cheetham, A.K.; Jang, H.M. Resolving the Physical Origin of Octahedral Tilting in Halide Perovskites. Chem. Mater. 2016, 28, 4259–4266. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Bristowe, N.C.; Bristowe, P.D.; Cheetham, A.K. Role of hydrogen bonding and its interplay with octahedral tilting in CH3NH3PbI3. Chem. Commun. 2015, 51, 6434–6437. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Lee, J.-H.; Kong, E.-H.; Jang, H.M. The nature of hydrogen-bonding interaction in the prototypic hybrid halide perovskite, tetragonal CH3NH3PbI3. Sci. Rep. 2016, 6, 21687. [Google Scholar] [CrossRef] [Green Version]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. Definition of the Pnictogen Bond: A Perspective. Inorganics 2022, 10, 149. [Google Scholar] [CrossRef]
- Resnati, G.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Krossing, I.; Legon, A.C.; Metrangolo, P.; Nicotra, F.; Rissanen, K.; Scheiner, S.; et al. Definition of the pnictogen bond (IUPAC Recommendations 202?). Pure Appl. Chem. 2023. Available online: https://iupac.org/wp-content/uploads/2023/02/PAC-REC-2020-1002.R4_PR20230210.pdf (accessed on 21 June 2023).
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. Definition of the tetrel bond. CrystEngComm 2023, 25, 1411–1423. [Google Scholar] [CrossRef]
- Varadwaj, P.R. Tetrel Bonding in Anion Recognition: A First-Principles Investigation. Molecules 2022, 27, 8449. [Google Scholar] [CrossRef]
- Domagała, M.; Lutyńska, A.; Palusiak, M. Extremely Strong Halogen Bond. The Case of a Double-Charge-Assisted Halogen Bridge. J. Phys. Chem. A 2018, 122, 5484–5492. [Google Scholar] [CrossRef] [PubMed]
- Lopes Jesus, A.J.; Redinha, J.S. Charge-Assisted Intramolecular Hydrogen Bonds in Disubstituted Cyclohexane Derivatives. J. Phys. Chem. A 2011, 115, 14069–14077. [Google Scholar] [CrossRef] [PubMed]
- Pakiari, A.H.; Eskandari, E. The chemical nature of very strong hydrogen bonds in some categories of compounds. J. Mol. Str. 2006, 759, 51–60. [Google Scholar] [CrossRef]
- Rybarczyk-Pirek, A.J. Co-crystal/salt crystal structure disorder of trichloroacetic acid–N-methylurea complex with double system of homo- and heteronuclear O–H···O/N–H···O hydrogen bonds: X-ray investigation, ab initio and DFT studies. Struct. Chem. 2012, 23, 1739–1749. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Suresh, C.H.; Remya, G.S.; Anjalikrishna, P.K. Molecular electrostatic potential analysis: A powerful tool to interpret and predict chemical reactivity. WIREs Comput. Mol. Sci. 2022, 12, e1601. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. The Molecular Electrostatic Potential: A Tool for Understanding and Predicting Molecular Interactions. In Molecular Orbital Calculations for Biological Systems; Sapse, A.-M., Ed.; Oxford University Press: Oxford, UK, 1998; pp. 49–84. [Google Scholar]
- Politzer, P.; Laurence, P.R.; Jayasuriya, K. Molecular electrostatic potentials: An effective tool for the elucidation of biochemical phenomena. Environ. Health Perspect. 1985, 61, 191–202. [Google Scholar] [CrossRef]
- Molecular Electrostatic Potentials: Concepts and Applications, 1st ed.; Murray, J.S.; Sen, K. (Eds.) Elsevier Science & Technology: Oxford, UK, 1996. [Google Scholar]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Hénon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef]
- Lefebvre, C.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. The Independent Gradient Model: A New Approach for Probing Strong and Weak Interactions in Molecules from Wave Function Calculations. ChemPhysChem 2018, 19, 724–735. [Google Scholar] [CrossRef]
- Leguy, A.M.A.; Frost, J.M.; McMahon, A.; Sakai, V.G.; Kochelmann, W.; Law, C.-H.; Li, X.; Foglia, F.; Walsh, A.; O’Regan, B.; et al. The dynamics of methylammonium ions in hybrid organic-inorganic perovskite solar cells. Nat.Commun. 2015, 6, 7124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Yoon, S.J.; Stamplecoskie, K.G.; Kamat, P.V. How lead halide complex chemistry dictates the composition of mixed halide perovskites. J. Phys. Chem. Lett. 2016, 7, 1368–1373. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Shi, Y.; Zou, C.; Lin, L.Y. MAPbBr3 First-Order Distributed Feedback Laser with High Stability. Adv. Photon. Res. 2023, 4, 2200071. [Google Scholar] [CrossRef]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [Green Version]
- Bone, R.G.A.; Bader, R.F.W. Identifying and analyzing intermolecular bonding interactions in van der Waals molecules. J. Phys. Chem. 1996, 100, 10892–10911. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Matta, C.F. Bonding to titanium. Inorg. Chem. 2001, 40, 5603–5611. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Weinhold, F.; Glendening, E.D. Comment on “Natural Bond Orbitals and the Nature of the Hydrogen Bond”. J. Phys. Chem. A 2018, 122, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutorial Example for Methylamine: Running an NBO Calculation. Available online: https://nbo7.chem.wisc.edu/tutorial.html (accessed on 10 June 2023).
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Chen, K.; Deng, X.; Goddard, R.; Tüysüz, H. Pseudomorphic Transformation of Organometal Halide Perovskite Using the Gaseous Hydrogen Halide Reaction. Chem. Mater. 2016, 28, 5530–5537. [Google Scholar] [CrossRef]
- López, C.A.; Martínez-Huerta, M.V.; Alvarez-Galván, M.C.; Kayser, P.; Gant, P.; Castellanos-Gomez, A.; Fernández-Díaz, M.T.; Fauth, F.; Alonso, J.A. Elucidating the Methylammonium (MA) Conformation in MAPbBr3 Perovskite with Application in Solar Cells. Inorg. Chem. 2017, 56, 14214–14219. [Google Scholar] [CrossRef] [Green Version]
- Weller, M.T.; Weber, O.J.; Henry, P.F.; Di Pumpo, A.M.; Hansen, T.C. Complete structure and cation orientation in the perovskite photovoltaic methylammonium lead iodide between 100 and 352 K. Chem. Commun. 2015, 51, 4180–4183. [Google Scholar]
- Butler, K.T. The chemical forces underlying octahedral tilting in halide perovskites. J. Mater. Chem. C 2018, 6, 12045–12051. [Google Scholar] [CrossRef]
- Young, J.; Rondinelli, J.M. Octahedral Rotation Preferences in Perovskite Iodides and Bromides. J. Phys. Chem. Lett. 2016, 7, 918–922. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comp. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. A direct MP2 gradient method. Chem. Phys. Lett. 1990, 166, 275–280. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Head-Gordon, T. Analytic MP2 frequencies without fifth-order storage. Theory and application to bifurcated hydrogen bonds in the water hexamer. Chem. Phys. Lett. 1994, 220, 122–128. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, J.S.; Lane, P.; Clark, T.; Politzer, P. σ-hole bonding: Molecules containing group VI atoms. J. Mol. Model. 2007, 13, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Resnati, G.; Politzer, P. Close contacts and noncovalent interactions in crystals. Faraday Discuss. 2017, 203, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Politzer, P. Interaction and Polarization Energy Relationships in σ-Hole and π-Hole Bonding. Crystals 2020, 10, 76. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen Bonding: A Halogen-Centered Noncovalent Interaction Yet to Be Understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. The Phosphorous Bond, or the Phosphorous-Centered Pnictogen Bond: The Covalently Bound Phosphorous Atom in Molecular Entities and Crystals as a Pnictogen Bond Donor. Molecules 2022, 27, 1487. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. The Nitrogen Bond, or the Nitrogen-Centered Pnictogen Bond: The Covalently Bound Nitrogen Atom in Molecular Entities and Crystals as a Pnictogen Bond Donor. Compounds 2022, 2, 80–110. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. The pnictogen bond forming ability of bonded bismuth atoms in molecular entities in the crystalline phase: A perspective. CrystEngComm 2023, 25, 1038–1052. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. σ-Hole Interactions: Perspectives and Misconceptions. Crystals 2017, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, A.; Marques, H.M.; Varadwaj, P.R. Is the Fluorine in Molecules Dispersive? Is Molecular Electrostatic Potential a Valid Property to Explore Fluorine-Centered Non-Covalent Interactions? Molecules 2019, 24, 379. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, A.; Varadwaj, P.R.; Jin, B.-Y. Fluorines in tetrafluoromethane as halogen bond donors: Revisiting address the nature of the fluorine’s σhole. Int. J. Quantum Chem. 2015, 115, 453–470. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. Revealing Factors Influencing the Fluorine-Centered Non-Covalent Interactions in Some Fluorine-Substituted Molecular Complexes: Insights from First-Principles Studies. ChemPhysChem 2018, 19, 1486–1499. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Does Chlorine in CH3Cl Behave as a Genuine Halogen Bond Donor? Crystals 2020, 10, 146. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R.; Varadwaj, A.; Jin, B.-Y. Halogen bonding interaction of chloromethane with several nitrogen donating molecules: Addressing the nature of the chlorine surface σ-hole. Phys. Chem. Chem. Phys. 2014, 16, 19573–19589. [Google Scholar] [CrossRef] [Green Version]
- Gatti, C.; Dessì, A.; Dallocchio, R.; Mamane, V.; Cossu, S.; Weiss, R.; Pale, P.; Aubert, E.; Peluso, P. Factors Impacting σ- and π-Hole Regions as Revealed by the Electrostatic Potential and Its Source Function Reconstruction: The Case of 4,4′-Bipyridine Derivatives. Molecules 2020, 25, 4409. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll, V. 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019. Available online: http://aim.tkgristmill.com(accessed on 21 June 2023).
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comp. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Multiwfn. Available online: http://sobereva.com/multiwfn/ (accessed on 14 April 2023).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ion Pairs | VS,max(X–Tt) | VS,max(X–Tt) | VS,max(X–Tt) | VS,min(Tt) | VS,max(N-C)/VS,max(C-N) | VS,min(X) | VS,min(X) |
|---|---|---|---|---|---|---|---|
| I3Pb···NH3CH3 | 26.1 | 26.1 | 26.1 | 22.2 | 38.2 | −18.0 | |
| I3Pb···CH3NH3 | 11.9 | 11.9 | 11.9 | 9.6 | 90.4 | −29.9 | |
| I3Pb···H3NCH3 | 23.4 | 23.4 | 19.4 | 18.3 | --- | −23.1 | −17.4 |
| Br3Pb···NH3CH3 | 26.6 | 26.6 | 26.6 | 22.2 | 36.4 | −21.2 | |
| Br3Pb···CH3NH3 | 10.4 | 10.4 | 10.4 | 7.7 | 88.3 | −34.1 | |
| Br3Pb···H3NCH3 | 23.0 | 23.0 | 19.1 | 17.4 | --- | −27.2 | −21.5 |
| Cl3Pb···NH3CH3 | 26.1 | 26.1 | 26.1 | 21.0 | 34.9 | −23.8 | |
| Cl3Pb···CH3NH3 | 8.3 | 8.3 | 8.3 | 5.1 | 86.6 | −37.0 | |
| Cl3Pb···H3NCH3 | 21.8 | 21.8 | 17.9 | 15.5 | --- | −30.5 | −26.9 |
| F3Pb···NH3CH3 | 22.8 | 22.8 | 22.8 | 14.8 | 27.4 | −37.6 | |
| F3Pb···CH3NH3 | −0.4 (4.1) | −0.4 (4.1) | −0.4 (4.1) | −5.7 (−4.2) | 79.6 (83.8) | −53.1 (−57.0) | |
| F3Pb···H3NCH3 | 16.1 | 16.1 | 12.5 | 6.8 | --- | −39.2 | −47.5 |
| I3Sn···NH3CH3 | 15.6 | 15.6 | 15.6 | 5.3 | 39.5 | −16.9 | |
| I3Sn···CH3NH3 | 0.5 | 0.5 | 0.5 | −7.9 | 91.4 | −30.0 | |
| I3Sn···H3NCH3 | 12.5 | 12.5 | 9.5 | 1.6 | --- | −22.2 | −17.2 |
| Br3Sn···NH3CH3 | 15.5 | 15.5 | 15.5 | 3.8 | 38.2 | −19.8 | |
| Br3Sn···CH3NH3 | −1.6 (3.6) | −1.6 (3.6) | −1.6 (3.6) | −11.1 (−10.1) | 89.8 (94.4) | −33.8 (−34.7) | |
| Br3Sn···H3NCH3 | 11.5 | 11.5 | 8.6 | −0.7 | --- | −25.8 | −20.9 |
| Cl3Sn···NH3CH3 | 14.6 | 14.6 | 14.6 | 1.7 | 37.2 | −22.1 | |
| Cl3Sn···CH3NH3 | −4.0 (1.1) | −4.0 (1.1) | −4.0 (1.1) | −14.8 (−14.2) | 88.5 (93.0) | −35.8 (−37.8) | |
| Cl3Sn···H3NCH3 | 10.1 | 10.1 | 7.2 | −3.6 | --- | −28.7 | --- |
| F3Sn···NH3CH3 | 11.5 | 11.5 | 11.5 | −5.4 | 31.6 | −34.6 | |
| F3Sn···CH3NH3 | −11.7 (−6.9) | −11.7 (−6.9) | −11.7 (−6.9) | −25.8 (−26.4) | 82.8 (---) | −50.8 (−54.4) | |
| F3Sn···H3NCH3 | 5.3 | 5.3 | 3.3 | −12.3 | −36.3 | −44.0 | |
| I3Ge···NH3CH3 | 7.0 | 7.0 | 7.0 | −3.0 | 40.6 | −17.3 | |
| I3Ge···CH3NH3 | −8.2 (−3.8) | −8.2 (−3.8) | −8.2 (−3.8) | −16.0 (−15.3) | 92.5 (97.1) | −30.9 (−31.1) | |
| I3Ge···H3NCH3 | 4.2 | 4.2 | 1.4 | −6.1 | --- | −22.2 | −17.8 |
| Br3Ge···NH3CH3 | 6.1 | 6.1 | 6.1 | −5.5 | 39.7 | −19.9 | |
| Br3Ge···CH3NH3 | −11.3 | −11.3 | −11.3 | −20.7 | 90.9 | −35.4 | |
| Br3Ge···H3NCH3 | 2.4 (7.4) | 2.4 (7.4) | −0.3 (4.3) | −9.6 (−8.9) | --- | −25.4 (−26.4) | −21.4 (−22.4) |
| Cl3Ge···NH3CH3 | 4.8 | 4.8 | 4.8 | −8.7 | 39.0 | −21.7 | |
| Cl3Ge···CH3NH3 | −14.2 | −14.2 | −14.2 | −25.3 | 89.7 | −37.9 | |
| Cl3Ge···H3NCH3 | 0.6 (5.9) | 0.6 (5.9) | −1.8 (2.7) | −13.4 (−13.1) | --- | −27.6 (−29.0) | −24.1 (−25.1) |
| F3Ge···NH3CH3 | −0.3 (4.7) | −0.3 (4.7) | −0.3 (4.7) | −19.2 (−19.9) | 35.4 (38.0) | −33.1 (−35.5) | |
| F3Ge···CH3NH3 | −23.8 | −23.8 | −23.8 | −39.5 | 85.2 | −50.3 | |
| F3Ge···H3NCH3 | −6.1 | −6.1 | −6.7 | −25.1 | --- | −41.1 | −35.3 |
| I3Si···NH3CH3 | −0.8 (3.3) | −0.8 (3.3) | −0.8 (3.3) | −15.8 (−16.0) | 41.8 (44.7) | −16.6 (−16.7) | |
| I3Si···CH3NH3 | −16.3 | −16.3 | −16.3 | −29.1 | 93.4 | −30.6 | |
| Br3Si···NH3CH3 | −3.1 (1.3) | −3.1 (1.3) | −3.1 (1.3) | −20.5 (−21.3) | 41.3 (44.2) | −19.2 (−19.7) | |
| Br3Si···CH3NH3 | −20.9 | −20.9 | −20.9 | −35.7 | 92.3 | −35.0 | |
| Cl3Si···NH3CH3 | −5.3 (−1.0) | −5.3 (−1.0) | −5.3 (−1.0) | −25.5 (−26.7) | 41.3 (44.2) | −21.0 (−21.7) | |
| Cl3Si···CH3NH3 | −24.6 | −24.6 | −24.6 | −41.9 | 91.7 | −37.6 | |
| Cl3Si···H3NCH3 | −9.8 | −9.8 | −10.8 | −29.8 | --- | −24.5 | −27.5 |
| F3Si···NH3CH3 | −12.6 | −12.6 | −12.6 | −41.2 | 40.8 | −32.1 | |
| F3Si···CH3NH3 | −35.0 | −35.0 | −35.0 | −60.0 | 89.6 | −50.6 | |
| F3Si···H3NCH3 | −17.70 | −17.70 | −15.0 | −45.8 | --- | −34.2 | −39.4 |
| System | Interaction Type | ΔE | ΔE(BSSE) |
|---|---|---|---|
| I3Pb···NH3CH3 | I···H(N) | −104.95 | −104.85 |
| I3Pb···CH3NH3 | I···H(C) | −76.42 | −76.33 |
| I3Pb···H3NCH3 | I···H(N), I···H(C) | −97.64 | −97.53 |
| Br3Pb···NH3CH3 | Br···H(N) | −111.04 | −110.66 |
| Br3Pb···CH3NH3 | Br···H(C) | −80.05 | −79.77 |
| Br3Pb···H3NCH3 | Br···H(N), Br···H(C) | −102.95 | −102.62 |
| Cl3Pb···NH3CH3 | Cl···H(N) | −115.95 | −115.66 |
| Cl3Pb···CH3NH3 | Cl···H(C) | −82.78 | −82.55 |
| Cl3Pb···H3NCH3 | Cl···H(N), Cl···H(C) | −107.24 | −106.99 |
| F3Pb···NH3CH3 | F···H(N) | −138.54 | −138.23 |
| F3Pb···CH3NH3 | F···H(C) | −94.39 | −94.18 |
| F3Pb···H3NCH3 | F···H(N), F···H(C) | −126.47 | −126.2 |
| I3Sn···NH3CH3 | I···H(N) | −103.12 | −103.01 |
| I3Sn···CH3NH3 | I···H(C) | −75.02 | −74.93 |
| I3Sn···H3NCH3 | I···H(N), I···H(C) | −96.07 | −95.96 |
| Br3Sn···NH3CH3 | Br···H(N) | −108.39 | −108.01 |
| Br3Sn···CH3NH3 | Br···H(C) | −78.17 | −77.89 |
| Br3Sn···H3NCH3 | Br···H(N), Br···H(C) | −100.76 | −100.44 |
| Cl3Sn···NH3CH3 | Cl···H(N) | −112.48 | −112.19 |
| Cl3Sn···CH3NH3 | Cl···H(C) | −80.43 | −80.2 |
| Cl3Sn···H3NCH3 | Cl···H(N), Cl···H(C) | −104.45 | −104.19 |
| F3Sn···NH3CH3 | F···H(N) | −131.44 | −131.11 |
| F3Sn···CH3NH3 | F···H(C) | −90.37 | −90.15 |
| F3Sn···H3NCH3 | F···H(N), F···H(C) | −121.08 | −120.79 |
| I3Ge···NH3CH3 | I···H(N) | −101.91 | −101.74 |
| I3Ge···CH3NH3 | I···H(C) | −74.2 | −74.05 |
| I3Ge···H3NCH3 | I···H(N), I···H(C) | −95.31 | −95.13 |
| Br3Ge···NH3CH3 | Br···H(N) | −106.63 | −106.22 |
| Br3Ge···CH3NH3 | Br···H(C) | −77.07 | −76.74 |
| Br3Ge···H3NCH3 | Br···H(N), Br···H(C) | −99.64 | −99.26 |
| Cl3Ge···NH3CH3 | Cl···H(N) | −110.15 | −109.82 |
| Cl3Ge···CH3NH3 | Cl···H(C) | −79.01 | −78.74 |
| Cl3Ge···H3NCH3 | Cl···H(N), Cl···H(C) | −102.88 | −102.56 |
| F3Ge···NH3CH3 | F···H(N) | −125.21 | −124.92 |
| F3Ge···CH3NH3 | F···H(C) | −87.26 | −87.04 |
| F3Ge···H3NCH3 | F···H(N), F···H(C) | −116.94 | −116.66 |
| I3Si···NH3CH3 | I···H(N) | −100.36 | −100.25 |
| I3Si···CH3NH3 | I···H(C) | −73.01 | −72.91 |
| Br3Si···NH3CH3 | Br···H(N) | −104.19 | −103.86 |
| Br3Si···CH3NH3 | Br···H(C) | −75.28 | −75.03 |
| Cl3Si···NH3CH3 | Cl···H(N) | −106.61 | −106.34 |
| Cl3Si···CH3NH3 | Cl···H(C) | −76.58 | −76.37 |
| Cl3Si···H3NCH3 | Cl···H(N), Cl···H(C) | −99.92 | −99.65 |
| F3Si···NH3CH3 | F···H(N) | −115.3 | −115.02 |
| F3Si···CH3NH3 | F···H(C) | −81.46 | −81.24 |
| F3Si···CH3NH3 | F···H(N), F···H(C) | −109.02 | −108.77 |
| System | Tetrel Bond | ρb | ∇2ρb | Hb | Pnictogen Bond | ρb | ∇2ρb | Hb |
|---|---|---|---|---|---|---|---|---|
| [CH3NH3+•PbI3−]2 | Pb···I | 0.0144 | 0.0272 | 0.0002 | N···I | 0.0058 | 0.0189 | 0.0009 |
| [CH3NH3+•PbBr3−]2 | Pb···Br | 0.0163 | 0.0368 | 0.0003 | N···Br | 0.0064 | 0.0230 | 0.0011 |
| [CH3NH3+•PbCl3−]2 | Pb···Cl | 0.0187 | 0.0496 | 0.0007 | N···Cl | 0.0066 | 0.0257 | 0.0013 |
| [CH3NH3+•PbF3−]2 | Pb···F | 0.0342 | 0.1373 | 0.0002 | N···F | 0.0062 | 0.0294 | 0.0016 |
| [CH3NH3+•SnI3−]2 | Sn···I | 0.0108 | 0.0191 | 0.0003 | N···I | 0.0070 | 0.0231 | 0.0010 |
| [CH3NH3+•SnBr3−]2 | Sn···Br | 0.0113 | 0.0234 | 0.0004 | N···Br | 0.0080 | 0.0293 | 0.0012 |
| [CH3NH3+•SnCl3−]2 | Sn···Cl | 0.0124 | 0.0292 | 0.0008 | N···Cl | 0.0086 | 0.0339 | 0.0015 |
| [CH3NH3+•SnF3−]2 | Sn···F | 0.0283 | 0.0965 | 0.00005 | N···F | 0.0094 | 0.0463 | 0.0023 |
| [CH3NH3+•GeI3−]2 | Ge···I | 0.0085 | 0.0193 | 0.0007 | N···I | 0.0077 | 0.0253 | 0.0010 |
| [CH3NH3+•GeBr3−]2 | Ge···Br | 0.0090 | 0.0174 | 0.0005 | N···Br | 0.0090 | 0.0326 | 0.0013 |
| [CH3NH3+•GeCl3−]2 | Ge···Cl | 0.0082 | 0.0211 | 0.0010 | N···Cl | 0.0096 | 0.0378 | 0.0016 |
| [CH3NH3+•GeF3−]2 | Ge···F | 0.0125 | 0.0415 | 0.0014 | N···F | 0.0116 | 0.0594 | 0.0027 |
| [CH3NH3+•SiI3−]2 | Si···I | 0.0068 | 0.0134 | 0.0005 | N···I | 0.0083 | 0.0274 | 0.0190 |
| [CH3NH3+•SiBr3−]2 | Si···Br | 0.0054 | 0.0125 | 0.0006 | N···Br | 0.0096 | 0.0351 | 0.0263 |
| [CH3NH3+•SiCl3−]2 | Si···Cl | 0.0026 | 0.0063 | 0.0004 | N···Cl | 0.0105 | 0.0418 | 0.0376 |
| [CH3NH3+•SiF3−]2 | H···Si b | 0.0228 | 0.0281 | −0.0018 | N···F | 0.0102 | 0.0496 | 0.0669 |
| System | Interaction Type a | ΔE | ΔE(BSSE) |
|---|---|---|---|
| [CH3NH3+•PbI3−]2 | Pb···I | −17.11 | −16.83 |
| [CH3NH3+•PbBr3−]2 | Pb···Br | −17.78 | −17.26 |
| [CH3NH3+•PbCl3−]2 | Pb···Cl | −18.26 | −17.85 |
| [CH3NH3+•PbF3−]2 | Pb···F | −25.21 | −24.73 |
| [CH3NH3+•SnI3−]2 | Sn···I | −13.80 | −13.51 |
| [CH3NH3+•SnBr3−]2 | Sn···Br | −13.40 | −12.87 |
| [CH3NH3+•SnCl3−]2 | Sn···Cl | −12.99 | −12.59 |
| [CH3NH3+•SnF3−]2 | Sn···F | −17.22 | −16.72 |
| [CH3NH3+•GeI3−]2 | Ge···I | −12.11 | −11.66 |
| [CH3NH3+•GeBr3−]2 | Ge···Br | −11.08 | −10.41 |
| [CH3NH3+•GeCl3−]2 | Ge···Cl | −10.01 | −9.50 |
| [CH3NH3+•GeF3−]2 | Ge···F | −9.09 | −8.60 |
| [CH3NH3+•SiI3−]2 | Si···I | −10.20 | −9.92 |
| [CH3NH3+•SiBr3−]2 | Si···Br | −8.81 | −8.33 |
| [CH3NH3+•SiCl3−]2 | Si···Cl | −7.49 | −7.15 |
| [CH3NH3+•SiF3−]2 | H···Si b | −14.48 | −14.35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. Methylammonium Tetrel Halide Perovskite Ion Pairs and Their Dimers: The Interplay between the Hydrogen-, Pnictogen- and Tetrel-Bonding Interactions. Int. J. Mol. Sci. 2023, 24, 10554. https://doi.org/10.3390/ijms241310554
Varadwaj PR, Varadwaj A, Marques HM, Yamashita K. Methylammonium Tetrel Halide Perovskite Ion Pairs and Their Dimers: The Interplay between the Hydrogen-, Pnictogen- and Tetrel-Bonding Interactions. International Journal of Molecular Sciences. 2023; 24(13):10554. https://doi.org/10.3390/ijms241310554
Chicago/Turabian StyleVaradwaj, Pradeep R., Arpita Varadwaj, Helder M. Marques, and Koichi Yamashita. 2023. "Methylammonium Tetrel Halide Perovskite Ion Pairs and Their Dimers: The Interplay between the Hydrogen-, Pnictogen- and Tetrel-Bonding Interactions" International Journal of Molecular Sciences 24, no. 13: 10554. https://doi.org/10.3390/ijms241310554