
Novel SPEA Superantigen Peptide Agonists and Peptide Agonist-TGFαL3 Conjugate. In Vitro Study of Their Growth-Inhibitory Effects for Targeted Cancer Immunotherapy


Abstract
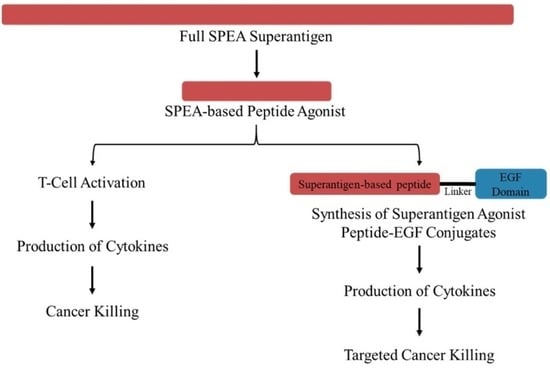
:
1. Introduction
2. Results
2.1. T-Cell Activity Assay
2.2. Production of Cytokines
2.3. Binding Assessment
2.4. MTT Assay
2.5. Tumor Killing Assay
3. Discussion
4. Method and Materials
4.1. Reagents, Antibodies, and Kits
4.2. T-Cell Activation Assay
4.3. Design of SPEA-Based Tumor-Targeted SAg Peptide
4.4. Production of Cytokines
4.5. Tumor Cell Binding Assay
4.6. MTT Assay
4.7. Assessment of Tumor Killing Ability
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jeanson, A.; Tomasini, P.; Souquet-Bressand, M.; Brandone, N.; Boucekine, M.; Grangeon, M.; Chaleat, S.; Khobta, N.; Milia, J.; Mhanna, L.; et al. Efficacy of Immune Checkpoint Inhibitors in KRAS-Mutant Non-Small Cell Lung Cancer (NSCLC). J. Thorac. Oncol. 2019, 14, 1095–1101. [Google Scholar] [CrossRef]
- Kuske, M.; Rauschenberg, R.; Garzarolli, M.; Meredyth-Stewart, M.; Beissert, S.; Troost, E.G.C.; Glitza, O.I.C.; Meier, F. Melanoma Brain Metastases: Local Therapies, Targeted Therapies, Immune Checkpoint Inhibitors and Their Combinations—Chances and Challenges. Am. J. Clin. Dermatol. 2018, 19, 529–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehrer, E.J.; Peterson, J.; Brown, P.D.; Sheehan, J.P.; Quiñones-Hinojosa, A.; Zaorsky, N.G.; Trifiletti, D.M. Treatment of brain metastases with stereotactic radiosurgery and immune checkpoint inhibitors: An international meta-analysis of individual patient data. Radiother. Oncol. 2019, 130, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Marrone, K.; Ying, W.; Naidoo, J. Immune-Related Adverse Events from Immune Checkpoint Inhibitors. Clin. Pharmacol. Ther. 2016, 100, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Drilon, A.; Lusque, A.B.; Mhanna, L.; Cortot, A.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- Mhanna, L.; Guibert, N.; Milia, J.; Mazieres, J. When to Consider Immune Checkpoint Inhibitors in Oncogene-Driven Non-Small Cell Lung Cancer? Curr. Treat. Options Oncol. 2019, 20, 60. [Google Scholar] [CrossRef] [Green Version]
- Robin, T.P.; Breeze, R.E.; Smith, D.E.; Rusthoven, C.G.; Lewis, K.D.; Gonzalez, R.; Brill, A.; Saiki, R.; Stuhr, K.; Gaspar, L.E.; et al. Immune checkpoint inhibitors and radiosurgery for newly diagnosed melanoma brain metastases. J. Neuro-Oncol. 2018, 140, 55–62. [Google Scholar] [CrossRef]
- Upadhrasta, S.; Elias, H.; Patel, K.; Zheng, L. Managing cardiotoxicity associated with immune checkpoint inhibitors. Chronic Dis. Transl. Med. 2019, 5, 6–14. [Google Scholar] [CrossRef]
- Zhang, Y.; Yao, Q.; Pan, Y.; Fang, X.; Xu, H.; Zhao, T.; Zhu, G.; Jiang, T.; Li, S.; Cao, H. Efficacy and Safety of PD-1/PD-L1 Checkpoint Inhibitors versus Anti-PD-1/PD-L1 Combined with Other Therapies for Tumors: A Systematic Review. Cancers 2023, 15, 682. [Google Scholar] [CrossRef]
- Zeng, H.; Xu, Q.; Wang, J.; Xu, X.; Luo, J.; Zhang, L.; Luo, C.; Ying, J.; Li, J. The effect of anti-PD-1/PD-L1 antibodies combined with VEGF receptor tyrosine kinase inhibitors versus bevacizumab in unresectable hepatocellular carcinoma. Front. Immunol. 2023, 14, 1073133. [Google Scholar] [CrossRef]
- Sjöstrand, M.; Sadelain, M. Driving CARs to new places: Locally produced BCMA CAR T cells to treat multiple myeloma. Haematologica 2023. [Google Scholar] [CrossRef]
- Dai, H.-P.; Kong, D.-Q.; Shen, H.-J.; Cui, W.; Wang, Q.; Li, Z.; Yin, J.; Kang, L.-Q.; Yu, L.; Wu, D.-P.; et al. CAR-T cell therapy followed by allogenic hematopoietic stem cell transplantation yielded comparable outcome between Ph like ALL and other high-risk ALL. Biomark. Res. 2023, 11, 19. [Google Scholar] [CrossRef]
- Leick, M.B.; Maus, M.V. Multiomics STEP up in correlative analysis of response to CAR T cells. Nat. Rev. Clin. Oncol. 2023, 20, 285–286. [Google Scholar] [CrossRef]
- Rini, S.S.; Kambayana, G. Autologous CD19-Targeted Chimeric Antigen Receptor (CAR)T-Cells as the Future of Systemic Lupus Erythematosus Treatment. Curr. Rheumatol. Rev. 2023, 19, 260–269. [Google Scholar] [CrossRef]
- Adabi, N.; Pordel, S.; Rezaee, M.A.; Shobeiri, F.S.; Shobeiri, S.S. Application of CAR-T cell technology in autoimmune diseases and human immunodeficiency virus infection treatment. J. Gene Med. 2023, 25, e3484. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, Q.; Zhou, S.; He, H.; Zhao, M.; Ma, W. Mesenchymal stem cell suppresses the efficacy of CAR-T toward killing lymphoma cells by modulating the microenvironment through stanniocalcin-1. Elife 2023, 12, e82934. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, J.; Qiao, L.; Chen, L.; Chen, C.; Zhang, H.; Luo, R.; Xiao, Y. Case report: Cryoablation as a novel bridging strategy prior to CAR-T cell therapy for B cell malignancies with bulky disease. Front. Oncol. 2023, 13, 1008828. [Google Scholar] [CrossRef]
- Nicod, C.; da Rocha, M.N.; Warda, W.; Roussel, X.; Haderbache, R.; Seffar, E.; Trad, R.; Bouquet, L.; Goncalves, M.; Bosdure, L.; et al. CAR-T cells targeting IL-1RAP produced in a closed semiautomatic system are ready for the first phase I clinical investigation in humans. Curr. Res. Transl. Med. 2023, 71, 103385. [Google Scholar] [CrossRef]
- Harrer, D.C.; Dörrie, J.; Schaft, N. CARs and Drugs: Pharmacological Ways of Boosting CAR-T-Cell Therapy. Int. J. Mol. Sci. 2023, 24, 2342. [Google Scholar] [CrossRef]
- Asokan, S.; Cullin, N.; Stein-Thoeringer, C.K.; Elinav, E. CAR-T Cell Therapy and the Gut Microbiota. Cancers 2023, 15, 794. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Abila, B.; Kamel, Y.M. CAR-T: What Is Next? Cancers 2023, 15, 663. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Llera, A.; Malchiodi, E.L.; Mariuzza, R.A. The structural basis of t cell activation by superantigens. Annu. Rev. Immunol. 1999, 17, 435–466. [Google Scholar] [CrossRef] [PubMed]
- Mehindate, K.; Thibodeau, J.; Dohlsten, M.; Kalland, T.; Sékaly, R.P.; Mourad, W. Cross-linking of major histocompatibility complex class II molecules by staphylococcal enterotoxin A superantigen is a requirement for inflammatory cytokine gene expression. J. Exp. Med. 1995, 182, 1573–1577. [Google Scholar] [CrossRef] [Green Version]
- Terman, D.S.; Serier, A.; Dauwalder, O.; Badiou, C.; Dutour, A.; Thomas, D.; Brun, V.; Bienvenu, J.; Etienne, J.; Vandenesch, F.; et al. Staphylococcal entertotoxins of the enterotoxin Gene cluster (egcSEs) induce nitrous oxide- and cytokine dependent tumor cell apoptosis in a broad panel of human tumor cells. Front. Cell. Infect. Microbiol. 2013, 3, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ejtehadifar, M.; Halabian, R.; Ghazavi, A.; Khansarinejad, B.; Mosayebi, G.; Fooladi, A.A.I. Bone marrow—mesenchymal stem cells impact on the U937 cells in the presence of staphylococcal enterotoxin B (SEB). Clin. Exp. Pharmacol. Physiol. 2018, 45, 849–858. [Google Scholar] [CrossRef]
- Hawkins, R.E.; Gore, M.; Shparyk, Y.; Bondar, V.; Gladkov, O.; Ganev, T.; Harza, M.; Polenkov, S.; Bondarenko, I.; Karlov, P.; et al. A Randomized Phase II/III Study of Naptumomab Estafenatox + IFNα versus IFNα in Renal Cell Carcinoma: Final Analysis with Baseline Biomarker Subgroup and Trend Analysis. Clin. Cancer Res. 2016, 22, 3172–3181. [Google Scholar] [CrossRef] [Green Version]
- Superantigens, superantigen-like proteins and superantigen derivatives for cancer treatment. Eur. Rev. Med. Pharmacol. 2021, 25, 1622–1630. [CrossRef]
- Golob-Urbanc, A.; Rajčević, U.; Strmšek, Z.; Jerala, R. Design of split superantigen fusion proteins for cancer immunotherapy. J. Biol. Chem. 2019, 294, 6294–6305. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zeng, L.; Zhao, Z.; Xie, Y.; Wang, S.; Zhang, J.; He, Y.; Zou, Z.; Zhang, J.; Tao, A. Construction, Expression, and Characterization of rSEA-EGF and In Vitro Evaluation of its Antitumor Activity Against Nasopharyngeal Cancer. Technol. Cancer Res. Treat. 2018, 17, 1533033818762910. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Xu, M.; Song, Y.; Su, Z.; Zhang, H.; Zhang, C. TNF-α produced by SEC2 mutant (SAM-3)-activated human T cells induces apoptosis of HepG2 cells. Appl. Microbiol. Biotechnol. 2016, 100, 2677–2684. [Google Scholar] [CrossRef]
- Eisen, T.; Hedlund, G.; Forsberg, G.; Hawkins, R. Naptumomab Estafenatox: Targeted Immunotherapy with a Novel Immunotoxin. Curr. Oncol. Rep. 2014, 16, 370. [Google Scholar] [CrossRef] [Green Version]
- Dohlsten, M.; Abrahmsén, L.; Björk, P.; Lando, P.A.; Hedlund, G.; Forsberg, G.; Brodin, T.; Gascoigne, N.R.; Förberg, C.; Lind, P. Monoclonal antibody-superantigen fusion proteins: Tumor-specific agents for T-cell-based tumor therapy. Proc. Natl. Acad. Sci. USA 1994, 91, 8945–8949. [Google Scholar] [CrossRef] [Green Version]
- Dohlsten, M.; Hedlund, G.; Akerblom, E.; Lando, P.A.; Kalland, T. Monoclonal antibody-targeted superantigens: A different class of anti-tumor agents. Proc. Natl. Acad. Sci. USA 1991, 88, 9287–9291. [Google Scholar] [CrossRef] [Green Version]
- Dohlsten, M.; Hansson, J.; Ohlsson, L.; Litton, M.; Kalland, T. Antibody-targeted superantigens are potent inducers of tumor-infiltrating T lymphocytes in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9791–9795. [Google Scholar] [CrossRef] [Green Version]
- Allen, T.M. Ligand-targeted therapeutics in anticancer therapy. Nat. Rev. Cancer 2002, 2, 750–763. [Google Scholar] [CrossRef]
- Wu, H.-C.; Chang, D.-K.; Huang, C.-T. Targeted Therapy for Cancer. J. Cancer Mol. 2006, 2, 57–66. [Google Scholar]
- Xu, Q.; Zhang, X.; Yue, J.; Liu, C.; Cao, C.; Zhong, H.; Ma, Q. Human TGFalpha-derived peptide TGFalphaL3 fused with superantigen for immunotherapy of EGFR-expressing tumours. BMC Biotechnol. 2010, 10, 91. [Google Scholar] [CrossRef] [Green Version]
- Grupka, N.L.; Lear-Kaul, K.C.; Kleinschmidt-DeMasters, B.K.; Singh, M. Epidermal growth factor receptor status in breast cancer metastases to the central nervous system. Comparison with HER-2/neu status. Arch. Pathol. Lab. Med. 2004, 128, 974–979. [Google Scholar] [CrossRef]
- Dua, R.; Zhang, J.; Nhonthachit, P.; Penuel, E.; Petropoulos, C.; Parry, G. EGFR over-expression and activation in high HER2, ER negative breast cancer cell line induces trastuzumab resistance. Breast Cancer Res. Treat. 2010, 122, 685–697. [Google Scholar] [CrossRef]
- Aziz, S.A.; Pervez, S.; Khan, S.; Kayani, N.; Rahbar, M.H. Epidermal growth factor receptor (EGFR) as a prognostic marker: An immunohistochemical study on 315 consecutive breast carcinoma patients. J. Pak. Med. Assoc. 2002, 52, 104–110. [Google Scholar]
- Agustoni, F.; Suda, K.; Yu, H.; Ren, S.; Rivard, C.J.; Ellison, K.; Caldwell, C.; Rozeboom, L.; Brovsky, K.; Hirsch, F.R. EGFR-directed monoclonal antibodies in combination with chemotherapy for treatment of non-small-cell lung cancer: An updated review of clinical trials and new perspectives in biomarkers analysis. Cancer Treat. Rev. 2018, 72, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Imani-Fooladi, A.A.; Yousefi, F.; Mousavi, S.F.; Amani, J. In Silico Design and Analysis of TGFalphaL3-SEB Fusion Protein as "a New Antitumor Agent" Candidate by Ligand-Targeted Superantigens Technique. Iran. J. Cancer Prev. 2014, 7, 152–164. [Google Scholar] [PubMed]
- Yousefi, F.; Mousavi, S.F.; Siadat, S.D.; Aslani, M.M.; Amani, J.; Rad, H.S.; Fooladi, A.A.I. Preparation and In Vitro Evaluation of Antitumor Activity of TGFαL3-SEB as a Ligand-Targeted Superantigen. Technol. Cancer Res. Treat. 2016, 15, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, F.; Siadat, S.D.; Saraji, A.A.; Hesaraki, S.; Aslani, M.M.; Mousavi, S.F.; Fooladi, A.A.I. Tagging staphylococcal enterotoxin B (SEB) with TGFaL3 for breast cancer therapy. Tumor Biol. 2016, 37, 5305–5316. [Google Scholar] [CrossRef] [PubMed]
- Miethke, T.; Duschek, K.; Wahl, C.; Heeg, K.; Wagner, H. Pathogenesis of the toxic shock syndrome: T cell mediated lethal shock caused by the superantigen TSST-1. Eur. J. Immunol. 1993, 23, 1494–1500. [Google Scholar] [CrossRef]
- Dama, P.; Ledoux, D.; Nys, M.; Vrindts, Y.; Groote, D.D.E.; Franchimont, P.; Lamy, M. Cytokine Serum Level During Severe Sepsis in Human IL-6 as a Marker of Severity. Ann. Surg. 1992, 215, 356–362. [Google Scholar] [CrossRef]
- Waage, A.; Brandtzaeg, P.; Halstensen, A.; Kierulf, P.; Espevik, T. The complex pattern of cytokines in serum from patients with meningococcal septic shock. Association between interleukin 6, interleukin 1, and fatal outcome. J. Exp. Med. 1989, 169, 333–338. [Google Scholar] [CrossRef]
- Hansson, J.; Ohlsson, L.; Persson, R.; Andersson, G.; Ilback, N.G.; Litton, M.J.; Kalland, T.; Dohlsten, M. Genetically engineered superantigens as tolerable antitumor agents. Proc. Natl. Acad. Sci. USA 1997, 94, 2489–2494. [Google Scholar] [CrossRef] [Green Version]
- Abrahmsen, L.; Dohlsten, M.; Segren, S.; Bjork, P.; Jonsson, E.; Kalland, T. Characterization of two distinct MHC class II binding sites in the superantigen staphylococcal enterotoxin A. EMBO J. 1995, 14, 2978–2986. [Google Scholar] [CrossRef]
- Dohlsten, M.; Lando, P.A.; Bjork, P.; Abrahmsen, L.; Ohlsson, L.; Lind, P.; Kalland, T. Immunotherapy of human colon cancer by antibody-targeted superantigens. Cancer Immunol. Immunother. CII 1995, 41, 162–168. [Google Scholar] [CrossRef]
- Liu, T.; Li, L.; Yin, L.; Yu, H.; Jing, H.; Liu, Y.; Kong, C.; Xu, M. Superantigen staphylococcal enterotoxin C1 inhibits the growth of bladder cancer. Biosci. Biotechnol. Biochem. 2017, 81, 1741–1746. [Google Scholar] [CrossRef] [Green Version]
- Shaw, D.M.; Connolly, N.B.; Patel, P.M.; Kilany, S.; Hedlund, G.; Nordle, O.; Forsberg, G.; Zweit, J.; Stern, P.L.; Hawkins, R.E. A phase II study of a 5T4 oncofoetal antigen tumour-targeted superantigen (ABR-214936) therapy in patients with advanced renal cell carcinoma. Br. J. Cancer 2007, 96, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Søgaard, M.; Hansson, J.; Litton, M.J.; Ohlsson, L.; Rosendahl, A.; Lando, P.A.; Antonsson, P.; Kalland, T.; Dohlsten, M. Antibody-targeted superantigens in cancer immunotherapy. Immunotechnology 1996, 2, 151–162. [Google Scholar] [CrossRef]
- Bashraheel, S.S.; AlQahtani, A.D.; Rashidi, F.B.; Al-Sulaiti, H.; Domling, A.; Orie, N.N.; Goda, S.K. Studies on vascular response to full superantigens and superantigen derived peptides: Possible production of novel superantigen variants with less vasodilation effect for tolerable cancer immunotherapy. Biomed. Pharmacother. 2019, 115, 108905. [Google Scholar] [CrossRef]
- Arad, G.; Levy, R.; Nasie, I.; Hillman, D.; Rotfogel, Z.; Barash, U.; Supper, E.; Shpilka, T.; Minis, A.; Kaempfer, R. Binding of Superantigen Toxins into the CD28 Homodimer Interface Is Essential for Induction of Cytokine Genes That Mediate Lethal Shock. PLoS Biol. 2011, 9, e1001149. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Sgagias, M.K.; Kasid, A.; Danforth, D.N. Interleukin-1α and Tumor Necrosis Factor-α (TNFα) Inhibit Growth and Induce TNF Messenger RNA in MCF-7 Human Breast Cancer Cells. Mol. Endocrinol. 1991, 5, 1740–1747. [Google Scholar] [CrossRef] [Green Version]
- Maund, S.L.; Barclay, W.W.; Hover, L.D.; Axanova, L.S.; Sui, G.; Hipp, J.D.; Fleet, J.C.; Thorburn, A.; Cramer, S.D. Interleukin-1α Mediates the Antiproliferative Effects of 1,25-Dihydroxyvitamin D3 in Prostate Progenitor/Stem Cells. Cancer Res. 2011, 71, 5276–5286. [Google Scholar] [CrossRef] [Green Version]
- Maund, S.L.; Shi, L.; Cramer, S.D. A Role for Interleukin-1 Alpha in the 1,25 Dihydroxyvitamin D3 Response in Mammary Epithelial Cells. PLoS ONE 2013, 8, e81367. [Google Scholar] [CrossRef] [Green Version]
- Le, T.T.; Skak, K.; Schroder, K.; Schroder, W.A.; Boyle, G.M.; Pierce, C.J.; Suhrbier, A. IL-1 Contributes to the Anti-Cancer Efficacy of Ingenol Mebutate. PLoS ONE 2016, 11, e0153975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oft, M. IL-10: Master Switch from Tumor-Promoting Inflammation to Antitumor Immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, R. Drug delivery and targeting. Nature 1998, 392 (Suppl. S6679), 5–10. [Google Scholar] [PubMed]
- Brannon-Peppas, L.; Blanchette, J.O. Nanoparticle and targeted systems for cancer therapy. Adv. Drug Deliv. Rev. 2004, 56, 1649–1659. [Google Scholar] [CrossRef]
- Chari, R.V.J.; Miller, M.L.; Widdison, W.C. Antibody-Drug Conjugates: An Emerging Concept in Cancer Therapy. Angew. Chem. Int. Ed. 2014, 53, 3796–3827. [Google Scholar] [CrossRef]
- Sharma, S.K.; Bagshawe, K.D. Antibody Directed Enzyme Prodrug Therapy (ADEPT): Trials and tribulations. Adv. Drug Deliv. Rev. 2017, 118, 2–7. [Google Scholar] [CrossRef]
- MacKay, J.A.; Chen, M.; McDaniel, J.R.; Liu, W.; Simnick, A.J.; Chilkoti, A. Self-assembling chimeric polypeptide–doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nat. Mater. 2009, 8, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Bazley, L.A.; Gullick, W.J. The epidermal growth factor receptor family. Endocr.-Relat. Cancer 2005, 12, S17–S27. [Google Scholar] [CrossRef]
- Arena, S.; Bellosillo, B.; Siravegna, G.; Martínez, A.; Cañadas, I.; Lazzari, L.; Ferruz, N.; Russo, M.; Misale, S.; González, I.; et al. Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clin. Cancer Res. 2015, 21, 2157–2166. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Jang, M.H.; Kim, E.J.; Kim, H.J.; Lee, H.J.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.-W.; Kim, I.A.; et al. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod. Pathol. 2014, 27, 1212–1222. [Google Scholar] [CrossRef] [Green Version]
- Azuma, K.; Ota, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; Kage, M.; et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann. Oncol. 2014, 25, 1935–1940. [Google Scholar] [CrossRef]
- Benelli, R.; Costa, D.; Salvini, L.; Tardito, S.; Tosetti, F.; Villa, F.; Zocchi, M.R.; Poggi, A. Targeting of colorectal cancer organoids with zoledronic acid conjugated to the anti-EGFR antibody cetuximab. J. Immunother. Cancer 2022, 10, e005660. [Google Scholar] [CrossRef]
- Lu, Y.; Huang, J.; Li, F.; Wang, Y.; Ding, M.; Zhang, J.; Yin, H.; Zhang, R.; Ren, X. EGFR-specific single-chain variable fragment antibody-conjugated Fe3O4/Au nanoparticles as an active MRI contrast agent for NSCLC. Magn. Reson. Mater. Physics Biol. Med. 2021, 34, 581–591. [Google Scholar] [CrossRef]
- Wu, S.-C.; Chen, Y.-J.; Wang, H.-C.; Chou, M.-Y.; Chang, T.-Y.; Yuan, S.-S.; Chen, C.-Y.; Hou, M.-F.; Hsu, J.T.-A.; Wang, Y.-M. Bispecific Antibody Conjugated Manganese-Based Magnetic Engineered Iron Oxide for Imaging of HER2/neu- and EGFR-Expressing Tumors. Theranostics 2016, 6, 118–130. [Google Scholar] [CrossRef]
- Leung, S.L.; Zha, Z.; Cohn, C.; Dai, Z.; Wu, X. Anti-EGFR antibody conjugated organic–inorganic hybrid lipid nanovesicles selectively target tumor cells. Colloids Surf. B Biointerfaces 2014, 121, 141–149. [Google Scholar] [CrossRef]
- Maleki, F.; Sadeghifard, N.; Hosseini, H.M.; Bakhtiyari, S.; Goleij, Z.; Behzadi, E.; Sedighian, H.; Fooladi, A.A.I. Growth-inhibitory effects of TGFαL3-SEB chimeric protein on colon cancer cell line. Biomed. Pharmacother. 2019, 110, 190–196. [Google Scholar] [CrossRef]
- Bashraheel, S.S.; Domling, A.; Goda, S.K. Update on targeted cancer therapies, single or in combination, and their fine tuning for precision medicine. Biomed. Pharmacother. 2020, 125, 110009. [Google Scholar] [CrossRef]
- Brandão, M.; Caparica, R.; Eiger, D.; de Azambuja, E. Biomarkers of response and resistance to PI3K inhibitors in estrogen receptor-positive breast cancer patients and combination therapies involving PI3K inhibitors. Ann. Oncol. 2019, 30, x27–x42. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, O.; Badalamenti, G.; De Summa, S.; Calabrese, A.; Argentiero, A.; Fucci, L.; Longo, V.; Galetta, D.; Perrotti, P.M.S.; Pinto, R.; et al. Molecular Characterization of a Long-Term Survivor Double Metastatic Non-Small Cell Lung Cancer and Pancreatic Ductal Adenocarcinoma Treated with Gefitinib in Combination with Gemcitabine Plus Nab-Paclitaxel and mFOLFOX6 as First and Second Line Therapy. Cancers 2019, 11, 749. [Google Scholar] [CrossRef] [Green Version]
- Kos, S.; Lopes, A.; Preat, V.; Cemazar, M.; Tratar, U.L.; Ucakar, B.; Vanvarenberg, K.; Sersa, G.; Vandermeulen, G. Intradermal DNA vaccination combined with dual CTLA-4 and PD-1 blockade provides robust tumor immunity in murine melanoma. PLoS ONE 2019, 14, e0217762. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Chan, C.K.; Weissman, I.L.; Kim, B.Y.; Hahn, S.M. Immune Priming of the Tumor Microenvironment by Radiation. Trends Cancer 2016, 2, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.; Rückert, M.; Deloch, L.; Rühle, P.F.; Derer, A.; Fietkau, R.; Gaipl, U.S. Immunomodulation by ionizing radiation-impact for design of radio-immunotherapies and for treatment of inflammatory diseases. Immunol. Rev. 2017, 280, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Mazloumi, Z.; Rafat, A.; Asl, K.D.; Charoudeh, H.N. A combination of telomerase inhibition and NK cell therapy increased breast cancer cell line apoptosis. Biochem. Biophys. Res. Commun. 2023, 640, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.M.; Zamani, S.; Elrayess, M.A.; Kafienah, W.; Younes, H.M. New Three-Dimensional Poly(decanediol-co-tricarballylate) Elastomeric Fibrous Mesh Fabricated by Photoreactive Electrospinning for Cardiac Tissue Engineering Applications. Polymers 2018, 10, 455. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SP2TGF | HRSSLVKNLQNIYFLYEGDPVTHENVKSVDQLLSHDLIYNGGSGSGGGVCHSGYVGARCEHADLL |
| TGFSP2 | VCHSGYVGARCEHADLLGGSGSGGGHRSSLVKNLQNIYFLYEGDPVTHENVKSVDQLLSHDLIYN |
| SP3TGF | NIYFLYEGDPVTHENVKSVDQLLSHDLIYNVSGPNYDKLKGGSGSGGGVCHSGYVGARCEHADLL |
| TGFSP3 | VCHSGYVGARCEHADLLGGSGSGGGNIYFLYEGDPVTHENVKSVDQLLSHDLIYNVSGPNYDKLK |
| SP15TGF | VTAQELDYKVRKYLTDNKQLYTNGPSKYETGYIKFIPKNKGGSGSGGGVCHSGYVGARCEHADLL |
| TGFSP15 | VCHSGYVGARCEHADLLGGSGSGGGVTAQELDYKVRKYLTDNKQLYTNGPSKYETGYIKFIPKNK |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bashraheel, S.S.; Goda, S.K. Novel SPEA Superantigen Peptide Agonists and Peptide Agonist-TGFαL3 Conjugate. In Vitro Study of Their Growth-Inhibitory Effects for Targeted Cancer Immunotherapy. Int. J. Mol. Sci. 2023, 24, 10507. https://doi.org/10.3390/ijms241310507
Bashraheel SS, Goda SK. Novel SPEA Superantigen Peptide Agonists and Peptide Agonist-TGFαL3 Conjugate. In Vitro Study of Their Growth-Inhibitory Effects for Targeted Cancer Immunotherapy. International Journal of Molecular Sciences. 2023; 24(13):10507. https://doi.org/10.3390/ijms241310507
Chicago/Turabian StyleBashraheel, Sara S., and Sayed K. Goda. 2023. "Novel SPEA Superantigen Peptide Agonists and Peptide Agonist-TGFαL3 Conjugate. In Vitro Study of Their Growth-Inhibitory Effects for Targeted Cancer Immunotherapy" International Journal of Molecular Sciences 24, no. 13: 10507. https://doi.org/10.3390/ijms241310507