

A Cell-Based Optimised Approach for Rapid and Efficient Gene Editing of Human Pluripotent Stem Cells
 , ,
, ,
Abstract
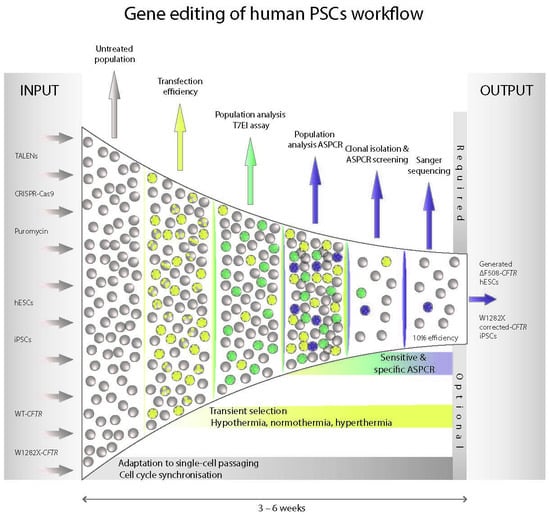
:
1. Introduction
2. Results
2.1. CFTR-Specific TALEN Efficiently Modified hESCs Using Hypothermia and Enrichment for Transfected Cells
2.2. Cell Cycle Synchronisation Prior to Nucleofection Was Tested for the Integration of the ΔF508 Mutation in hESCs
2.3. Clonal Expansion and Direct ASPCR Screening Enabled the Isolation of a ΔF508 Clone
2.4. CRISPR-Cas9 Efficiently Modified the CFTR Locus in Patient-Derived iPSC Lines
2.5. Identification of Clones Containing the Corrected W1282X Mutation in 8K, 4D, and P20801 iPSC Lines
3. Discussion
4. Materials and Methods
4.1. Gene Editing Tools
4.2. Cell Culture
4.3. Cell Cycle Synchronisation
4.4. Nucleofection and Temperature Conditions
4.5. Transient Puromycin Selection
4.6. Clonal Isolation
4.7. Genomic DNA Extraction
4.8. T7 Endonuclease I Assay
4.9. Sanger Sequencing Analysis
4.10. Immunohistochemistry and Confocal Microscopy
4.11. Immunostaining and Flow Cytometry
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bibikova, M.; Golic, M.; Golic, K.G.; Carroll, D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 2002, 161, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Tan, S.Y.; Qiao, G.J.; Barlow, K.A.; Wang, J.B.; Xia, D.F.; Meng, X.D.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, U143–U149. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Morbitzer, R.; Lutge, F.; Dannemann, N.; Lahaye, T.; Cathomen, T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011, 39, 9283–9293. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Lee, Y.K.; Schaefer, E.A.; Peters, D.T.; Veres, A.; Kim, K.; Kuperwasser, N.; Motola, D.L.; Meissner, T.B.; Hendriks, W.T.; et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell 2013, 12, 238–251. [Google Scholar] [CrossRef] [Green Version]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Bobis-Wozowicz, S.; Osiak, A.; Rahman, S.H.; Cathomen, T. Targeted genome editing in pluripotent stem cells using zinc-finger nucleases. Methods 2011, 53, 339–346. [Google Scholar] [CrossRef]
- Schwank, G.; Koo, B.K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional Repair of CFTR by CRISPR/Cas9 in Intestinal Stem Cell Organoids of Cystic Fibrosis Patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.P.; Chin, S.; Xia, S.; Garner, J.; Bear, C.E.; Rossant, J. Efficient generation of functional CFTR-expressing airway epithelial cells from human pluripotent stem cells. Nat. Protoc. 2015, 10, 363–381. [Google Scholar] [CrossRef] [PubMed]
- Adewumi, O.; Aflatoonian, B.; Ahrlund-Richter, L.; Amit, M.; Andrews, P.W.; Beighton, G.; Bello, P.A.; Benvenisty, N.; Berry, L.S.; Bevan, S.; et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat. Biotechnol. 2007, 25, 803–816. [Google Scholar] [PubMed]
- Eckford, P.D.W.; McCormack, J.; Munsie, L.; He, G.; Stanojevic, S.; Pereira, S.L.; Ho, K.; Avolio, J.; Bartlett, C.; Yang, J.Y.; et al. The CF Canada-Sick Kids Program in individual CF therapy: A resource for the advancement of personalized medicine in CF. J. Cyst. Fibros. 2018, 18, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Boyle, M.P.; Bell, S.C.; Konstan, M.W.; McColley, S.A.; Rowe, S.M.; Rietschel, E.; Huang, X.H.; Waltz, D.; Patel, N.R.; Rodman, D.; et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: A phase 2 randomised controlled trial. Lancet Respir. Med. 2014, 2, 527–538. [Google Scholar] [CrossRef]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Durmowicz, A.G.; Lim, R.; Rogers, H.; Rosebraugh, C.J.; Chowdhury, B.A. The U.S. Food and Drug Administration’s Experience with Ivacaftor in Cystic Fibrosis. Establishing Efficacy Using In Vitro Data in Lieu of a Clinical Trial. Ann. Am. Thorac. Soc. 2018, 15, 1–2. [Google Scholar] [CrossRef]
- Bosch, B.; De Boeck, K. Searching for a cure for cystic fibrosis. A 25-year quest in a nutshell. Eur. J. Pediatr. 2016, 175, 1–8. [Google Scholar] [CrossRef]
- Strug, L.J.; Stephenson, A.L.; Panjwani, N.; Harris, A. Recent advances in developing therapeutics for cystic fibrosis. Hum. Mol. Genet. 2018, 27, R173–R186. [Google Scholar] [CrossRef] [Green Version]
- Cuevas-Ocana, S.; Laselva, O.; Avolio, J.; Nenna, R. The era of CFTR modulators: Improvements made and remaining challenges. Breathe 2020, 16, 200016. [Google Scholar] [CrossRef]
- Zwaka, T.P.; Thomson, J.A. Homologous recombination in human embryonic stem cells. Nat. Biotechnol. 2003, 21, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Yusa, K. Seamless genome editing in human pluripotent stem cells using custom endonuclease-based gene targeting and the piggyBac transposon. Nat. Protoc. 2013, 8, 2061–2078. [Google Scholar] [CrossRef] [PubMed]
- Crane, A.M.; Kramer, P.; Bui, J.H.; Chung, W.J.; Li, X.S.; Gonzalez-Garay, M.L.; Hawkins, F.; Liao, W.; Mora, D.; Choi, S.; et al. Targeted Correction and Restored Function of the CFTR Gene in Cystic Fibrosis Induced Pluripotent Stem Cells. Stem Cell Rep. 2015, 4, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firth, A.L.; Menon, T.; Parker, G.S.; Qualls, S.J.; Lewis, B.M.; Ke, E.; Dargitz, C.T.; Wright, R.; Khanna, A.; Gage, F.H.; et al. Functional Gene Correction for Cystic Fibrosis in Lung Epithelial Cells Generated from Patient iPSCs. Cell Rep. 2015, 12, 1385–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, J.D.; Bateup, H.S.; Hockemeyer, D.F. Establishment of Genome-edited Human Pluripotent Stem Cell Lines: From Targeting to Isolation. JoVE-J. Vis. Exp. 2016, 108, e53583. [Google Scholar]
- Suzuki, S.; Sargent, R.G.; Illek, B.; Fischer, H.; Esmaeili-Shandiz, A.; Yezzi, M.J.; Lee, A.; Yang, Y.; Kim, S.; Renz, P.; et al. TALENs Facilitate Single-step Seamless SDF Correction of F508del CFTR in Airway Epithelial Submucosal Gland Cell-derived CF-iPSCs. Mol. Ther. Nucleic Acids 2016, 5, e273. [Google Scholar] [CrossRef] [Green Version]
- Kondrashov, A.; Duc Hoang, M.; Smith, J.G.W.; Bhagwan, J.R.; Duncan, G.; Mosqueira, D.; Munoz, M.B.; Vo, N.T.N.; Denning, C. Simplified Footprint-Free Cas9/CRISPR Editing of Cardiac-Associated Genes in Human Pluripotent Stem Cells. Stem Cells Dev. 2018, 27, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.H.; Guell, M.; Byrne, S.; Yang, J.L.; De Los Angeles, A.; Mali, P.; Aach, J.; Kim-Kiselak, C.; Briggs, A.W.; Rios, X.; et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013, 41, 9049–9061. [Google Scholar] [CrossRef]
- Arias-Fuenzalida, J.; Jarazo, J.; Qing, X.; Walter, J.; Gomez-Giro, G.; Nickels, S.L.; Zaehres, H.; Scholer, H.R.; Schwamborn, J.C. FACS-Assisted CRISPR-Cas9 Genome Editing Facilitates Parkinson’s Disease Modeling. Stem Cell Rep. 2017, 9, 1423–1431. [Google Scholar] [CrossRef] [Green Version]
- Haupt, A.; Grancharova, T.; Arakaki, J.; Fuqua, M.A.; Roberts, B.; Gunawardane, R.N. Endogenous Protein Tagging in Human Induced Pluripotent Stem Cells Using CRISPR/Cas9. JoVE-J. Vis. Exp. 2018, 138, e58130. [Google Scholar]
- Lonowski, L.A.; Narimatsu, Y.; Riaz, A.; Delay, C.E.; Yang, Z.; Niola, F.; Duda, K.; Ober, E.A.; Clausen, H.; Wandall, H.H.; et al. Genome editing using FACS enrichment of nuclease-expressing cells and indel detection by amplicon analysis. Nat. Protoc. 2017, 12, 581–603. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yang, L.H.; Grishin, D.; Rios, X.; Ye, L.Y.; Hu, Y.; Li, K.; Zhang, D.H.; Church, G.M.; Pu, W.T. Efficient, footprint-free human iPSC genome editing by consolidation of Cas9/CRISPR and piggyBac technologies. Nat. Protoc. 2017, 12, 88–103. [Google Scholar] [CrossRef] [Green Version]
- Steyer, B.; Bu, Q.; Cory, E.; Jiang, K.; Duong, S.; Sinha, D.; Steltzer, S.; Gamm, D.; Chang, Q.; Saha, K. Scarless Genome Editing of Human Pluripotent Stem Cells via Transient Puromycin Selection. Stem Cell Rep. 2018, 10, 642–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasaka, R.; Matsumoto, M.; Okada, M.; Sasaki, H.; Kanie, K.; Kii, H.; Uozumi, T.; Kiyota, Y.; Honda, H.; Kato, R. Visualization of morphological categories of colonies for monitoring of effect on induced pluripotent stem cell culture status. Regen. Ther. 2017, 6, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Ugozzoli, L.; Wallace, R.B. Application of an allele-specific polymerase chain reaction to the direct determination of ABO blood group genotypes. Genomics 1992, 12, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajpai, R.; Lesperance, J.; Kim, M.; Terskikh, A.V. Efficient propagation of single cells Accutase-dissociated human embryonic stem cells. Mol. Reprod. Dev. 2008, 75, 818–827. [Google Scholar] [CrossRef]
- Chen, K.G.; Mallon, B.S.; Hamilton, R.S.; Kozhich, O.A.; Park, K.; Hoeppner, D.J.; Robey, P.G.; McKay, R.D.G. Non-colony type monolayer culture of human embryonic stem cells. Stem Cell Res. 2012, 9, 237–248. [Google Scholar] [CrossRef]
- Hohenstein, K.A.; Pyle, A.D.; Chern, J.Y.; Lock, L.F.; Donovan, P.J. Nucleofection mediates high-efficiency stable gene knockdown and transgene expression in human embryonic stem cells. Stem Cells 2008, 26, 1436–1443. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.L.; Wang, Y.J.; Huang, H.; Chen, B.Y.; Chen, X.J.; Hu, J.D.; Chang, T.; Lin, R.J.; Yee, J.K. Precise Gene Modification Mediated by TALEN and Single-Stranded Oligodeoxynucleotides in Human Cells. PLoS ONE 2014, 9, e93575. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.Q.; Pruett-Miller, S.M.; Huang, Y.P.; Gjoka, M.; Duda, K.; Taunton, J.; Collingwood, T.N.; Frodin, M.; Davis, G.D. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat. Methods 2011, 8, U753–U796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyon, Y.; Choi, V.M.; Xia, D.F.; Vo, T.D.; Gregory, P.D.; Holmes, M.C. Transient cold shock enhances zinc-finger nuclease-mediated gene disruption. Nat. Methods. 2010, 7, 459–460. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Alzubi, J.; Fine, E.J.; Morbitzer, R.; Cradick, T.J.; Lahaye, T.; Bao, G.; Cathomen, T. TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res. 2014, 42, 6762–6773. [Google Scholar] [CrossRef] [Green Version]
- Remy, S.; Tesson, L.; Menoret, S.; Usal, C.; De Cian, A.; Thepenier, V.; Thinard, R.; Baron, D.; Charpentier, M.; Renaud, J.B.; et al. Efficient gene targeting by homology-directed repair in rat zygotes using TALE nucleases. Genome Res. 2014, 24, 1371–1383. [Google Scholar] [CrossRef] [Green Version]
- Xiang, G.; Zhang, X.; An, C.; Cheng, C.; Wang, H. Temperature effect on CRISPR-Cas9 mediated genome editing. J. Genet. Genom. 2017, 44, 199–205. [Google Scholar] [CrossRef]
- Lin, S.; Staahl, B.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 2014, 3, e04766. [Google Scholar] [CrossRef]
- Yang, D.; Scavuzzo, M.A.; Chmielowiec, J.; Sharp, R.; Bajic, A.; Borowiak, M. Enrichment of G2/M cell cycle phase in human pluripotent stem cells enhances HDR-mediated gene repair with customizable endonucleases. Sci. Rep. 2016, 6, 21264. [Google Scholar] [CrossRef] [Green Version]
- Saleh-Gohari, N.; Helleday, T. Conservative homologous recombination preferentially repairs DNA double-strand breaks in the S phase of the cell cycle in human cells. Nucleic Acids Res. 2004, 32, 3683–3688. [Google Scholar] [CrossRef] [Green Version]
- Karanam, K.; Kafri, R.; Loewer, A.; Lahav, G. Quantitative Live Cell Imaging Reveals a Gradual Shift between DNA Repair Mechanisms and a Maximal Use of HR in Mid S Phase. Mol. Cell 2012, 47, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Yiangou, L.; Grandy, R.A.; Morell, C.M.; Tomaz, R.A.; Osnato, A.; Kadiwala, J.; Muraro, D.; Garcia-Bernardo, J.; Nakanoh, S.; Bernard, W.G.; et al. Method to Synchronize Cell Cycle of Human Pluripotent Stem Cells without Affecting Their Fundamental Characteristics. Stem Cell Rep. 2019, 12, 165–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phadnis, S.M.; Loewke, N.O.; Dimov, I.K.; Pai, S.; Amwake, C.E.; Solgaard, O.; Baer, T.M.; Chen, B.; Pera, R.A.R. Dynamic and social behaviors of human pluripotent stem cells. Sci. Rep. 2015, 5, 14209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- University-of-Pennsylvania. Available online: https://www.seas.upenn.edu/~diamond/iPS%20P20801%20Cell%20line%20description.pdf (accessed on 12 June 2023).
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef]
- Sampaziotis, F.; de Brito, M.C.; Madrigal, P.; Bertero, A.; Saeb-Parsy, K.; Soares, F.A.C.; Schrumpf, E.; Melum, E.; Karlsen, T.H.; Bradley, J.A.; et al. Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat. Biotechnol. 2015, 33, 845–852. [Google Scholar] [CrossRef] [Green Version]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Drick, N.; Sahabian, A.; Pongpamorn, P.; Merkert, S.; Gohring, G.; Welte, T.; Martin, U.; Olmer, R. Generation of a NKX2.1—p63 double transgenic knock-in reporter cell line from human induced pluripotent stem cells (MHHi006-A-4). Stem Cell Res. 2020, 42, 101659. [Google Scholar] [CrossRef]
- Gu, B.; Posfai, E.; Rossant, J. Efficient generation of targeted large insertions by microinjection into two-cell-stage mouse embryos. Nat. Biotechnol. 2018, 36, 632–637. [Google Scholar] [CrossRef]
- Jain, S.; Shukla, S.; Yang, C.; Zhang, M.; Fatma, Z.; Lingamaneni, M.; Abesteh, S.; Lane, S.T.; Xiong, X.; Wang, Y.; et al. TALEN outperforms Cas9 in editing heterochromatin target sites. Nat. Commun. 2021, 12, 606. [Google Scholar] [CrossRef]
- Kim, Y.; Kweon, J.; Kim, A.; Chon, J.K.; Yoo, J.Y.; Kim, H.J.; Kim, S.; Lee, C.; Jeong, E.; Chung, E.; et al. A library of TAL effector nucleases spanning the human genome. Nat. Biotechnol. 2013, 31, 251–258. [Google Scholar] [CrossRef]
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016, 533, 125–129. [Google Scholar] [CrossRef]
- Aushev, M.; Koller, U.; Mussolino, C.; Cathomen, T.; Reichelt, J. Traceless Targeting and Isolation of Gene-Edited Immortalized Keratinocytes from Epidermolysis Bullosa Simplex Patients. Mol. Ther. Methods Clin. Dev. 2017, 6, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Kwart, D.; Paquet, D.; Teo, S.; Tessier-Lavigne, M. Precise and efficient scarless genome editing in stem cells using CORRECT. Nat. Protoc. 2017, 12, 329–354. [Google Scholar] [CrossRef]
- Villa-Diaz, L.G.; Garcia-Perez, J.L.; Krebsbach, P.H. Enhanced Transfection Efficiency of Human Embryonic Stem Cells by the Incorporation of DNA Liposomes in Extracellular Matrix. Stem Cells Dev. 2010, 19, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Stratigopoulos, G.; De Rosa, M.C.; LeDuc, C.A.; Leibel, R.L.; Doege, C.A. DMSO increases efficiency of genome editing at two non-coding loci. PLoS ONE 2018, 13, e0198637. [Google Scholar] [CrossRef]
- Liang, X.; Potter, J.; Kumar, S.; Zou, Y.; Quintanilla, R.; Sridharan, M.; Carte, J.; Chen, W.; Roark, N.; Ranganathan, S.; et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 2015, 208, 44–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nii, T.; Kohara, H.; Marumoto, T.; Sakuma, T.; Yamamoto, T.; Tani, K. Single-Cell-State Culture of Human Pluripotent Stem Cells Increases Transfection Efficiency. BioRes. Open Access 2016, 5, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Ruan, J.; Hirai, H.; Yang, D.; Ma, L.; Hou, X.; Jiang, H.; Wei, H.; Rajagopalan, C.; Mou, H.; Wang, G.; et al. Efficient Gene Editing at Major CFTR Mutation Loci. Mol. Ther. Nucleic Acids 2019, 16, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedell, V.M.; Wang, Y.; Campbell, J.M.; Poshusta, T.L.; Starker, C.G.; Krug, R.G., 2nd; Tan, W.; Penheiter, S.G.; Ma, A.C.; Leung, A.Y.; et al. In vivo genome editing using a high-efficiency TALEN system. Nature 2012, 491, 114–118. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Bao, Z.; Xiong, X.; Zhao, H. SunnyTALEN: A second-generation TALEN system for human genome editing. Biotechnol. Bioeng. 2014, 111, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Mackley, V.A.; Rao, A.; Chong, A.T.; Dewitt, M.A.; Corn, J.E.; Murthy, N. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. eLife 2017, 6, e25312. [Google Scholar] [CrossRef]
- Ma, M.; Zhuang, F.F.; Hu, X.B.; Wang, B.L.; Wen, X.Z.; Ji, J.F.; Xi, J.J. Efficient generation of mice carrying homozygous double-floxp alleles using the Cas9-Avidin/Biotin-donor DNA system. Cell Res. 2017, 27, 578–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veres, A.; Gosis, B.S.; Ding, Q.; Collins, R.; Ragavendran, A.; Brand, H.; Erdin, S.; Cowan, C.A.; Talkowski, M.E.; Musunuru, K. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell 2014, 15, 27–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Janssen, J.M.; Liu, J.; Maggio, I.; ‘t Jong, A.E.J.; Mikkers, H.M.M.; Goncalves, M. In trans paired nicking triggers seamless genome editing without double-stranded DNA cutting. Nat. Commun. 2017, 8, 657. [Google Scholar] [CrossRef] [Green Version]
- Morbitzer, R.; Elsaesser, J.; Hausner, J.; Lahaye, T. Assembly of custom TALE-type DNA binding domains by modular cloning. Nucleic Acids Res. 2011, 39, 5790–5799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Ueno, M.; Kamiya, D.; Nishiyama, A.; Matsumura, M.; Wataya, T.; Takahashi, J.B.; Nishikawa, S.; Nishikawa, S.; Muguruma, K.; et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 2007, 25, 681–686. [Google Scholar] [CrossRef]
- Gauthaman, K.; Fong, C.Y.; Bongso, A. Effect of ROCK Inhibitor Y-27632 on Normal and Variant Human Embryonic Stem Cells (hESCs) In Vitro: Its Benefits in hESC Expansion. Stem Cell Rev. Rep. 2010, 6, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Guschin, D.Y.; Waite, A.J.; Katibah, G.E.; Miller, J.C.; Holmes, M.C.; Rebar, E.J. A rapid and general assay for monitoring endogenous gene modification. Methods Mol. Biol. 2010, 649, 247–256. [Google Scholar]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Transfection Method | Nuclease | Integrated Selection Cassette | FACS | Number of Screened Clones | Efficiency | Time Required | References and Comments |
|---|---|---|---|---|---|---|---|---|
| iPSCs | N | reTALENs, CRISPR-Cas9 | No | Yes | 100 | 0.6–1.7% | 3 weeks | Clones reported using re-TALENs [28] |
| Only heterozygous clones | ||||||||
| hESCs | E | TALEN | No | Yes | 192 | 1.60% | Less than 1 month | Ding et al. [6] |
| iPSCs | N | CRISPR-Cas9 | eGFP or dTOMATO | Yes | 24 | 1st transfection (2.2–3.8%) + 2nd transfection (2.2–6.5%) | More than 2 weeks, not specified | Clones derived from correctly modified ‘polyclones’ previously identified by FACS [29] |
| hESCs | N | ZFN, TALEN | Puromycin | No | 24 | Not specified, but two rounds of transfection | ~3 months | Only heterozygous clones [22] |
| iPSCs | N | CRISPR-Cas9 | eGFP-puromycin | No | 36 | 1st transfection (16.7%) + 2nd transfection (1–88.1%) | Not specified, but 2 rounds of colony isolation and screening | Firth et al. [24] |
| hESCs | E | ZFN, TALEN, CRISPR-Cas9 | Puromycin-eGFP | No | 150–412 | 87–96% of eGFP+ clones | 20–24 days to identify integrated selection cassette | Only PCR verified modifications [25] |
| 2 rounds of transfection | ||||||||
| iPSCs | N | TALEN | No | No | 30 | 20% after 6 enrichment cycles | Not specified, but 6 enrichment cycles from 1st observation of correction (day 9 after transfection) to isolation of clones | Each cycle consisted of cell dissociation, seeding as clumps and PCR screening [26] |
| hESCs, iPSCs | E | CRISPR-Cas9 | No | Yes | 1st isolation (96–192) + 2nd isolation (384–96) | 1st transfection (2.9–9.6%) + 2nd transfection (7.7–15.4%) | ~3 months | 2 rounds of transfection, to remove the introduced mutations in the PAM [65] |
| hESCs, iPSCs | N, FuGene HD transfection | CRISPR-Cas9 | Puromycin | No | 1st isolation (11) or 2nd isolation (12) | 1st transfection (18.2%) or 2nd transfection (41.6%) | 35 days, but 2 rounds of transfection | The authors suggest performing only the 2nd clonal screening [27] |
| hESCs, iPSCs | N | TALEN, CRISPR-Cas9 | No | No | 24–238 | 1–10% | ~3–6 weeks | The method described here |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuevas-Ocaña, S.; Yang, J.Y.; Aushev, M.; Schlossmacher, G.; Bear, C.E.; Hannan, N.R.F.; Perkins, N.D.; Rossant, J.; Wong, A.P.; Gray, M.A. A Cell-Based Optimised Approach for Rapid and Efficient Gene Editing of Human Pluripotent Stem Cells. Int. J. Mol. Sci. 2023, 24, 10266. https://doi.org/10.3390/ijms241210266
Cuevas-Ocaña S, Yang JY, Aushev M, Schlossmacher G, Bear CE, Hannan NRF, Perkins ND, Rossant J, Wong AP, Gray MA. A Cell-Based Optimised Approach for Rapid and Efficient Gene Editing of Human Pluripotent Stem Cells. International Journal of Molecular Sciences. 2023; 24(12):10266. https://doi.org/10.3390/ijms241210266
Chicago/Turabian StyleCuevas-Ocaña, Sara, Jin Ye Yang, Magomet Aushev, George Schlossmacher, Christine E. Bear, Nicholas R. F. Hannan, Neil D. Perkins, Janet Rossant, Amy P. Wong, and Michael A. Gray. 2023. "A Cell-Based Optimised Approach for Rapid and Efficient Gene Editing of Human Pluripotent Stem Cells" International Journal of Molecular Sciences 24, no. 12: 10266. https://doi.org/10.3390/ijms241210266