
Synaptamide Ameliorates Hippocampal Neurodegeneration and Glial Activation in Mice with Traumatic Brain Injury
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
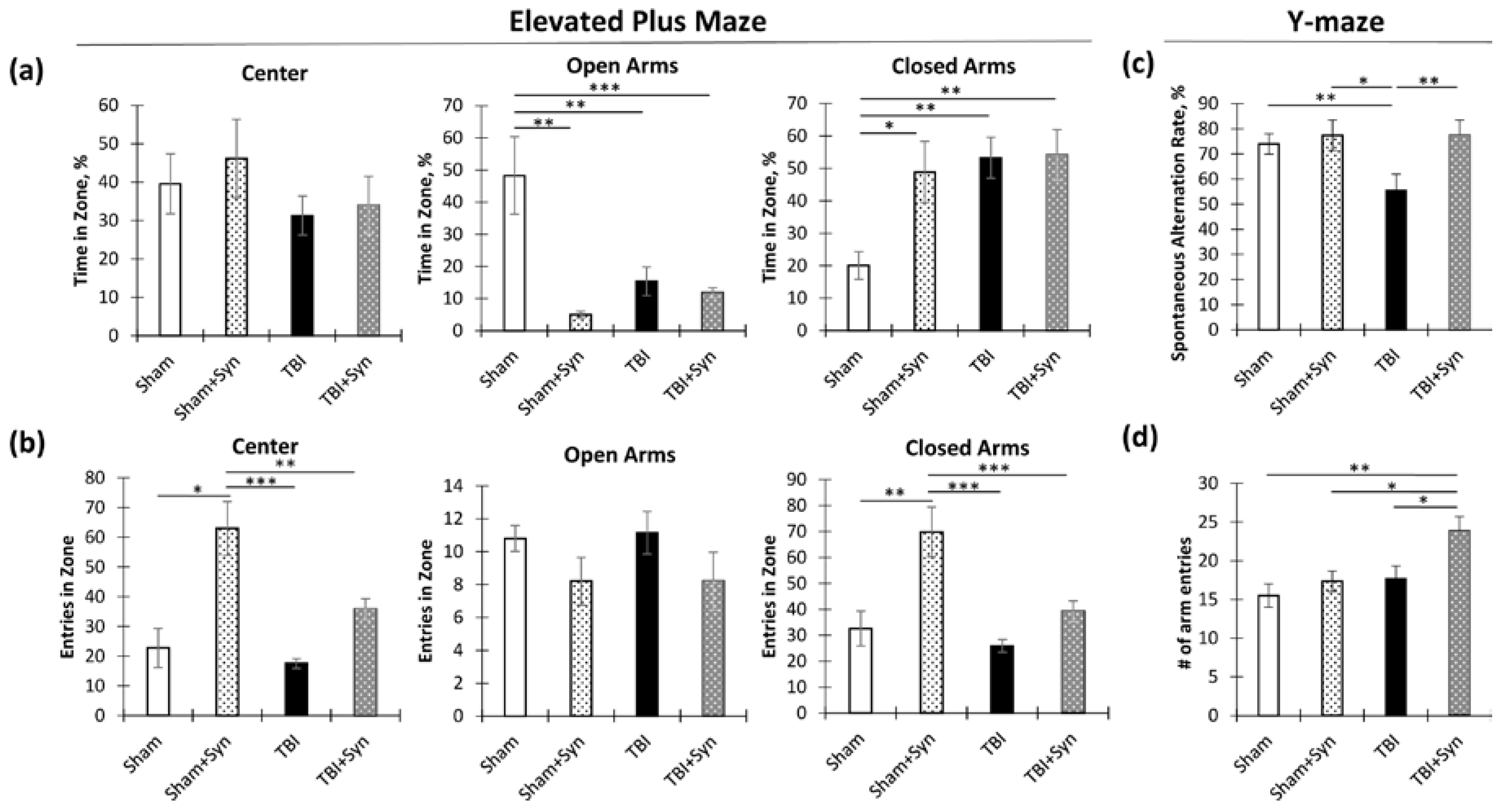
2.1. The Impact of Synaptamide Treatment on Behavioral Changes in TBI
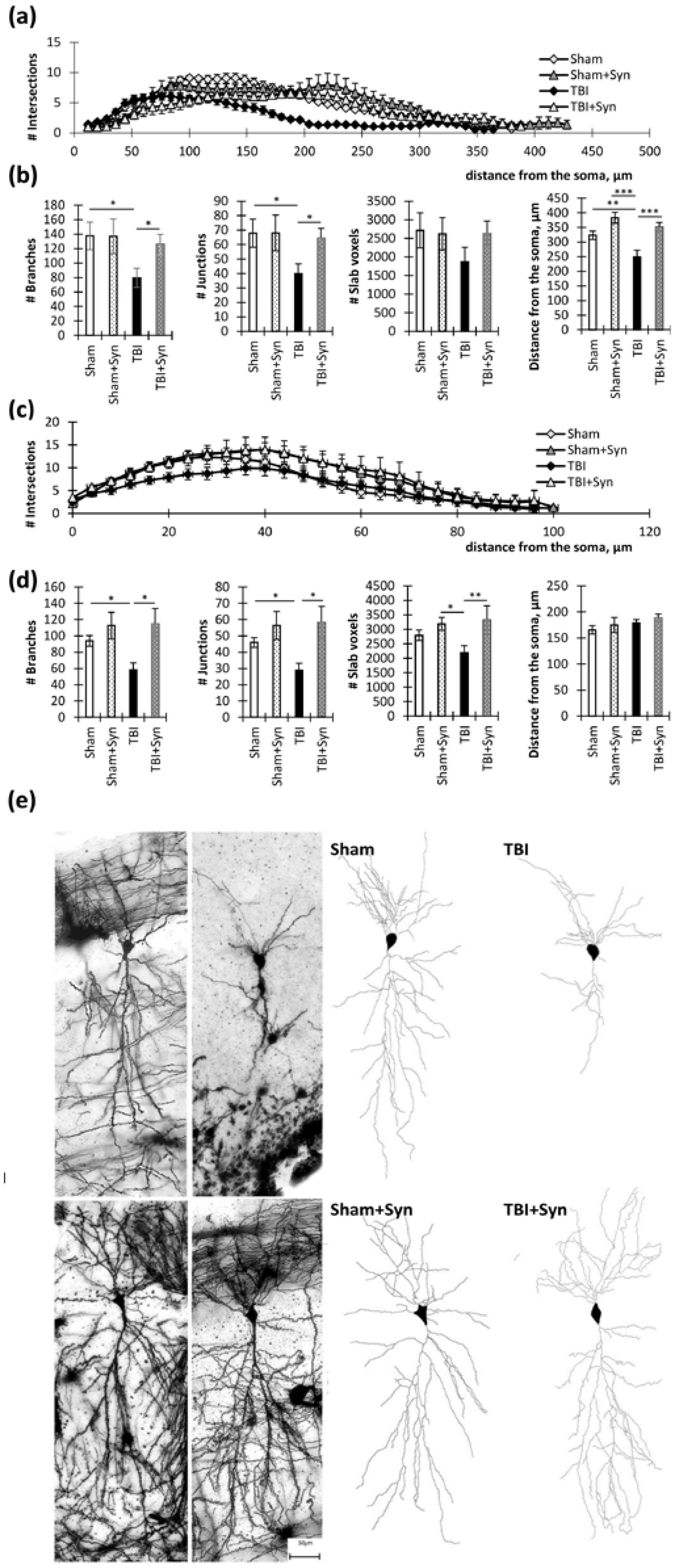
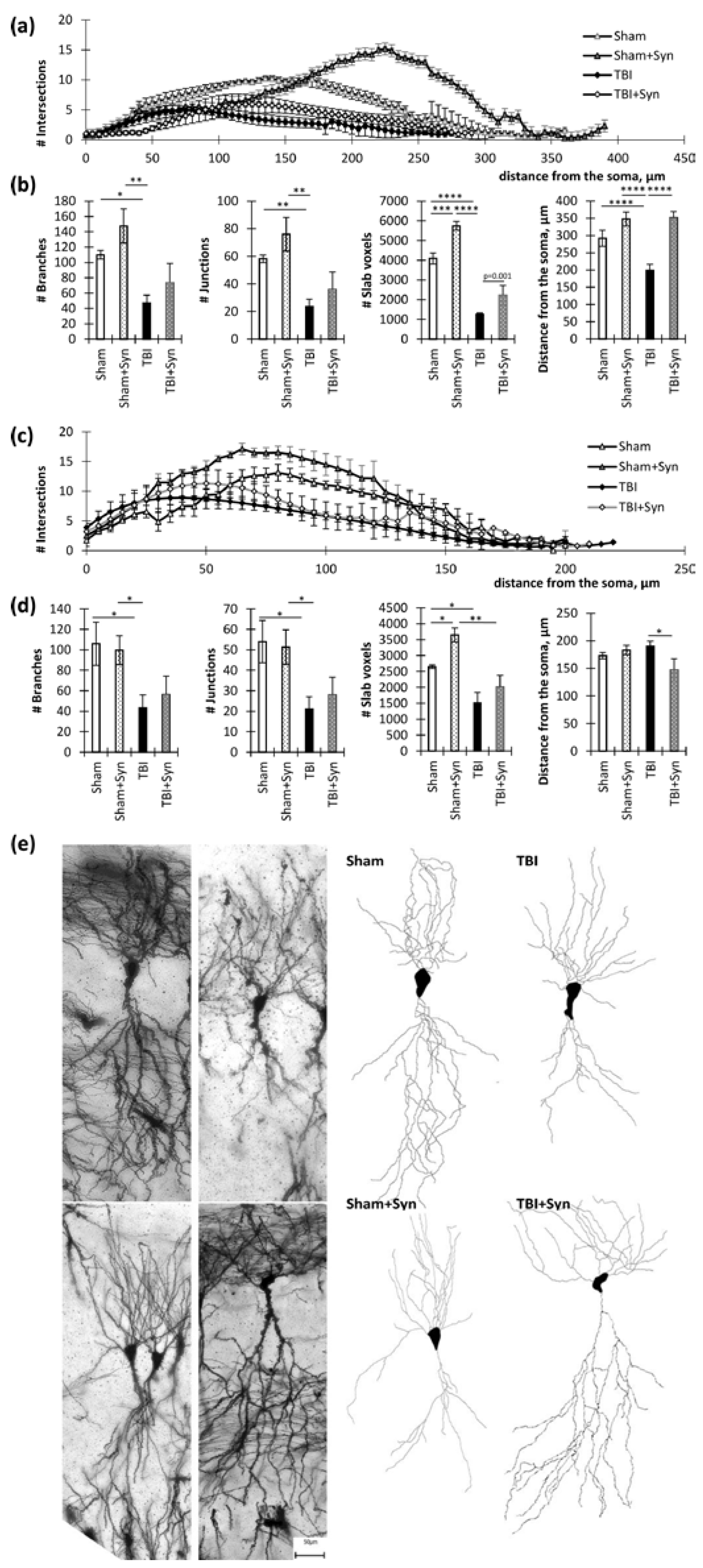
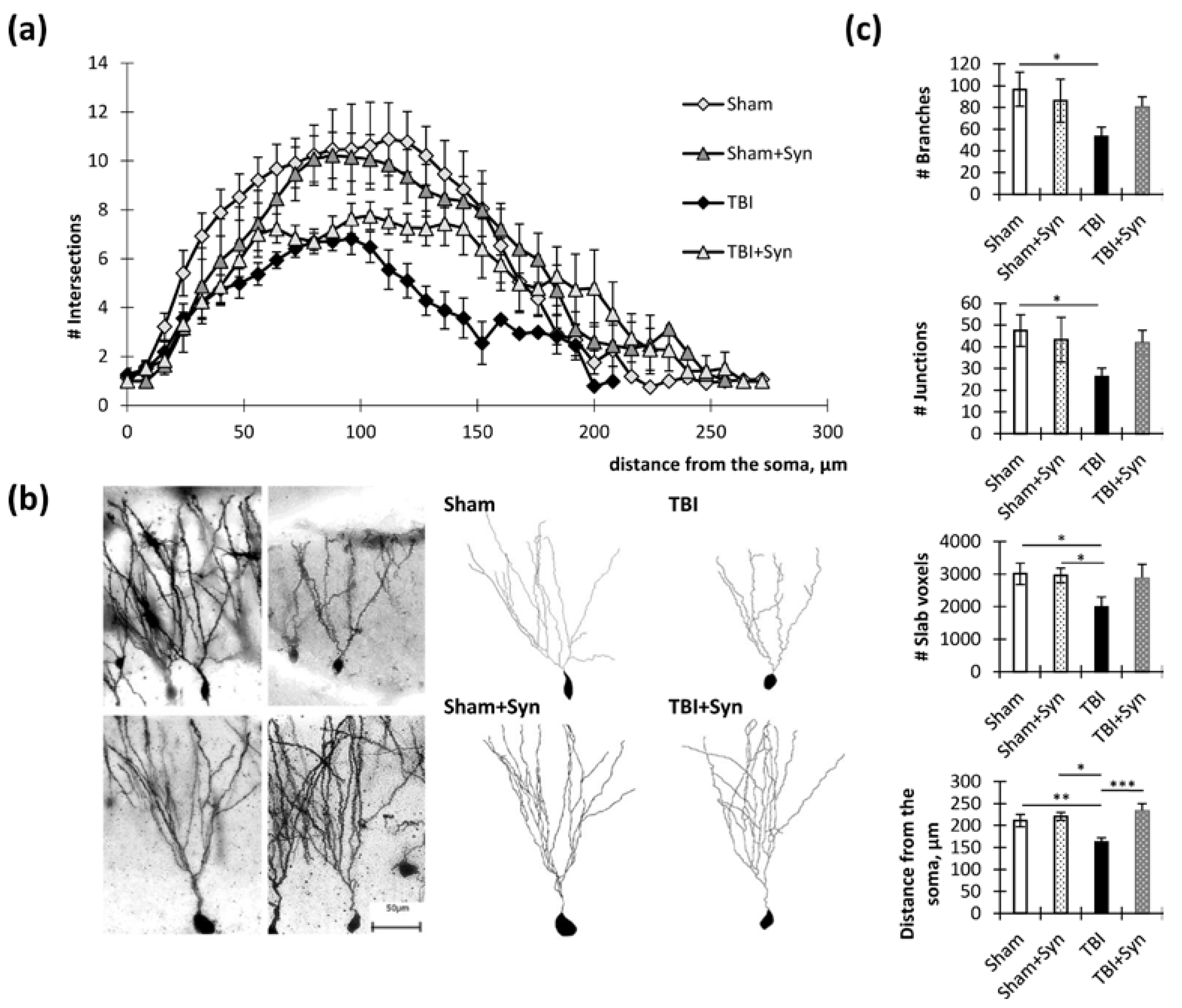
2.2. Influence of TBI and Synaptamide Treatment on the Morphology of Hippocampal Neurons
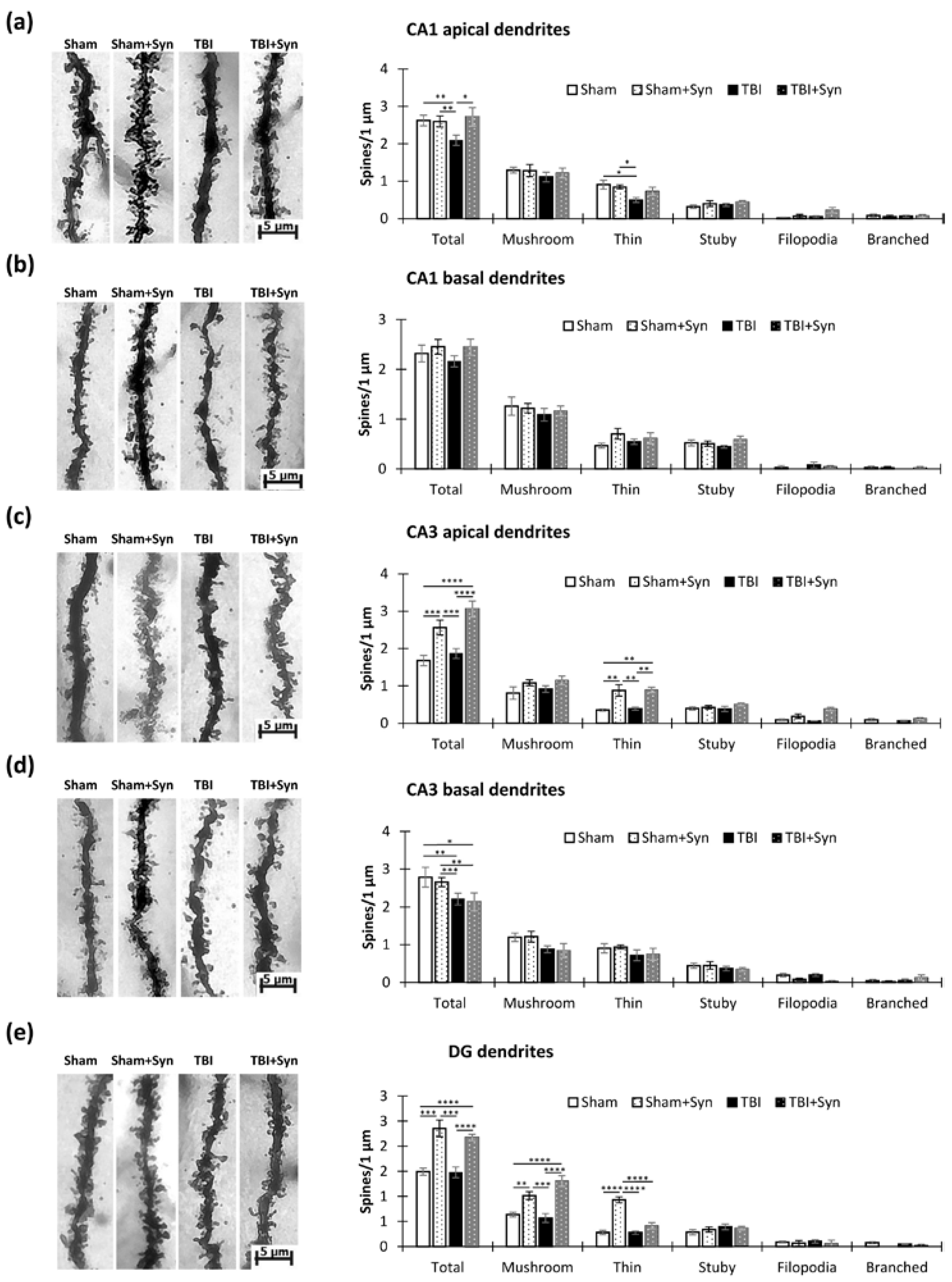
2.3. The Impact of Trauma and Treatment on Dendritic Spine Morphology
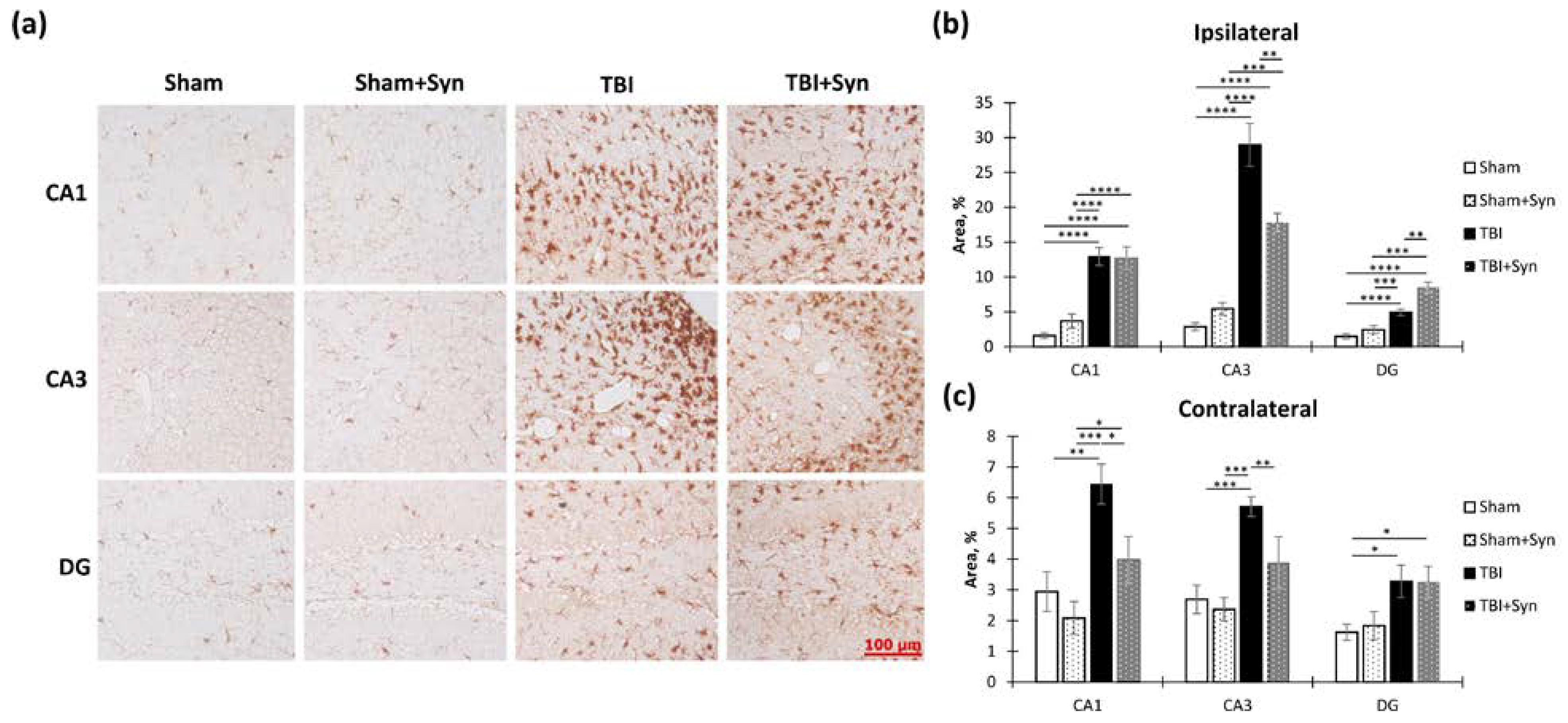
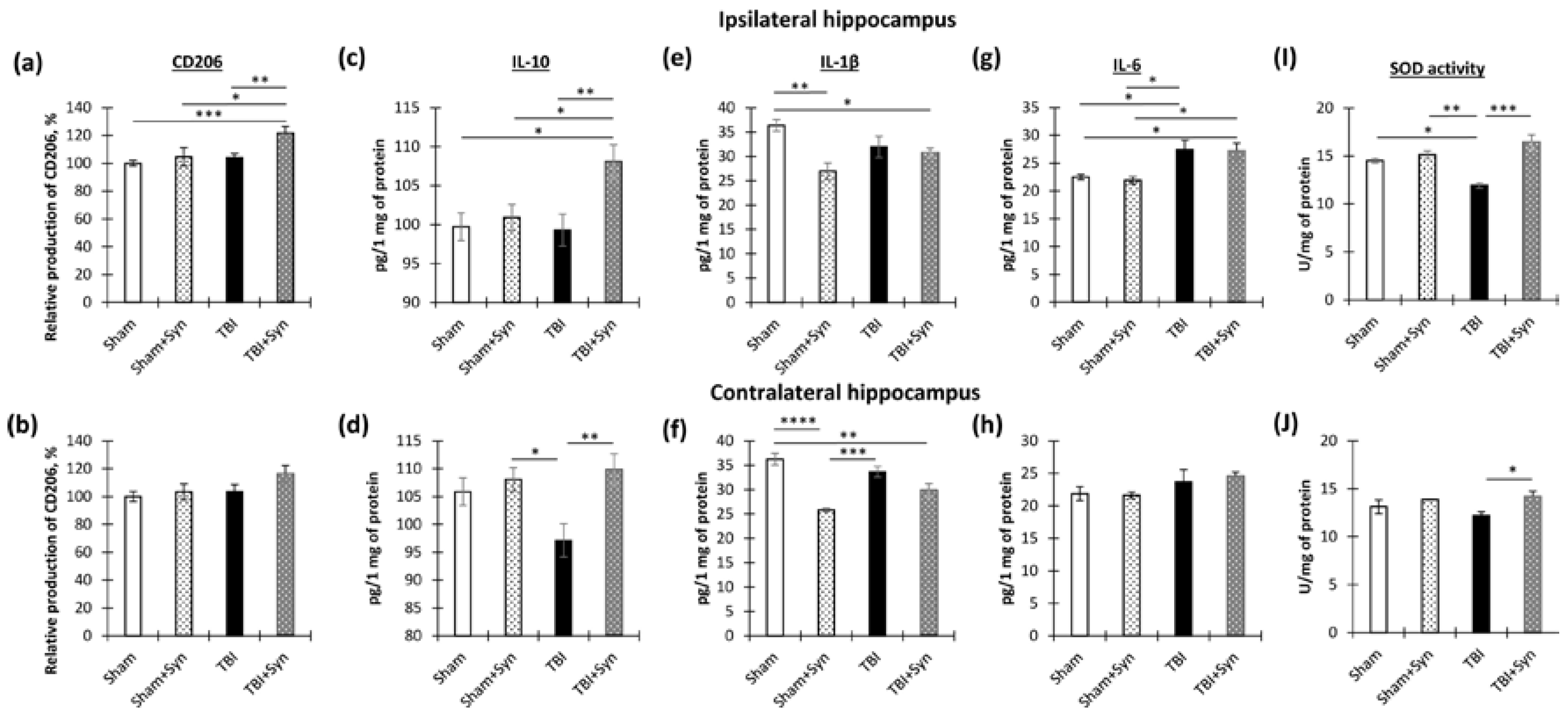
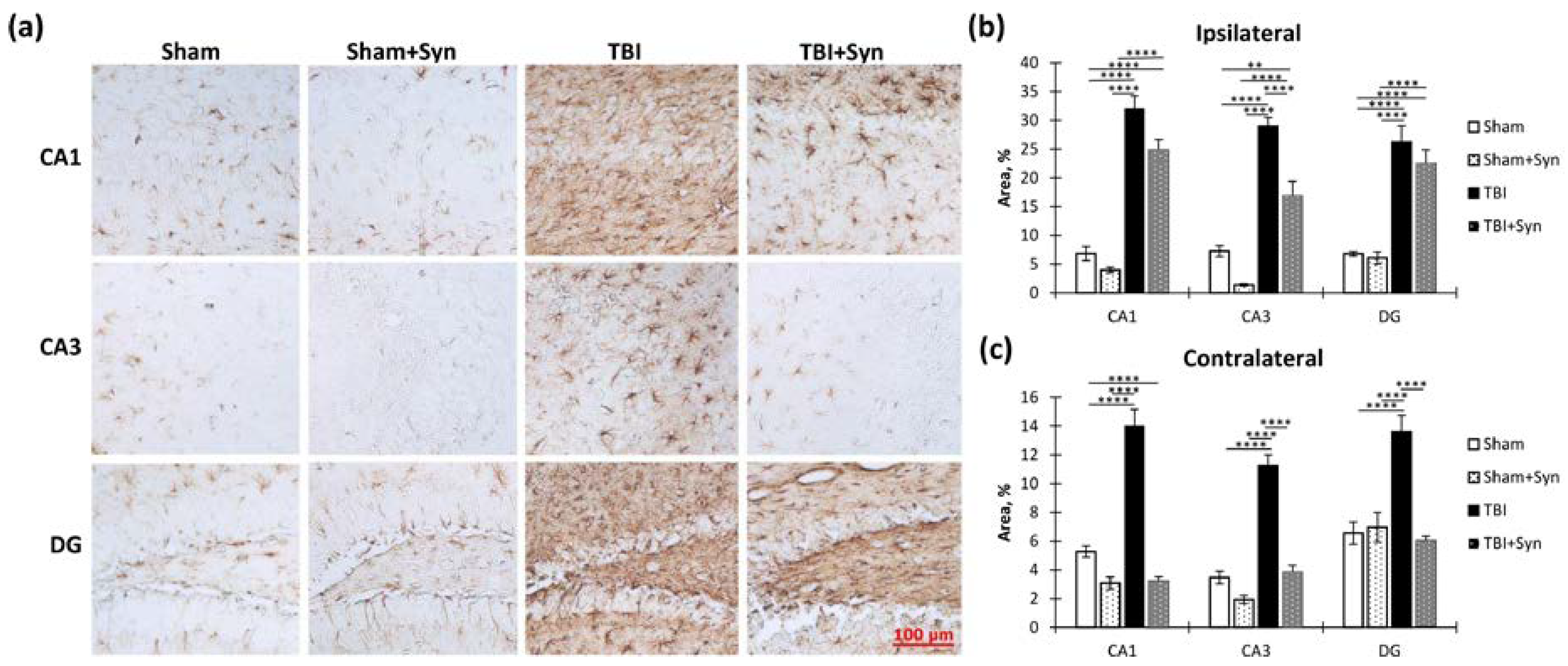
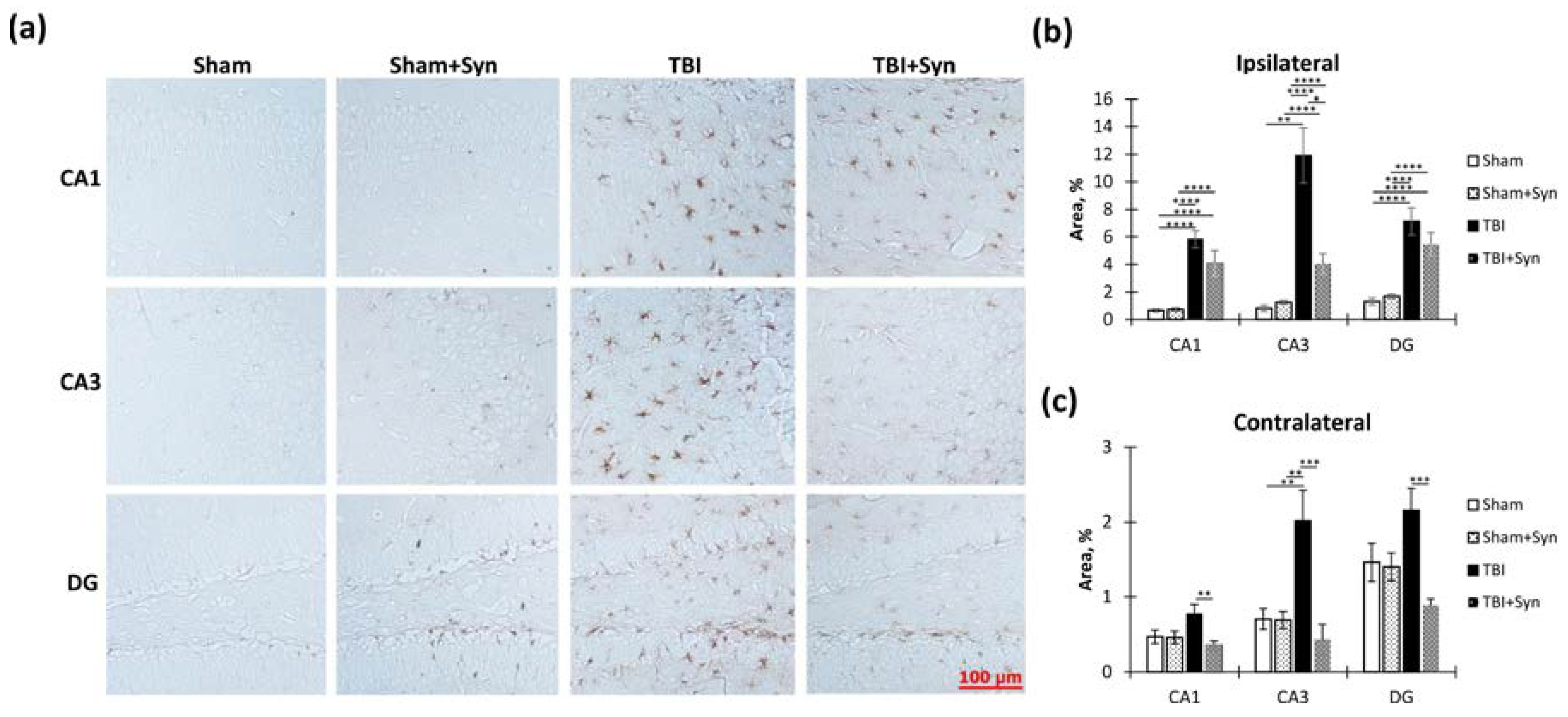
2.4. The Effect of TBI and Synaptamide Treatment on the State of Hippocampal Microglia and the Production of Pro- and Anti-Inflammatory Factors
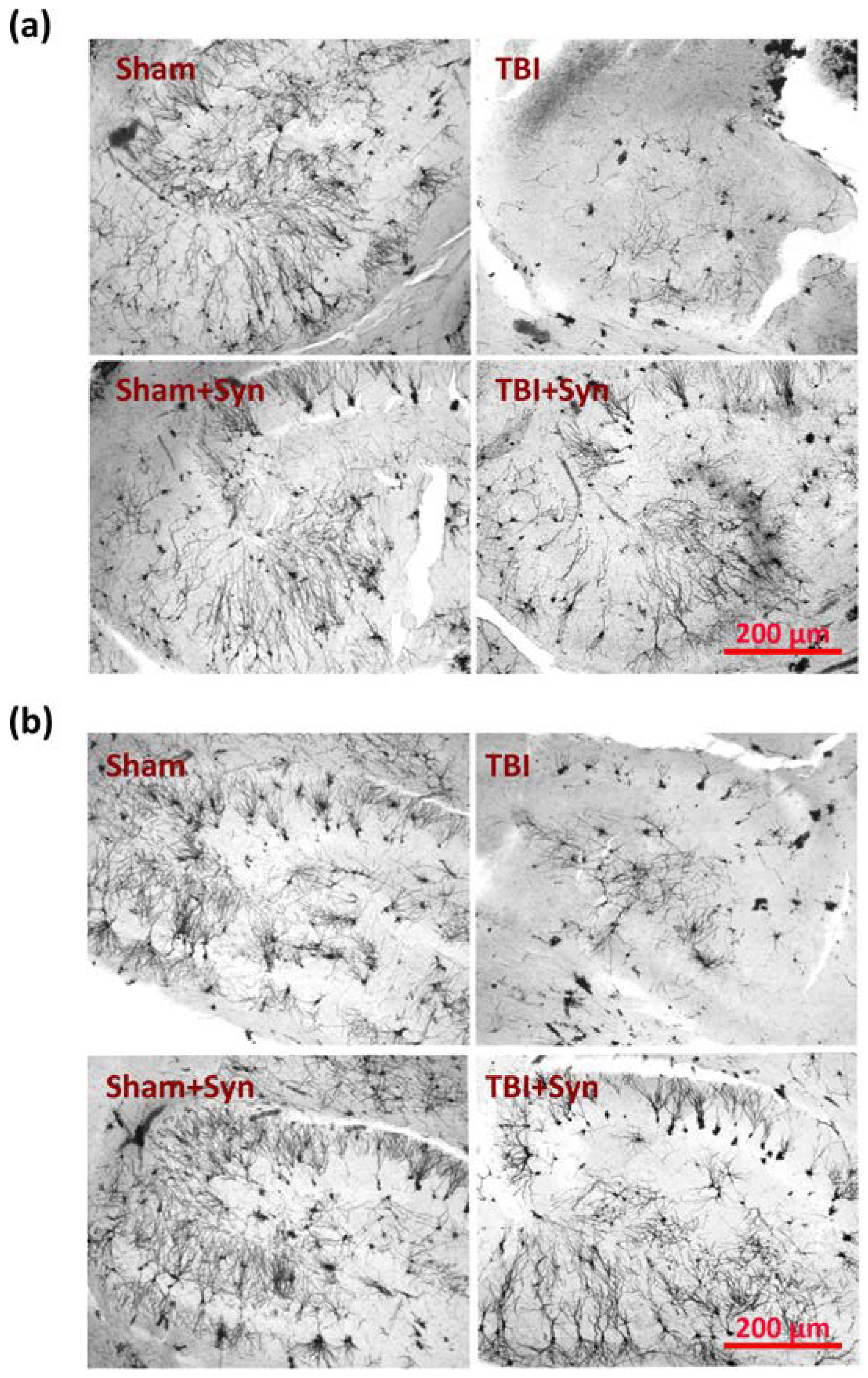
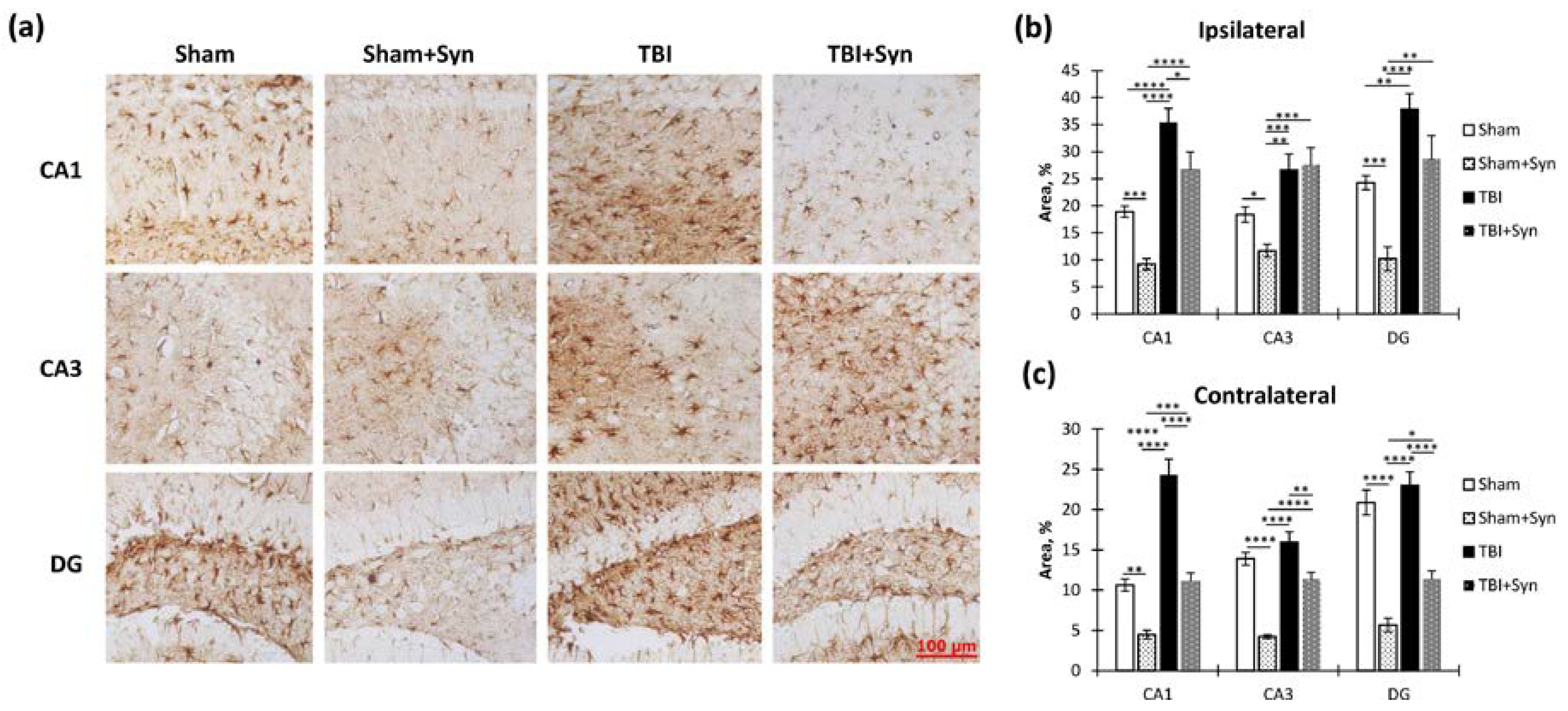
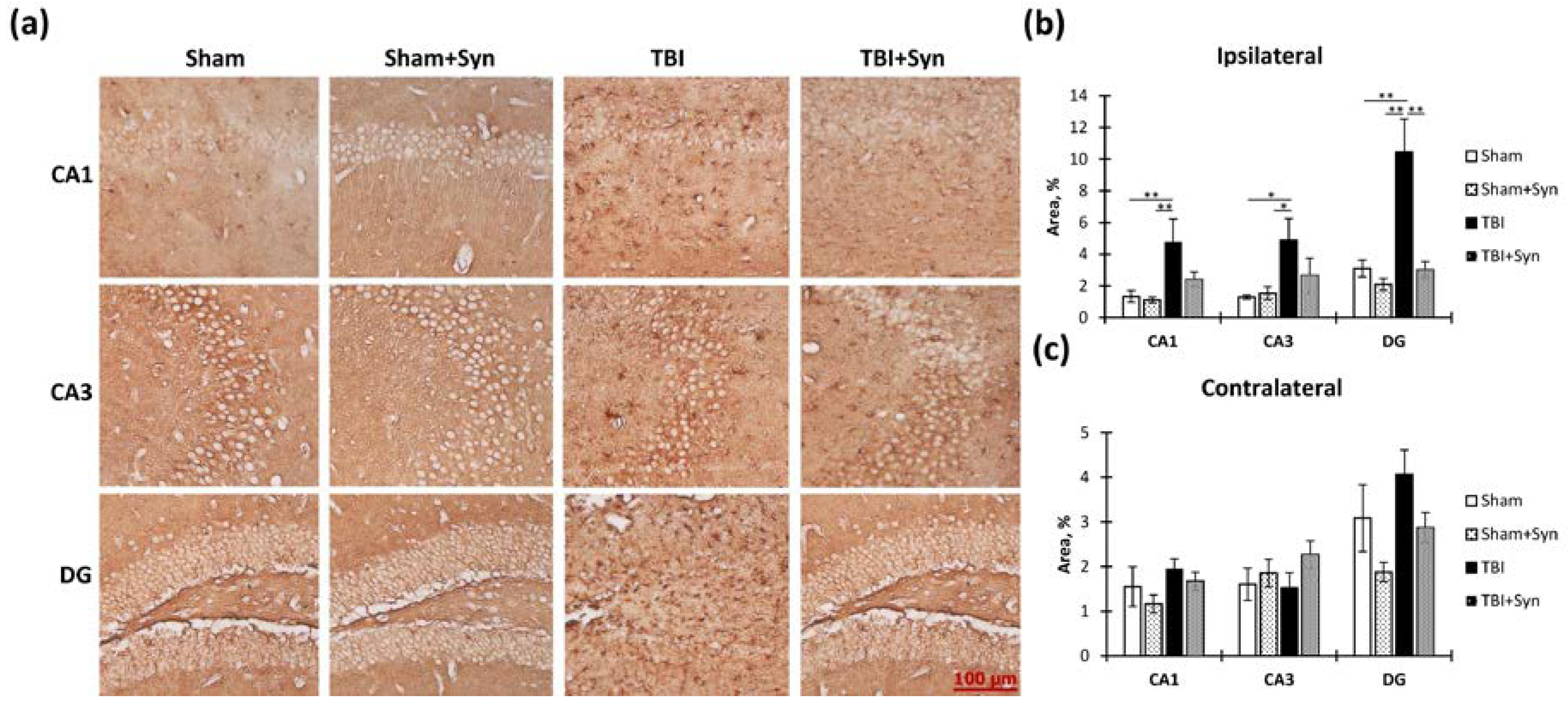
2.5. The Effect of TBI and Synaptamide Treatment on the State of Hippocampal Astroglia
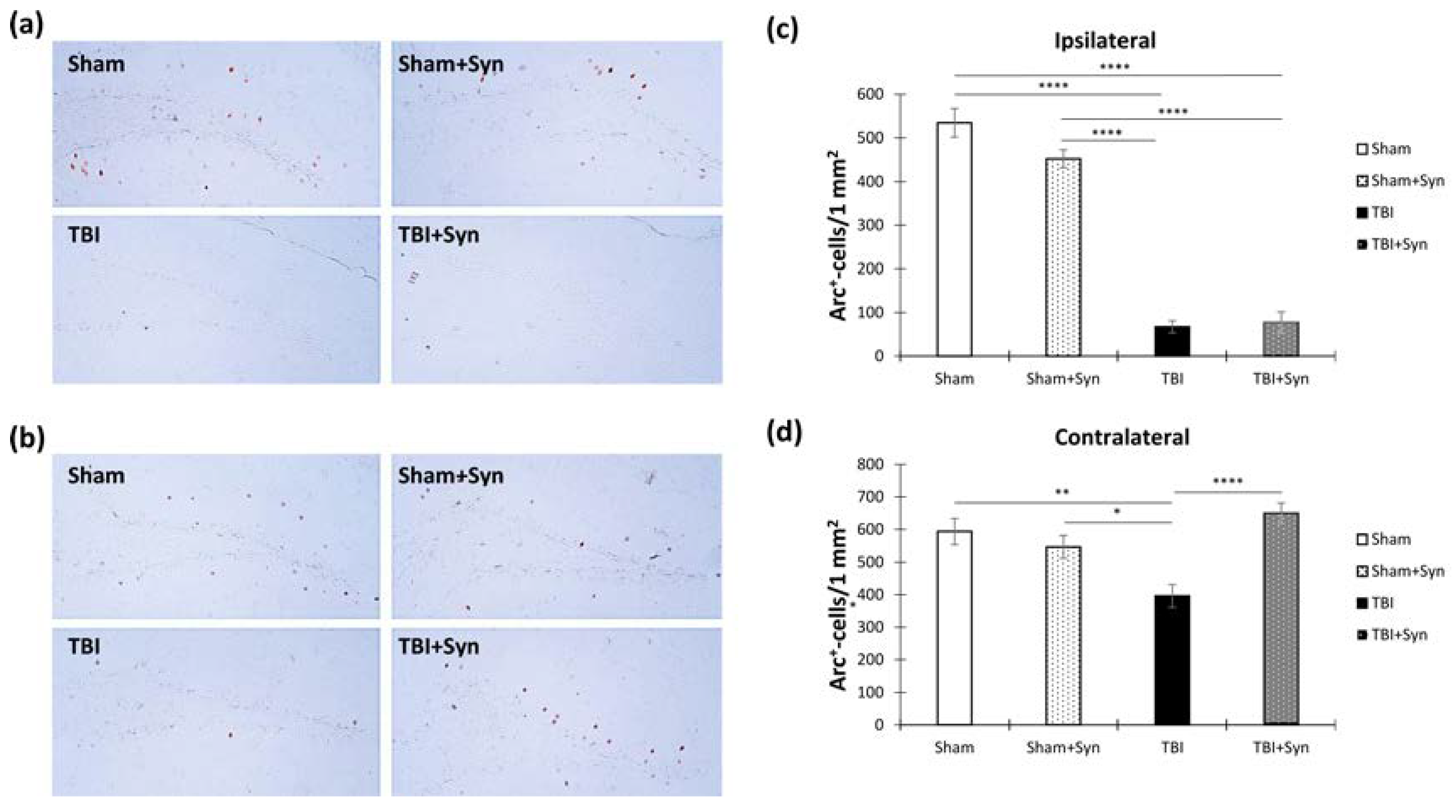
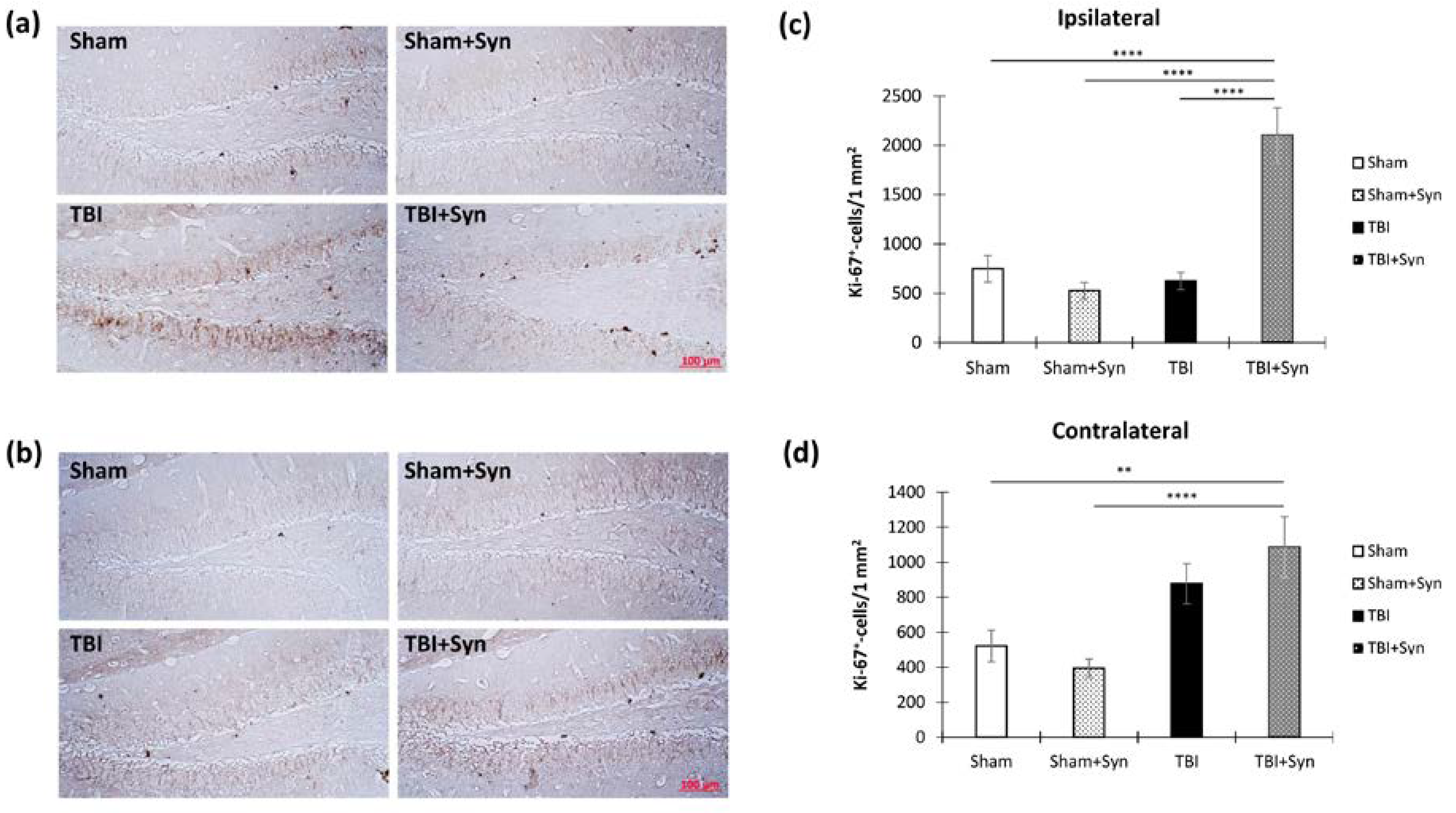
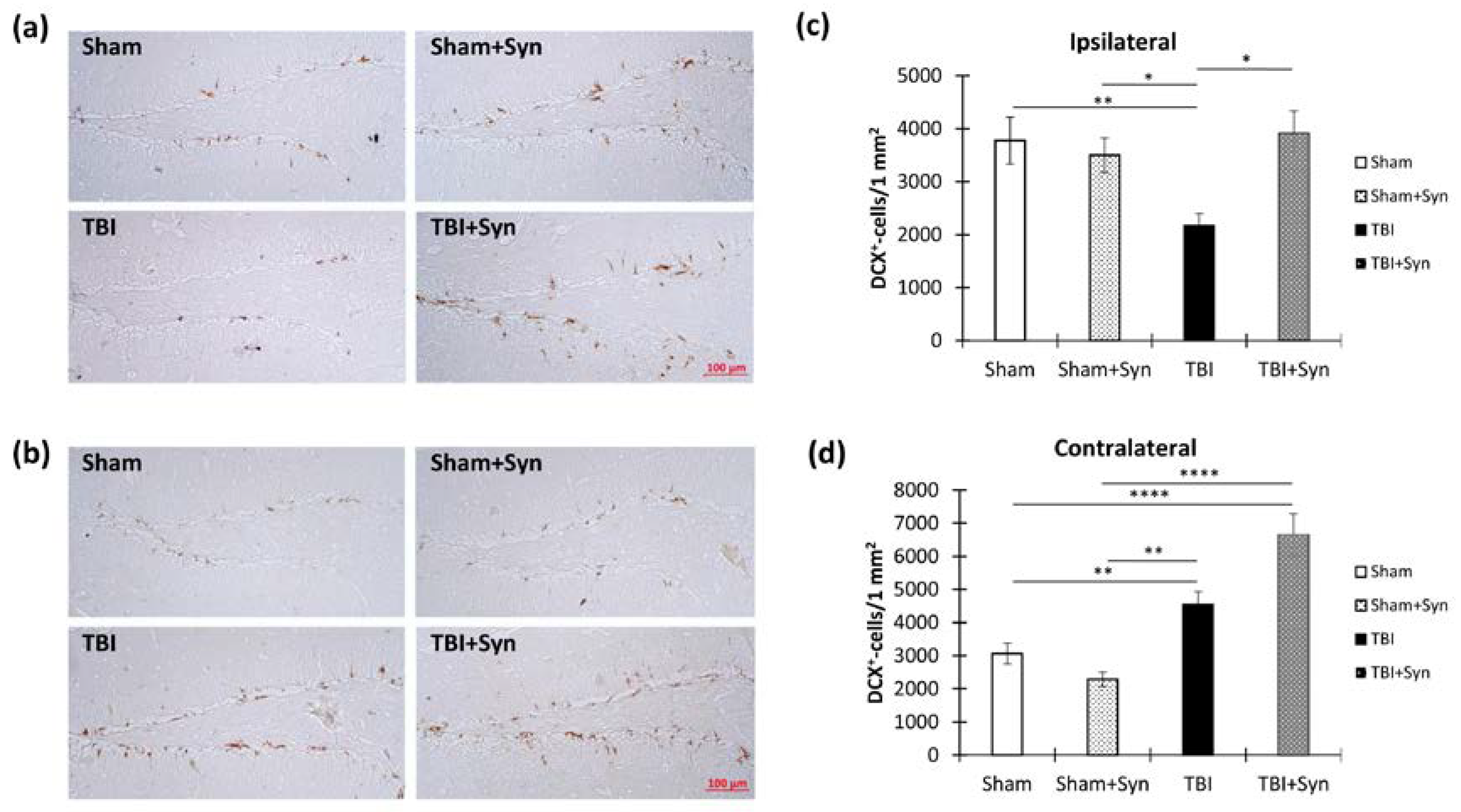
2.6. TBI and Synaptamide Effect on Hippocampal Arc-Protein Production and Adult Neurogenesis
2.7. Anti-Apoptotic Activities of Synaptamide in TBI
3. Discussion
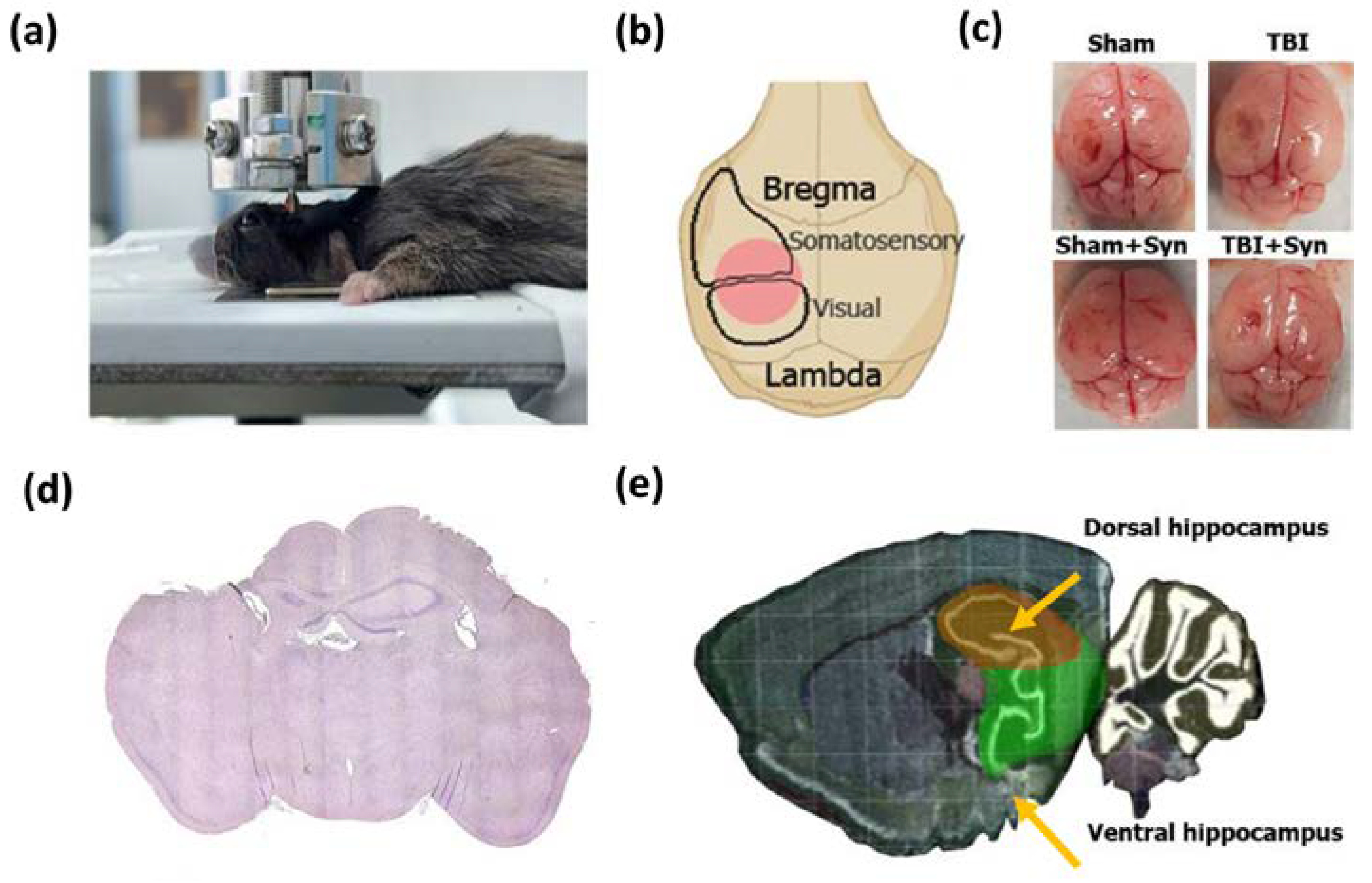
4. Materials and Methods
4.1. Animals
4.2. Surgery and Treatment
4.3. Behavioral Studies
4.3.1. Elevated plus Maze
4.3.2. Y-Maze
4.4. Golgi–Cox Staining
4.5. Sholl Analysis
4.6. Immunohistochemical Studies
4.7. ELISA
4.8. Evaluation of Superoxide Dismutase Activity
4.9. Statistical Analysis
5. Conclusions
6. Limitations
- The design of the study allowed us to study the effects of synaptamide in TBI in the short term, but did not address the long-term consequences.
- In the study of the production of pro- and anti-inflammatory microglia markers, we used mixed cell populations (whole-hippocampus homogenate) rather than isolated microglia to specifically study transcriptomic factors.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maas, A.I.R.; Menon, D.K.; Manley, G.T.; Abrams, M.; Åkerlund, C.; Andelic, N.; Aries, M.; Bashford, T.; Bell, M.J.; Bodien, Y.G.; et al. InTBIR Participants and Investigators. Traumatic brain injury: Progress and challenges in prevention, clinical care, and research. Lancet Neurol. 2022, 21, 1004–1060. [Google Scholar] [CrossRef] [PubMed]
- Voormolen, D.C.; Haagsma, J.A.; Polinder, S.; Maas, A.I.R.; Steyerberg, E.W.; Vuleković, P.; Sewalt, C.A.; Gravesteijn, B.Y.; Covic, A.; Andelic, N.; et al. Post-concussion symptoms in complicated vs. uncomplicated mild traumatic brain injury patients at three and six months post-injury: Results from the CENTER-TBI study. J. Clin. Med. 2019, 8, 1921. [Google Scholar] [CrossRef] [Green Version]
- Shirazi, R.S.; Vyssotski, M.; Lagutin, K.; Thompson, D.; MacDonald, C.; Luscombe, V.; Glass, M.; Parker, K.; Gowing, E.K.; Williams, D.B.G.; et al. Neuroprotective activity of new Δ3-N-acylethanolamines in a focal ischemia stroke model. Lipids 2022, 57, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Ignatowska-Jankowska, B.M.; Baillie, G.L.; Kinsey, S.; Crowe, M.; Ghosh, S.; Owens, R.A.; Damaj, I.M.; Poklis, J.; Wiley, J.L.; Zanda, M.; et al. A cannabinoid CB1 receptor-positive allosteric modulator reduces neuropathic pain in the mouse with no psychoactive effects. Neuropsychopharmacology 2015, 40, 2948–2959. [Google Scholar] [CrossRef] [Green Version]
- Tsuboi, K.; Uyama, T.; Okamoto, Y.; Ueda, N. Endocannabinoids and related N-acyl ethanolamines: Biological activities and metabolism. Inflamm. Regen. 2018, 38, 28. [Google Scholar] [CrossRef]
- Katz, P.S.; Sulzer, J.K.; Impastato, R.A.; Teng, S.X.; Rogers, E.K.; Molina, P.E. Endocannabinoid degradation inhibition improves neurobehavioral function, blood-brain barrier integrity, and neuroinflammation following mild traumatic brain injury. J. Neurotrauma 2015, 32, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Spector, A.A. N-Docosahexaenoylethanolamine: A neurotrophic and neuroprotective metabolite of docosahexaenoic acid. Mol. Asp. Med. 2018, 64, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kevala, K.; Aflaki, E.; Marugan, J.; Kim, H.Y. GPR110 ligands reduce chronic optic tract gliosis and visual deficit following repetitive mild traumatic brain injury in mice. J. Neuroinflamm. 2021, 18, 157. [Google Scholar] [CrossRef]
- Bertagna, N.B.; Dos Santos, P.G.C.; Queiroz, R.M.; Fernandes, G.J.D.; Cruz, F.C.; Miguel, T.T. Involvement of the ventral, but not dorsal, hippocampus in anxiety-like behaviors in mice exposed to the elevated plus maze: Participation of CRF1 receptor and PKA pathway. Pharmacol. Rep. 2021, 73, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri, S.; Fathollahi, Y.; Javan, M.; Shojaei, A.; Asgari, A.; Mirnajafi-Zadeh, J. Effect of low-frequency stimulation on impaired spontaneous alternation behavior of kindled rats in Y-maze test. Epilepsy Res. 2016, 126, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Jin, J.L.; Ge, H.M.; Yin, K.L.; Chen, X.; Han, L.J.; Chen, Y.; Qian, L.; Li, X.X.; Xu, Y. Malibatol A regulates microglia M1/M2 polarization in experimental stroke in a PPARγ-dependent manner. J. Neuroinflamm. 2015, 12, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladak, A.A.; Enam, S.A.; Ibrahim, M.T. A review of the molecular mechanisms of traumatic brain injury. World Neurosurg. 2019, 131, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Lebkuechner, I.; Wilhelmsson, U.; Möllerström, E.; Pekna, M.; Pekny, M. Heterogeneity of Notch signaling in astrocytes and the effects of GFAP and vimentin deficiency. J. Neurochem. 2015, 135, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Abdelhak, A.; Foschi, M.; Abu-Rumeileh, S.; Yue, J.K.; D’Anna, L.; Huss, A.; Oeckl, P.; Ludolph, A.C.; Kuhle, J.; Petzold, A.; et al. Blood GFAP as an emerging biomarker in brain and spinal cord disorders. Nat. Rev. Neurol. 2022, 18, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.J.; Davies, H.A.; Silva, A.T.; De Souza, I.E.; Peddie, C.J.; Colyer, F.M.; Lancashire, C.L.; Fine, A.; Errington, M.L.; Bliss, T.V.; et al. Long-term potentiation in the rat dentate gyrus is associated with enhanced Arc/Arg3.1 protein expression in spines, dendrites and glia. Eur. J. Neurosci. 2005, 21, 2384–2396. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G. Adult Neurogenesis: Stem Cells and Neuronal Development in the Adult Brain; Oxford University Press: New York, NY, USA, 2006. [Google Scholar]
- Semënov, M.V. Adult hippocampal neurogenesis is a developmental process involved in cognitive development. Front. Neurosci. 2019, 13, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redell, J.B.; Maynard, M.E.; Underwood, E.L.; Vita, S.M.; Dash, P.K.; Kobori, N. Traumatic brain injury and hippocampal neurogenesis: Functional implications. Exp. Neurol. 2020, 331, 113372. [Google Scholar] [CrossRef]
- Lozano, D.; Gonzales-Portillo, G.S.; Acosta, S.; de la Pena, I.; Tajiri, N.; Kaneko, Y.; Borlongan, C.V. Neuroinflammatory responses to traumatic brain injury: Etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr. Dis. Treat. 2015, 11, 97–106. [Google Scholar]
- Wang, X.; Gao, X.; Michalski, S.; Zhao, S.; Chen, J. Traumatic brain injury severity affects neurogenesis in adult mouse hippocampus. J. Neurotrauma 2016, 33, 721–733. [Google Scholar] [CrossRef]
- Iljazi, A.; Ashina, H.; Al-Khazali, H.M.; Lipton, R.B.; Ashina, M.; Schytz, H.W.; Ashina, S. Post-traumatic stress disorder after traumatic brain injury—A systematic review and meta-analysis. Neurol. Sci. 2020, 41, 2737–2746. [Google Scholar] [CrossRef]
- Wohleb, E.S. Neuron–microglia interactions in mental health disorders: “for better, and for worse”. Front. Immunol. 2016, 7, 544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacialli, P.; Palladino, A.; Lucini, C. Role of brain-derived neurotrophic factor during the regenerative response after traumatic brain injury in adult zebrafish. Neural Regen. Res. 2018, 13, 941–944. [Google Scholar] [CrossRef]
- Ricci, G.; Volpi, L.; Pasquali, L.; Petrozzi, L.; Siciliano, G. Astrocyte-neuron interactions in neurological disorders. J. Biol. Phys. 2009, 35, 317–336. [Google Scholar] [CrossRef] [Green Version]
- Park, T.; Chen, H.; Kim, H.Y. GPR110 (ADGRF1) mediates anti-inflammatory effects of N-docosahexaenoylethanolamine. J. Neuroinflamm. 2019, 16, 225. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Spector, A.A. Synaptamide, an endocannabinoid-like derivative of docosahexaenoic acid with cannabinoid-independent function. Prostaglandins Leukot. Essent. Fatty Acids 2013, 88, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Starinets, A.; Tyrtyshnaia, A.; Manzhulo, I. Anti-Inflammatory Activity of Synaptamide in the Peripheral Nervous System in a Model of Sciatic Nerve Injury. Int. J. Mol. Sci. 2023, 24, 6273. [Google Scholar] [CrossRef] [PubMed]
- Paton, K.F.; Shirazi, R.; Vyssotski, M.; Kivell, B.M. N-docosahexaenoyl ethanolamine (synaptamide) has antinociceptive effects in male mice. Eur. J. Pain 2020, 24, 1990–1998. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Spector, A.A.; Xiong, Z.M. A synaptogenic amide N-docosahexaenoylethanolamide promotes hippocampal development. Prostaglandins Other Lipid Mediat. 2011, 96, 114–120. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Huang, B.X.; Spector, A.A. Molecular and Signaling Mechanisms for Docosahexaenoic Acid-Derived Neurodevelopment and Neuroprotection. Int. J. Mol. Sci. 2022, 23, 4635. [Google Scholar] [CrossRef]
- Morganti-Kossmann, M.C.; Semple, B.D.; Hellewell, S.C.; Bye, N.; Ziebell, J.M. The complexity of neuroinflammation consequent to traumatic brain injury: From research evidence to potential treatments. Acta Neuropathol. 2019, 137, 731–755. [Google Scholar] [CrossRef]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziebell, J.M.; Morganti-Kossmann, M.C. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 2010, 7, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Zhang, J.; Hu, X.; Zhang, L.; Mao, L.; Jiang, X.; Liou, A.K.; Leak, R.K.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J. Cereb. Blood Flow Metab. 2013, 33, 1864–1874. [Google Scholar] [CrossRef] [Green Version]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial activation in traumatic brain injury. Front. Aging Neurosci. 2017, 9, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Italiani, P.; Boraschi, D. From monocytes to M1/M2 macrophages: Phenotypical vs. functional differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Shao, A.; Yao, Y.; Tu, S.; Deng, Y.; Zhang, J. Dual roles of astrocytes in plasticity and reconstruction after traumatic brain injury. Cell Commun. Signal. 2020, 18, 62. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Song, G.; Chuang, H.; Chiu, C.; Abdelmaksoud, A.; Ye, Y.; Zhao, L. Portrait of glial scar in neurological diseases. Int. J. Immunopathol. Pharmacol. 2018, 31, 2058738418801406. [Google Scholar] [CrossRef] [Green Version]
- Ekmark-Lewén, S.; Lewén, A.; Israelsson, C.; Li, G.L.; Farooque, M.; Olsson, Y.; Ebendal, T.; Hillered, L. Vimentin and GFAP responses in astrocytes after contusion trauma to the murine brain. Restor. Neurol. Neurosci. 2010, 28, 311–321. [Google Scholar] [CrossRef]
- Sarkis, G.A.; Lees-Gayed, N.; Banoub, J.; Abbatielo, S.E.; Robertson, C.; Haskins, W.E.; Yost, R.A.; Wang, K.K.W. Generation and release of neurogranin, vimentin, and MBP proteolytic peptides, following traumatic brain injury. Mol. Neurobiol. 2022, 59, 731–747. [Google Scholar] [CrossRef]
- van Landeghem, F.K.; Maier-Hauff, K.; Jordan, A.; Hoffmann, K.T.; Gneveckow, U.; Scholz, R.; Thiesen, B.; Brück, W.; von Deimling, A. Post-mortem studies in glioblastoma patients treated with thermotherapy using magnetic nanoparticles. Biomaterials 2009, 30, 52–57. [Google Scholar] [CrossRef]
- Song, H.J.; Stevens, C.F.; Gage, F.H. Neural stem cells from adult hippocampus develop essential properties of functional CNS neurons. Nat. Neurosci. 2002, 5, 438–445. [Google Scholar] [CrossRef]
- Wilhelmsson, U.; Li, L.; Pekna, M.; Berthold, C.H.; Blom, S.; Eliasson, C.; Renner, O.; Bushong, E.; Ellisman, M.; Morgan, T.E.; et al. Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J. Neurosci. 2004, 24, 5016–5021. [Google Scholar] [CrossRef] [Green Version]
- Wilhelmsson, U.; Pozo-Rodrigalvarez, A.; Kalm, M.; de Pablo, Y.; Widestrand, Å.; Pekna, M.; Pekny, M. The role of GFAP and vimentin in learning and memory. Biol. Chem. 2019, 400, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Chirumamilla, S.; Sun, D.; Bullock, M.R.; Colello, R.J. Traumatic brain injury-induced cell proliferation in the adult mammalian central nervous system. J. Neurotrauma 2002, 19, 693–703. [Google Scholar] [CrossRef]
- Butler, C.R.; Boychuk, J.A.; Smith, B.N. Effects of rapamycin treatment on neurogenesis and synaptic reorganization in the dentate gyrus after controlled cortical impact injury in mice. Front. Syst. Neurosci. 2015, 9, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuberger, E.J.; Swietek, B.; Corrubia, L.; Prasanna, A.; Santhakumar, V. Enhanced dentate neurogenesis after brain injury undermines long-term neurogenic potential and promotes seizure susceptibility. Stem Cell Rep. 2017, 9, 972–984. [Google Scholar] [CrossRef] [Green Version]
- Blaiss, C.A.; Yu, T.S.; Zhang, G.; Chen, J.; Dimchev, G.; Parada, L.F.; Powell, C.M.; Kernie, S.G. Temporally specified genetic ablation of neurogenesis impairs cognitive recovery after traumatic brain injury. J. Neurosci. 2011, 31, 4906–4916. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.; Miao, Z.; Ye, Y.; Zhao, P.; Fan, L.; Bao, Z.; Tu, Y.; Li, C.; Chao, H.; Xu, X.; et al. Curcumin alleviates neuroinflammation, enhances hippocampal neurogenesis and improves spatial memory after traumatic brain injury. Brain Res. Bull. 2020, 162, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, T.; Takumi, T. Postsynaptic density proteins and their involvement in neurodevelopmental disorders. J. Biochem. 2018, 163, 447–455. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Zhu, J.; Wang, Y.H.; Hang, C.H. Arc silence aggravates traumatic neuronal injury via mGluR1-mediated ER stress and necroptosis. Cell Death Dis. 2020, 11, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilman, C.P.; Mattson, M.P. Do apoptotic mechanisms regulate synaptic plasticity and growth-cone motility? Neuromol. Med. 2002, 2, 197–214. [Google Scholar] [CrossRef]
- Parellada, E.; Gassó, P. Glutamate and microglia activation as a driver of dendritic apoptosis: A core pathophysiological mechanism to understand schizophrenia. Transl. Psychiatry 2021, 11, 271. [Google Scholar] [CrossRef]
- Mattson, M.P.; Keller, J.N.; Begley, J.G. Evidence for synaptic apoptosis. Exp. Neurol. 1998, 153, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Lewén, A.; Fujimura, M.; Sugawara, T.; Matz, P.; Copin, J.C.; Chan, P.H. Oxidative stress-dependent release of mitochondrial cytochrome c after traumatic brain injury. J. Cereb. Blood Flow Metab. 2001, 21, 914–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Aravind, A.; Pfister, B.J.; Chandra, N.; Haorah, J. Animal models of traumatic brain injury and assessment of injury severity. Mol. Neurobiol. 2019, 56, 5332–5345. [Google Scholar] [CrossRef]
- Tyrtyshnaia, A.; Bondar, A.; Konovalova, S.; Manzhulo, I. Synaptamide improves cognitive functions and neuronal plasticity in neuropathic pain. Int. J. Mol. Sci. 2021, 22, 12779. [Google Scholar] [CrossRef] [PubMed]
















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyrtyshnaia, A.; Manzhulo, O.; Manzhulo, I. Synaptamide Ameliorates Hippocampal Neurodegeneration and Glial Activation in Mice with Traumatic Brain Injury. Int. J. Mol. Sci. 2023, 24, 10014. https://doi.org/10.3390/ijms241210014
Tyrtyshnaia A, Manzhulo O, Manzhulo I. Synaptamide Ameliorates Hippocampal Neurodegeneration and Glial Activation in Mice with Traumatic Brain Injury. International Journal of Molecular Sciences. 2023; 24(12):10014. https://doi.org/10.3390/ijms241210014
Chicago/Turabian StyleTyrtyshnaia, Anna, Olga Manzhulo, and Igor Manzhulo. 2023. "Synaptamide Ameliorates Hippocampal Neurodegeneration and Glial Activation in Mice with Traumatic Brain Injury" International Journal of Molecular Sciences 24, no. 12: 10014. https://doi.org/10.3390/ijms241210014