
The Role of the Endocannabinoid System in Binge Eating Disorder

Abstract
:1. The Endocannabinoid System and Food Intake
1.1. The Endocannabinoid System
1.2. The Endocannabinoid System Regulates Food Intake
2. Binge Eating Disorder
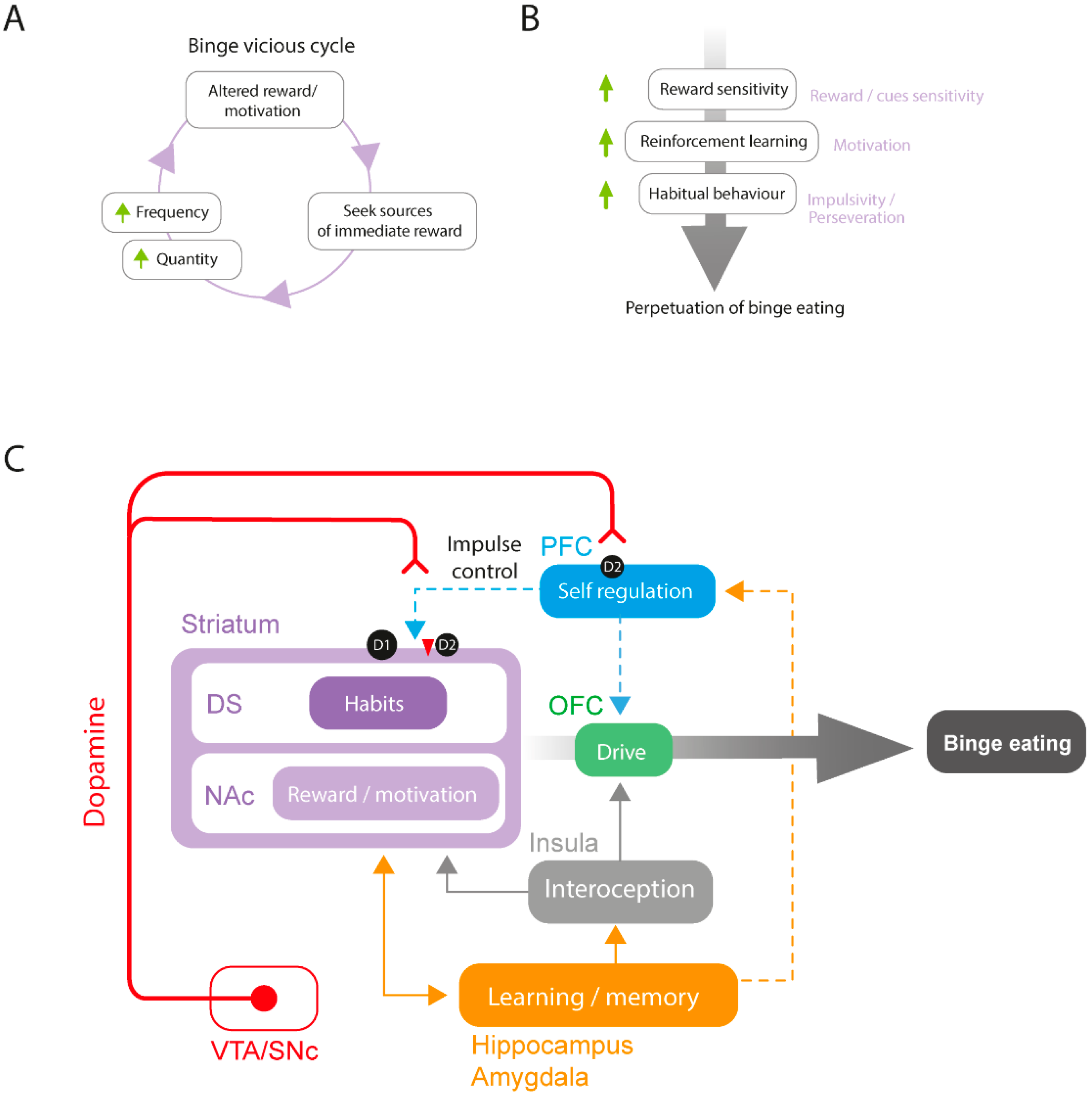
2.1. The Neurobiology of BED
2.2. Human Studies
2.3. Animal Studies
2.4. A Proposed Model for Altered Dopamine Activity and Reward Circuit Connectivity in BED
3. Binge Eating Disorder and the Endocannabinoid System
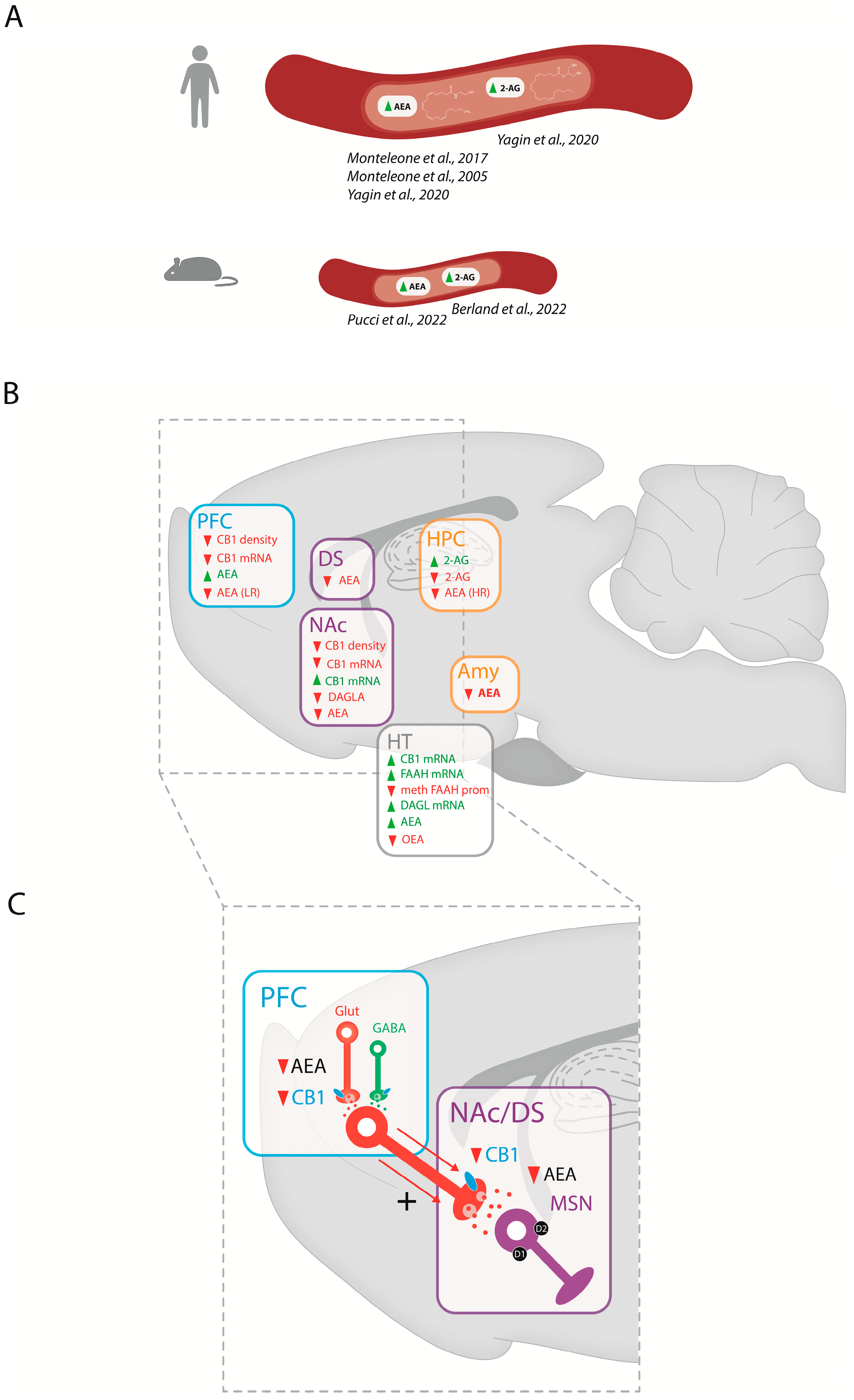
3.1. Endocannabinoid Levels in BED
3.2. Catabolic and Anabolic Cannabinoid Enzymes in BED
3.2.1. Transcriptional Regulations of the Cannabinoid Enzymes in BED
3.2.2. Genetic Animal Models Targeting the Endocannabinoid Enzymes to Study Food Intake and Addictive-like Eating Behaviors

{kind=link}
{kind=link}
| Target | Reference | Regimen | Body Weight | ||
|---|---|---|---|---|---|
| CB1 Full KO | Ledent, 1999 | [128] | Chow | Similar | |
| Zimmer, 1999 | [129] | Chow | Similar | ||
| Varvel, 2002 | [130] | Chow | Reduced | ||
| Di Marzo, 2001 | [131] | Chow | |||
| Fasting–refeeding | |||||
| Cota, 2003 | [132] | Chow | Reduced | ||
| Song, 2011 | [133] | Chow | Similar | ||
| Bellochio, 2010, 2013 | [134,135] | Fasting–refeeding | |||
| Massa, 2010 | [136] | Chow | Reduced | ||
| HFD | Reduced | ||||
| Liu, 2012 | [137] | Chow | Similar | ||
| HFD 14 wk | Reduced | ||||
| Osei-Hyiaman, 2005, 2008 | [138,139] | Chow | Reduced | ||
| HFD 14 wk | Reduced | ||||
| Yoshida, 2019 | [126] | HFD | Reduced | ||
| Soria-Gomez, 2014 # | [140] | Fasting–refeeding | |||
| Ravinet Trillou, 2003 | [141] | HFD 6 wk | Reduced | ||
| Ravinet Trillou, 2004 | [142] | Chow | Reduced | ||
| Free-choice chow + HFD | Reduced | ||||
| Bura, 2010 | [143] | Free-choice HFD + sweet solution | Reduced | ||
| Brommage, 2008 | [144] | HFD | Reduced | ||
| Powell, 2015 | [145] | Chow | Reduced | ||
| HFD | Reduced | ||||
| Quarta, 2010 | [146] | Chow 12 wk | Reduced | ||
| HFD 12 wk | Reduced | ||||
| Poncelet, 2003 | [147] | Chow | |||
| Two-bottle choice sucrose 5% | |||||
| Sanchis Segura, 2004 | [148] | Two-bottle choice sucrose 5% | |||
| Operant sucrose 5% | |||||
| CB1 Full KO | Ward, 2005 | [149] | Operant sweet food | ||
| Operant corn oil | |||||
| Guegan, 2013 | [150] | Operant chow/fasted | |||
| Operant highly palatable food | |||||
| Operant highly isocaloric palatable food | |||||
| Mancino, 2015 | [151] | Operant chocolate pellet | Reduced | ||
| Bi, 2020 | [50] | Operant sucrose 5% | |||
| CB1 cKO nervous system | CamK-CB1KO | Quarta, 2010 | [146] | Chow 12 wk | Reduced |
| Fasting–refeeding | |||||
| HFD 12 wk | Reduced | ||||
| Bellochio, 2013 | [135] | Fasting–refeeding | |||
| Glu-CB1KO | Lafenetre, 2009 | [152] | Repeated exposure to novel palatable food | ||
| Bellochio, 2010, 2013 | [134,135] | Fasting–refeeding | |||
| Exposure to palatable food in fed animals | Similar | ||||
| Domingo-Rodriguez, 2020 | [153] | Operant food addiction | |||
| Ruiz de Azu,a 2021 | [154] | LFD | Similar | ||
| HFD 12 wk | Reduced | ||||
| Operant chocolate pellet following LFD/HFD | Reduced | ||||
| Free-choice LFD + HFD | Reduced | ||||
| Soria-Gomez, 2014 | [140] | Fasting–refeeding | |||
| Glu CB1 RS | Soria-Gomez, 2014 | [140] | |||
| GABA-CB1KO | Lafenetre, 2009 | [152] | Repeated exposure to novel palatable food | ||
| CB1 cKO nervous system | Bellochio, 2010, 2013 | [134,135] | Fasting-refeeding | ||
| Exposure to palatable food in fed animals | Similar | ||||
| Massa, 210 | [136] | Chow | Similar | ||
| HFD | Similar for 10 wk, then reduced | ||||
| Glu/GABA CB1KO | Bellochio, 2010 | [134] | Fasting–refeeding | ||
| TPH2-CB1KO | Bellochio, 2013 | [135] | Fasting–refeeding | ||
| SF1-CB1KO | Bellochio 2013 | [135] | Fasting–refeeding | ||
| Cardinal, 2014 | [155] | Chow | Similar | ||
| HFD 8 wk | Increased | ||||
| Sim-CB1KO | Bellochio, 2013 | [135] | Fasting–refeeding | ||
| Cardinal, 2015 | [156] | chow | Similar | ||
| HFD 12 wk | Reduced | ||||
| Hyp-CB1KO | Cardinal, 2012 | [157] | Chow | Reduced | |
| Fasting–refeeding | Similar | ||||
| Thy1-CB1KO | Pang, 2011 | [158] | LFD | Reduced | |
| HFD 12 wk | Reduced | ||||
| CB1 cKO periphery | htgCB1KO | Liu, 2012 | [137] | Chow | Similar |
| HFD 14 wk | Reduced | ||||
| LCB1KO | Osei-Hyiaman, 2008 | [139] | chow | Similar | |
| HFD 14 wk | Similar | ||||
| intCB1KO | Avalos, 2020 | [159] | Western diet preference | ||
| CB2KO | Agudo, 2010 | [160] | Chow | Increased | |
| HFD 8 wk | Reduced | ||||
| Deveaux, 2009 | [161] | HFD 15 wk | Reduced | ||
| Flake, 2012 | [162] | Chow | Increased | ||
| Pradier, 2015 | [163] | Chow | Increased | ||
| Bi, 2020 | [50] | Operant sucrose 5% | |||
| Garcia Blanco 2023 | [164] | Operant chocolate pellet | Similar | ||
| CB2xP | Romero-Zerbo, 2012 | [165] | Fasting–refeeding | Reduced | |
| Garcia Blanco, 2023 | [164] | Operant chocolate pellet | Reduced | ||
| Enzymes | DAGLKO | Gao, 2010 | [166] | Chow | Reduced |
| Powell, 2015 | [145] | Chow | Reduced | ||
| NAPE Pld | Powell, 2015 | [145] | Chow | Similar | |
| MAGLKO | Chanda, 2010 | [123] | Chow | Reduced | |
| Taschler, 2011 | [125] | Chow | Similar | ||
| HFD 10 wk | Similar | ||||
| Fasting–refeeding | |||||
| Douglass, 2015 | [124] | LFD | Reduced | ||
| HFD | Reduced | ||||
| iMGL | Chon, 2012 | [127] | Chow | Similar | |
| HFD 3 wk | Increased | ||||
| FAAH | Cravatt, 2001 | [167] | Chow | Similar | |
| Tourino, 2010 | [168] | Chow | Increased | ||
| HFD 12 wk | Increased | ||||
| Operant chow | |||||
| Operant chocolate | |||||
| Operant fat |
3.3. Cannabinoid Receptors in BED
3.3.1. Transcriptional Regulations of Cannabinoid Receptors
3.3.2. Genetic Animal Models Targeting the Cannabinoid Receptors to Study Food Intake and Addictive-like Eating Behaviors
- Full knockout of CB1 receptors
- Conditional CB1 knockout mice in the central nervous system
- Conditional CB1 knockout mice in the periphery
- Knockout of CB2 receptors
4. Discussion on the Role of the ECS in BED
4.1. Peripheral Endocannabinoid Regulations in BED
4.2. Central ECS Regulations and Brain Structures Involved in BED
4.2.1. Central Regulations
4.2.2. PFC-Striatal Connections
4.2.3. HPC and Amygdala
4.2.4. Hypothalamus
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- UNODC. World Drug Report; UNODC: Vienna, Austria, 2022. [Google Scholar]
- OFDT. Drugs and Addictions, Key Data 2022; OFDT: Paris, France, 2022; pp. 2803–9408. [Google Scholar]
- Lau, B.K.; Cota, D.; Cristino, L.; Borgland, S.L. Endocannabinoid modulation of homeostatic and non-homeostatic feeding circuits. Neuropharmacology 2017, 124, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Freund, T.F.; Katona, I.; Piomelli, D. Role of endogenous cannabinoids in synaptic signaling. Physiol. Rev. 2003, 83, 1017–1066. [Google Scholar] [CrossRef]
- Navarrete, M.; Diez, A.; Araque, A. Astrocytes in endocannabinoid signalling. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130599. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, R.; Berrendero, F.; Ozaita, A.; Robledo, P. Neurochemical basis of cannabis addiction. Neuroscience 2011, 181, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.X.; Peng, X.Q.; Li, X.; Song, R.; Zhang, H.Y.; Liu, Q.R.; Yang, H.J.; Bi, G.H.; Li, J.; Gardner, E.L. Brain cannabinoid CB(2) receptors modulate cocaine’s actions in mice. Nat. Neurosci. 2011, 14, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kim, J. Neuronal expression of CB2 cannabinoid receptor mRNAs in the mouse hippocampus. Neuroscience 2015, 311, 253–267. [Google Scholar] [CrossRef]
- Onaivi, E.S.; Ishiguro, H.; Gong, J.P.; Patel, S.; Perchuk, A.; Meozzi, P.A.; Myers, L.; Mora, Z.; Tagliaferro, P.; Gardner, E.; et al. Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann. N. Y. Acad. Sci. 2006, 1074, 514–536. [Google Scholar] [CrossRef]
- Stempel, A.V.; Stumpf, A.; Zhang, H.Y.; Ozdogan, T.; Pannasch, U.; Theis, A.K.; Otte, D.M.; Wojtalla, A.; Racz, I.; Ponomarenko, A.; et al. Cannabinoid Type 2 Receptors Mediate a Cell Type-Specific Plasticity in the Hippocampus. Neuron 2016, 90, 795–809. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Gao, M.; Shen, H.; Bi, G.H.; Yang, H.J.; Liu, Q.R.; Wu, J.; Gardner, E.L.; Bonci, A.; Xi, Z.X. Expression of functional cannabinoid CB2 receptor in VTA dopamine neurons in rats. Addict. Biol. 2017, 22, 752–765. [Google Scholar] [CrossRef]
- Chadwick, V.L.; Rohleder, C.; Koethe, D.; Leweke, F.M. Cannabinoids and the endocannabinoid system in anxiety, depression, and dysregulation of emotion in humans. Curr. Opin. Psychiatry 2020, 33, 20–42. [Google Scholar] [CrossRef]
- Maldonado, R.; Cabanero, D.; Martin-Garcia, E. The endocannabinoid system in modulating fear, anxiety, and stress. Dialogues Clin. Neurosci. 2020, 22, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Befort, K. Interactions of the opioid and cannabinoid systems in reward: Insights from knockout studies. Front. Pharmacol. 2015, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Panagis, G.; Mackey, B.; Vlachou, S. Cannabinoid Regulation of Brain Reward Processing with an Emphasis on the Role of CB1 Receptors: A Step Back into the Future. Front. Psychiatry 2014, 5, 92. [Google Scholar] [CrossRef]
- Zimmer, A. Genetic Manipulation of the Endocannabinoid System. Handb. Exp. Pharmacol. 2015, 231, 129–183. [Google Scholar] [CrossRef]
- Scherma, M.; Fattore, L.; Castelli, M.P.; Fratta, W.; Fadda, P. The role of the endocannabinoid system in eating disorders: Neurochemical and behavioural preclinical evidence. Curr. Pharm. Des. 2014, 20, 2089–2099. [Google Scholar] [CrossRef]
- Ferrario, C.R.; Labouebe, G.; Liu, S.; Nieh, E.H.; Routh, V.H.; Xu, S.; O’Connor, E.C. Homeostasis Meets Motivation in the Battle to Control Food Intake. J. Neurosci. 2016, 36, 11469–11481. [Google Scholar] [CrossRef]
- Morton, G.J.; Meek, T.H.; Schwartz, M.W. Neurobiology of food intake in health and disease. Nat. Rev. Neurosci. 2014, 15, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, A.; Dunnett, S.B. Dopamine neuron systems in the brain: An update. Trends Neurosci. 2007, 30, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Wise, R.A.; Baler, R. The dopamine motive system: Implications for drug and food addiction. Nat. Rev. Neurosci. 2017, 18, 741–752. [Google Scholar] [CrossRef]
- Nieh, E.H.; Matthews, G.A.; Allsop, S.A.; Presbrey, K.N.; Leppla, C.A.; Wichmann, R.; Neve, R.; Wildes, C.P.; Tye, K.M. Decoding neural circuits that control compulsive sucrose seeking. Cell 2015, 160, 528–541. [Google Scholar] [CrossRef]
- Jennings, J.H.; Ung, R.L.; Resendez, S.L.; Stamatakis, A.M.; Taylor, J.G.; Huang, J.; Veleta, K.; Kantak, P.A.; Aita, M.; Shilling-Scrivo, K.; et al. Visualizing hypothalamic network dynamics for appetitive and consummatory behaviors. Cell 2015, 160, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.A.; Basiri, M.L.; McHenry, J.A.; Kosyk, O.; Otis, J.M.; van den Munkhof, H.E.; Bryois, J.; Hubel, C.; Breen, G.; Guo, W.; et al. Obesity remodels activity and transcriptional state of a lateral hypothalamic brake on feeding. Science 2019, 364, 1271–1274. [Google Scholar] [CrossRef]
- Brown, R.M.; James, M.H. Binge eating, overeating and food addiction: Approaches for examining food overconsumption in laboratory rodents. Prog. Neuropsychopharmacol. Biol. Psychiatry 2023, 123, 110717. [Google Scholar] [CrossRef] [PubMed]
- Milano, W.; Tecce, M.; Capasso, A. The role of endocannabinoids in the control of eating disorders. Dis. Disord. 2017, 1, 1–6. [Google Scholar] [CrossRef]
- Levichev, A.; Faumont, S.; Berner, R.Z.; Purcell, Z.; White, A.M.; Chicas-Cruz, K.; Lockery, S.R. The conserved endocannabinoid anandamide modulates olfactory sensitivity to induce hedonic feeding in C. elegans. Curr. Biol. 2023, 33, 1625–1639.e1624. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.L. Cannabis: Effects on hunger and thirst. Behav. Biol. 1975, 15, 255–281. [Google Scholar] [CrossRef]
- Di Marzo, V. Endocannabinoids: An appetite for fat. Proc. Natl. Acad. Sci. USA 2011, 108, 12567–12568. [Google Scholar] [CrossRef]
- Williams, C.M.; Rogers, P.J.; Kirkham, T.C. Hyperphagia in pre-fed rats following oral delta9-THC. Physiol. Behav. 1998, 65, 343–346. [Google Scholar] [CrossRef]
- Di Marzo, V.; Matias, I. Endocannabinoid control of food intake and energy balance. Nat. Neurosci. 2005, 8, 585–589. [Google Scholar] [CrossRef]
- Meye, F.J.; Adan, R.A. Feelings about food: The ventral tegmental area in food reward and emotional eating. Trends Pharmacol. Sci. 2014, 35, 31–40. [Google Scholar] [CrossRef]
- Kirkham, T.C.; Williams, C.M.; Fezza, F.; Di Marzo, V. Endocannabinoid levels in rat limbic forebrain and hypothalamus in relation to fasting, feeding and satiation: Stimulation of eating by 2-arachidonoyl glycerol. Br. J. Pharmacol. 2002, 136, 550–557. [Google Scholar] [CrossRef]
- Novelle, M.G.; Dieguez, C. Unravelling the role and mechanism of adipokine and gastrointestinal signals in animal models in the nonhomeostatic control of energy homeostasis: Implications for binge eating disorder. Eur. Eat. Disord. Rev. 2018, 26, 551–568. [Google Scholar] [CrossRef]
- Satta, V.; Scherma, M.; Piscitelli, F.; Usai, P.; Castelli, M.P.; Bisogno, T.; Fratta, W.; Fadda, P. Limited Access to a High Fat Diet Alters Endocannabinoid Tone in Female Rats. Front. Neurosci. 2018, 12, 40. [Google Scholar] [CrossRef]
- Brown, J.D.; Karimian Azari, E.; Ayala, J.E. Oleoylethanolamide: A fat ally in the fight against obesity. Physiol. Behav. 2017, 176, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Micioni Di Bonaventura, M.V.; Gallelli, C.A.; Koczwara, J.B.; Smeets, D.; Giusepponi, M.E.; De Ceglia, M.; Friuli, M.; Micioni Di Bonaventura, E.; Scuderi, C.; et al. Oleoylethanolamide decreases frustration stress-induced binge-like eating in female rats: A novel potential treatment for binge eating disorder. Neuropsychopharmacology 2020, 45, 1931–1941. [Google Scholar] [CrossRef] [PubMed]
- Colombo, G.; Agabio, R.; Diaz, G.; Lobina, C.; Reali, R.; Gessa, G.L. Appetite suppression and weight loss after the cannabinoid antagonist SR 141716. Life Sci. 1998, 63, PL113–PL117. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, P.J.; Winston, K.; Swezey, L.; Wisniecki, A.; Aberman, J.; Tardif, D.J.; Betz, A.J.; Ishiwari, K.; Makriyannis, A.; Salamone, J.D. The cannabinoid CB1 antagonists SR 141716A and AM 251 suppress food intake and food-reinforced behavior in a variety of tasks in rats. Behav. Pharmacol. 2003, 14, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Parylak, S.L.; Cottone, P.; Sabino, V.; Rice, K.C.; Zorrilla, E.P. Effects of CB1 and CRF1 receptor antagonists on binge-like eating in rats with limited access to a sweet fat diet: Lack of withdrawal-like responses. Physiol. Behav. 2012, 107, 231–242. [Google Scholar] [CrossRef]
- Scherma, M.; Fattore, L.; Satta, V.; Businco, F.; Pigliacampo, B.; Goldberg, S.R.; Dessi, C.; Fratta, W.; Fadda, P. Pharmacological modulation of the endocannabinoid signalling alters binge-type eating behaviour in female rats. Br. J. Pharmacol. 2013, 169, 820–833. [Google Scholar] [CrossRef]
- Moore, C.F.; Schlain, G.S.; Mancino, S.; Sabino, V.; Cottone, P. A behavioral and pharmacological characterization of palatable diet alternation in mice. Pharmacol. Biochem. Behav. 2017, 163, 1–8. [Google Scholar] [CrossRef]
- Bermudez-Silva, F.J.; Viveros, M.P.; McPartland, J.M.; Rodriguez de Fonseca, F. The endocannabinoid system, eating behavior and energy homeostasis: The end or a new beginning? Pharmacol. Biochem. Behav. 2010, 95, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Samat, A.; Tomlinson, B.; Taheri, S.; Thomas, G.N. Rimonabant for the treatment of obesity. Recent Pat. Cardiovasc. Drug Discov. 2008, 3, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Farrimond, J.A.; Whalley, B.J.; Williams, C.M. Cannabinol and cannabidiol exert opposing effects on rat feeding patterns. Psychopharmacology 2012, 223, 117–129. [Google Scholar] [CrossRef]
- Ignatowska-Jankowska, B.; Jankowski, M.M.; Swiergiel, A.H. Cannabidiol decreases body weight gain in rats: Involvement of CB2 receptors. Neurosci. Lett. 2011, 490, 82–84. [Google Scholar] [CrossRef]
- Sofia, R.D.; Knobloch, L.C. Comparative effects of various naturally occurring cannabinoids on food, sucrose and water consumption by rats. Pharmacol. Biochem. Behav. 1976, 4, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Brady, K.T.; Balster, R.L. The effects of delta 9-tetrahydrocannabinol alone and in combination with cannabidiol on fixed-interval performance in rhesus monkeys. Psychopharmacology 1980, 72, 21–26. [Google Scholar] [CrossRef]
- Hiltunen, A.J.; Jarbe, T.U.; Kamkar, M.R.; Archer, T. Behaviour in rats maintained by low differential reinforcement rate: Effects of delta 1-tetrahydrocannabinol, cannabinol and cannabidiol, alone and in combination. Neuropharmacology 1989, 28, 183–189. [Google Scholar] [CrossRef]
- Bi, G.H.; Galaj, E.; He, Y.; Xi, Z.X. Cannabidiol inhibits sucrose self-administration by CB1 and CB2 receptor mechanisms in rodents. Addict. Biol. 2020, 25, e12783. [Google Scholar] [CrossRef]
- Schlicker, E.; Kathmann, M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol. Sci. 2001, 22, 565–572. [Google Scholar] [CrossRef]
- Thoeni, S.; Loureiro, M.; O’Connor, E.C.; Luscher, C. Depression of Accumbal to Lateral Hypothalamic Synapses Gates Overeating. Neuron 2020, 107, 158–172.e4. [Google Scholar] [CrossRef]
- Gaetani, S.; Kaye, W.H.; Cuomo, V.; Piomelli, D. Role of endocannabinoids and their analogues in obesity and eating disorders. Eat. Weight. Disord. 2008, 13, e42–e48. [Google Scholar] [CrossRef] [PubMed]
- Association, A.P. The Diagnostic and Statistical Manual of Mental Disorders (DSM-V), 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Guerdjikova, A.I.; Mori, N.; Casuto, L.S.; McElroy, S.L. Update on Binge Eating Disorder. Med. Clin. N. Am. 2019, 103, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Levallius, J.; Monell, E.; Birgegard, A.; Clinton, D.; Forsen Mantilla, E. Binge Eating and Addictive-Like Behaviours in Males and Females. Psychol. Rep. 2022, 125, 148–166. [Google Scholar] [CrossRef]
- Milano, W.; De Biasio, V.; Di Munzio, W.; Foggia, G.; Capasso, A. Obesity: The New Global Epidemic Pharmacological Treatment, Opportunities and Limits for Personalized Therapy. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 1232–1243. [Google Scholar] [CrossRef]
- Aguera, Z.; Lozano-Madrid, M.; Mallorqui-Bague, N.; Jimenez-Murcia, S.; Menchon, J.M.; Fernandez-Aranda, F. A review of binge eating disorder and obesity. Neuropsychiatry 2021, 35, 57–67. [Google Scholar] [CrossRef]
- Kessler, R.M.; Hutson, P.H.; Herman, B.K.; Potenza, M.N. The neurobiological basis of binge-eating disorder. Neurosci. Biobehav. Rev. 2016, 63, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Geisler, C.E.; Hayes, M.R. Metabolic Hormone Action in the VTA: Reward-Directed Behavior and Mechanistic Insights. Physiol. Behav. 2023, 268, 114236. [Google Scholar] [CrossRef]
- Spierling, S.; de Guglielmo, G.; Kirson, D.; Kreisler, A.; Roberto, M.; George, O.; Zorrilla, E.P. Insula to ventral striatal projections mediate compulsive eating produced by intermittent access to palatable food. Neuropsychopharmacology 2020, 45, 579–588. [Google Scholar] [CrossRef]
- Juarascio, A.S.; Srivastava, P.; Manasse, S.M.; Wilkinson, M.L.; Felonis, C.R.; Drexler, S.A. Reward retraining: A pilot randomized controlled trial of a novel treatment approach for transdiagnostic binge eating. Int. J. Eat. Disord. 2023, 56, 662–670. [Google Scholar] [CrossRef]
- Berridge, K.C.; Robinson, T.E. Liking, wanting, and the incentive-sensitization theory of addiction. Am. Psychol. 2016, 71, 670–679. [Google Scholar] [CrossRef]
- Leenaerts, N.; Jongen, D.; Ceccarini, J.; Van Oudenhove, L.; Vrieze, E. The neurobiological reward system and binge eating: A critical systematic review of neuroimaging studies. Int. J. Eat. Disord. 2022, 55, 1421–1458. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Aranda, F.; Karwautz, A.; Treasure, J. Food addiction: A transdiagnostic construct of increasing interest. Eur. Eat. Disord. Rev. 2018, 26, 536–540. [Google Scholar] [CrossRef]
- Lindgren, E.; Gray, K.; Miller, G.; Tyler, R.; Wiers, C.E.; Volkow, N.D.; Wang, G.J. Food addiction: A common neurobiological mechanism with drug abuse. Front. Biosci. 2018, 23, 811–836. [Google Scholar] [CrossRef]
- Haynos, A.F.; Camchong, J.; Pearson, C.M.; Lavender, J.M.; Mueller, B.A.; Peterson, C.B.; Specker, S.; Raymond, N.; Lim, K.O. Resting State Hypoconnectivity of Reward Networks in Binge Eating Disorder. Cereb. Cortex 2021, 31, 2494–2504. [Google Scholar] [CrossRef] [PubMed]
- Geliebter, A.; Benson, L.; Pantazatos, S.P.; Hirsch, J.; Carnell, S. Greater anterior cingulate activation and connectivity in response to visual and auditory high-calorie food cues in binge eating: Preliminary findings. Appetite 2016, 96, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.R.; Ku, J.; Lee, J.H.; Lee, H.; Jung, Y.C. Functional and effective connectivity of anterior insula in anorexia nervosa and bulimia nervosa. Neurosci. Lett. 2012, 521, 152–157. [Google Scholar] [CrossRef]
- Schienle, A.; Schafer, A.; Hermann, A.; Vaitl, D. Binge-eating disorder: Reward sensitivity and brain activation to images of food. Biol. Psychiatry 2009, 65, 654–661. [Google Scholar] [CrossRef]
- Simon, J.J.; Skunde, M.; Walther, S.; Bendszus, M.; Herzog, W.; Friederich, H.C. Neural signature of food reward processing in bulimic-type eating disorders. Soc. Cogn. Affect. Neurosci. 2016, 11, 1393–1401. [Google Scholar] [CrossRef]
- Broft, A.; Shingleton, R.; Kaufman, J.; Liu, F.; Kumar, D.; Slifstein, M.; Abi-Dargham, A.; Schebendach, J.; Van Heertum, R.; Attia, E.; et al. Striatal dopamine in bulimia nervosa: A PET imaging study. Int. J. Eat. Disord. 2012, 45, 648–656. [Google Scholar] [CrossRef]
- Majuri, J.; Joutsa, J.; Johansson, J.; Voon, V.; Alakurtti, K.; Parkkola, R.; Lahti, T.; Alho, H.; Hirvonen, J.; Arponen, E.; et al. Dopamine and Opioid Neurotransmission in Behavioral Addictions: A Comparative PET Study in Pathological Gambling and Binge Eating. Neuropsychopharmacology 2017, 42, 1169–1177. [Google Scholar] [CrossRef]
- Wang, G.J.; Geliebter, A.; Volkow, N.D.; Telang, F.W.; Logan, J.; Jayne, M.C.; Galanti, K.; Selig, P.A.; Han, H.; Zhu, W.; et al. Enhanced striatal dopamine release during food stimulation in binge eating disorder. Obesity 2011, 19, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Shivacharan, R.S.; Rolle, C.E.; Barbosa, D.A.N.; Cunningham, T.N.; Feng, A.; Johnson, N.D.; Safer, D.L.; Bohon, C.; Keller, C.; Buch, V.P.; et al. Pilot study of responsive nucleus accumbens deep brain stimulation for loss-of-control eating. Nat. Med. 2022, 28, 1791–1796. [Google Scholar] [CrossRef] [PubMed]
- Voon, V. Cognitive biases in binge eating disorder: The hijacking of decision making. CNS Spectr. 2015, 20, 566–573. [Google Scholar] [CrossRef]
- Westwater, M.L.; Murley, A.G.; Diederen, K.M.J.; Carpenter, T.A.; Ziauddeen, H.; Fletcher, P.C. Characterizing cerebral metabolite profiles in anorexia and bulimia nervosa and their associations with habitual behavior. Transl. Psychiatry 2022, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Giel, K.E.; Teufel, M.; Junne, F.; Zipfel, S.; Schag, K. Food-Related Impulsivity in Obesity and Binge Eating Disorder-A Systematic Update of the Evidence. Nutrients 2017, 9, 170. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.J.; Francis, H.M. The hippocampus and the regulation of human food intake. Psychol. Bull. 2017, 143, 1011–1032. [Google Scholar] [CrossRef]
- Stice, E.; Yokum, S.; Rohde, P.; Gau, J.; Shaw, H. Evidence that a novel transdiagnostic eating disorder treatment reduces reward region response to the thin beauty ideal and high-calorie binge foods. Psychol. Med. 2021, 53, 2252–2262. [Google Scholar] [CrossRef]
- Nakamura, Y.; Koike, S. Association of Disinhibited Eating and Trait of Impulsivity With Insula and Amygdala Responses to Palatable Liquid Consumption. Front. Syst. Neurosci. 2021, 15, 647143. [Google Scholar] [CrossRef]
- Kelley, A.E.; Baldo, B.A.; Pratt, W.E.; Will, M.J. Corticostriatal-hypothalamic circuitry and food motivation: Integration of energy, action and reward. Physiol. Behav. 2005, 86, 773–795. [Google Scholar] [CrossRef]
- Kelley, A.E.; Schiltz, C.A.; Landry, C.F. Neural systems recruited by drug- and food-related cues: Studies of gene activation in corticolimbic regions. Physiol. Behav. 2005, 86, 11–14. [Google Scholar] [CrossRef]
- Quansah Amissah, R.; Chometton, S.; Calvez, J.; Guevremont, G.; Timofeeva, E.; Timofeev, I. Differential Expression of DeltaFosB in Reward Processing Regions Between Binge Eating Prone and Resistant Female Rats. Front. Syst. Neurosci. 2020, 14, 562154. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, E.B.; Culbert, K.M.; Gradl, D.R.; Richardson, K.A.; Klump, K.L.; Sisk, C.L. Differential mesocorticolimbic responses to palatable food in binge eating prone and binge eating resistant female rats. Physiol. Behav. 2015, 152, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Halpern, C.H.; Tekriwal, A.; Santollo, J.; Keating, J.G.; Wolf, J.A.; Daniels, D.; Bale, T.L. Amelioration of binge eating by nucleus accumbens shell deep brain stimulation in mice involves D2 receptor modulation. J. Neurosci. 2013, 33, 7122–7129. [Google Scholar] [CrossRef]
- Bocarsly, M.E.; Hoebel, B.G.; Paredes, D.; von Loga, I.; Murray, S.M.; Wang, M.; Arolfo, M.P.; Yao, L.; Diamond, I.; Avena, N.M. GS 455534 selectively suppresses binge eating of palatable food and attenuates dopamine release in the accumbens of sugar-bingeing rats. Behav. Pharmacol. 2014, 25, 147–157. [Google Scholar] [CrossRef]
- Hajnal, A.; Norgren, R. Repeated access to sucrose augments dopamine turnover in the nucleus accumbens. Neuroreport 2002, 13, 2213–2216. [Google Scholar] [CrossRef]
- Inoue, K.; Kiriike, N.; Okuno, M.; Fujisaki, Y.; Kurioka, M.; Iwasaki, S.; Yamagami, S. Prefrontal and striatal dopamine metabolism during enhanced rebound hyperphagia induced by space restriction--a rat model of binge eating. Biol. Psychiatry 1998, 44, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Avena, N.M.; Hoebel, B.G. Daily bingeing on sugar repeatedly releases dopamine in the accumbens shell. Neuroscience 2005, 134, 737–744. [Google Scholar] [CrossRef]
- Yu, Y.; Miller, R.; Groth, S.W. A literature review of dopamine in binge eating. J. Eat. Disord. 2022, 10, 11. [Google Scholar] [CrossRef]
- Corwin, R.L.; Wojnicki, F.H.; Zimmer, D.J.; Babbs, R.K.; McGrath, L.E.; Olivos, D.R.; Mietlicki-Baase, E.G.; Hayes, M.R. Binge-type eating disrupts dopaminergic and GABAergic signaling in the prefrontal cortex and ventral tegmental area. Obesity 2016, 24, 2118–2125. [Google Scholar] [CrossRef]
- Heal, D.J.; Hallam, M.; Prow, M.; Gosden, J.; Cheetham, S.; Choi, Y.K.; Tarazi, F.; Hutson, P. Dopamine and mu-opioid receptor dysregulation in the brains of binge-eating female rats—Possible relevance in the psychopathology and treatment of binge-eating disorder. J. Psychopharmacol. 2017, 31, 770–783. [Google Scholar] [CrossRef]
- Bello, N.T.; Lucas, L.R.; Hajnal, A. Repeated sucrose access influences dopamine D2 receptor density in the striatum. Neuroreport 2002, 13, 1575–1578. [Google Scholar] [CrossRef] [PubMed]
- Colantuoni, C.; Schwenker, J.; McCarthy, J.; Rada, P.; Ladenheim, B.; Cadet, J.L.; Schwartz, G.J.; Moran, T.H.; Hoebel, B.G. Excessive sugar intake alters binding to dopamine and mu-opioid receptors in the brain. Neuroreport 2001, 12, 3549–3552. [Google Scholar] [CrossRef] [PubMed]
- Spangler, R.; Wittkowski, K.M.; Goddard, N.L.; Avena, N.M.; Hoebel, B.G.; Leibowitz, S.F. Opiate-like effects of sugar on gene expression in reward areas of the rat brain. Mol. Brain Res. 2004, 124, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Cordner, Z.A.; Boersma, G.; Moran, T.H. Cognitive impairment and gene expression alterations in a rodent model of binge eating disorder. Physiol. Behav. 2017, 180, 78–90. [Google Scholar] [CrossRef]
- Everitt, B.J.; Belin, D.; Economidou, D.; Pelloux, Y.; Dalley, J.W.; Robbins, T.W. Review. Neural mechanisms underlying the vulnerability to develop compulsive drug-seeking habits and addiction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 3125–3135. [Google Scholar] [CrossRef]
- Anastasio, N.C.; Stutz, S.J.; Price, A.E.; Davis-Reyes, B.D.; Sholler, D.J.; Ferguson, S.M.; Neumaier, J.F.; Moeller, F.G.; Hommel, J.D.; Cunningham, K.A. Convergent neural connectivity in motor impulsivity and high-fat food binge-like eating in male Sprague-Dawley rats. Neuropsychopharmacology 2019, 44, 1752–1761. [Google Scholar] [CrossRef]
- Cano, A.M.; Murphy, E.S.; Lupfer, G. Delay discounting predicts binge-eating in Wistar rats. Behav. Process. 2016, 132, 1–4. [Google Scholar] [CrossRef]
- Vickers, S.P.; Goddard, S.; Brammer, R.J.; Hutson, P.H.; Heal, D.J. Investigation of impulsivity in binge-eating rats in a delay-discounting task and its prevention by the d-amphetamine prodrug, lisdexamfetamine. J. Psychopharmacol. 2017, 31, 784–797. [Google Scholar] [CrossRef]
- Gabbott, P.L.; Warner, T.A.; Jays, P.R.; Salway, P.; Busby, S.J. Prefrontal cortex in the rat: Projections to subcortical autonomic, motor, and limbic centers. J. Comp. Neurol. 2005, 492, 145–177. [Google Scholar] [CrossRef]
- Koob, G.F.; Volkow, N.D. Neurobiology of addiction: A neurocircuitry analysis. Lancet Psychiatry 2016, 3, 760–773. [Google Scholar] [CrossRef]
- George, O.; Koob, G.F. Individual differences in prefrontal cortex function and the transition from drug use to drug dependence. Neurosci. Biobehav. Rev. 2010, 35, 232–247. [Google Scholar] [CrossRef] [PubMed]
- Graybiel, A.M.; Rauch, S.L. Toward a neurobiology of obsessive-compulsive disorder. Neuron 2000, 28, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Christoffel, D.J.; Walsh, J.J.; Heifets, B.D.; Hoerbelt, P.; Neuner, S.; Sun, G.; Ravikumar, V.K.; Wu, H.; Halpern, C.H.; Malenka, R.C. Input-specific modulation of murine nucleus accumbens differentially regulates hedonic feeding. Nat. Commun. 2021, 12, 2135. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; Aizenstein, H.; Mazurkewicz, L.; Fudge, J.; Frank, G.K.; Putnam, K.; Bailer, U.F.; Fischer, L.; Kaye, W.H. Altered insula response to taste stimuli in individuals recovered from restricting-type anorexia nervosa. Neuropsychopharmacology 2008, 33, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Fudge, J.L.; Breitbart, M.A.; Danish, M.; Pannoni, V. Insular and gustatory inputs to the caudal ventral striatum in primates. J. Comp. Neurol. 2005, 490, 101–118. [Google Scholar] [CrossRef]
- Kelley, A.E.; Bakshi, V.P.; Haber, S.N.; Steininger, T.L.; Will, M.J.; Zhang, M. Opioid modulation of taste hedonics within the ventral striatum. Physiol. Behav. 2002, 76, 365–377. [Google Scholar] [CrossRef]
- Hsu, T.M.; Hahn, J.D.; Konanur, V.R.; Noble, E.E.; Suarez, A.N.; Thai, J.; Nakamoto, E.M.; Kanoski, S.E. Hippocampus ghrelin signaling mediates appetite through lateral hypothalamic orexin pathways. eLife 2015, 4, e11190. [Google Scholar] [CrossRef]
- Kanoski, S.E.; Grill, H.J. Hippocampus Contributions to Food Intake Control: Mnemonic, Neuroanatomical, and Endocrine Mechanisms. Biol. Psychiatry 2017, 81, 748–756. [Google Scholar] [CrossRef]
- Will, M.J.; Franzblau, E.B.; Kelley, A.E. The amygdala is critical for opioid-mediated binge eating of fat. Neuroreport 2004, 15, 1857–1860. [Google Scholar] [CrossRef]
- Johnson, P.M.; Kenny, P.J. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat. Neurosci. 2010, 13, 635–641. [Google Scholar] [CrossRef]
- Navarro, D.; Gasparyan, A.; Navarrete, F.; Torregrosa, A.B.; Rubio, G.; Marin-Mayor, M.; Acosta, G.B.; Garcia-Gutierrez, M.S.; Manzanares, J. Molecular Alterations of the Endocannabinoid System in Psychiatric Disorders. Int. J. Mol. Sci. 2022, 23, 4764. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, P.; Tortorella, A.; Martiadis, V.; Di Filippo, C.; Canestrelli, B.; Maj, M. The cDNA 385C to A missense polymorphism of the endocannabinoid degrading enzyme fatty acid amide hydrolase (FAAH) is associated with overweight/obesity but not with binge eating disorder in overweight/obese women. Psychoneuroendocrinology 2008, 33, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, P.; Matias, I.; Martiadis, V.; De Petrocellis, L.; Maj, M.; Di Marzo, V. Blood levels of the endocannabinoid anandamide are increased in anorexia nervosa and in binge-eating disorder, but not in bulimia nervosa. Neuropsychopharmacology 2005, 30, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, A.M.; Piscitelli, F.; Dalle Grave, R.; El Ghoch, M.; Di Marzo, V.; Maj, M.; Monteleone, P. Peripheral Endocannabinoid Responses to Hedonic Eating in Binge-Eating Disorder. Nutrients 2017, 9, 1377. [Google Scholar] [CrossRef]
- Yagin, N.L.; Aliasgari, F.; Alizadeh, M.; Aliasgharzadeh, S.; Mahdavi, R. Comparison of endocannabinoids levels, FAAH gene polymorphisms, and appetite regulatory substances in women with and without binge eating disorder: A cross- sectional study. Nutr. Res. 2020, 83, 86–93. [Google Scholar] [CrossRef]
- Berland, C.; Castel, J.; Terrasi, R.; Montalban, E.; Foppen, E.; Martin, C.; Muccioli, G.G.; Luquet, S.; Gangarossa, G. Identification of an endocannabinoid gut-brain vagal mechanism controlling food reward and energy homeostasis. Mol. Psychiatry 2022, 27, 2340–2354. [Google Scholar] [CrossRef]
- Pucci, M.; D’Addario, C.; Micioni Di Bonaventura, E.; Mercante, F.; Annunzi, E.; Fanti, F.; Sergi, M.; Botticelli, L.; Einaudi, G.; Cifani, C.; et al. Endocannabinoid System Regulation in Female Rats with Recurrent Episodes of Binge Eating. Int. J. Mol. Sci. 2022, 23, 15228. [Google Scholar] [CrossRef]
- De Sa Nogueira, D.; Bourdy, R.; Filliol, D.; Awad, G.; Andry, V.; Goumon, Y.; Olmstead, M.C.; Befort, K. Binge sucrose-induced neuroadaptations: A focus on the endocannabinoid system. Appetite 2021, 164, 105258. [Google Scholar] [CrossRef]
- Valverde, O.; Karsak, M.; Zimmer, A. Analysis of the endocannabinoid system by using CB1 cannabinoid receptor knockout mice. Handb. Exp. Pharmacol. 2005, 117–145. [Google Scholar] [CrossRef]
- Chanda, P.K.; Gao, Y.; Mark, L.; Btesh, J.; Strassle, B.W.; Lu, P.; Piesla, M.J.; Zhang, M.Y.; Bingham, B.; Uveges, A.; et al. Monoacylglycerol lipase activity is a critical modulator of the tone and integrity of the endocannabinoid system. Mol. Pharmacol. 2010, 78, 996–1003. [Google Scholar] [CrossRef]
- Douglass, J.D.; Zhou, Y.X.; Wu, A.; Zadroga, J.A.; Gajda, A.M.; Lackey, A.I.; Lang, W.; Chevalier, K.M.; Sutton, S.W.; Zhang, S.P.; et al. Global deletion of MGL in mice delays lipid absorption and alters energy homeostasis and diet-induced obesity. J. Lipid. Res. 2015, 56, 1153–1171. [Google Scholar] [CrossRef] [PubMed]
- Taschler, U.; Radner, F.P.; Heier, C.; Schreiber, R.; Schweiger, M.; Schoiswohl, G.; Preiss-Landl, K.; Jaeger, D.; Reiter, B.; Koefeler, H.C.; et al. Monoglyceride lipase deficiency in mice impairs lipolysis and attenuates diet-induced insulin resistance. J. Biol. Chem. 2011, 286, 17467–17477. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Kita, Y.; Tokuoka, S.M.; Hamano, F.; Yamazaki, M.; Sakimura, K.; Kano, M.; Shimizu, T. Monoacylglycerol lipase deficiency affects diet-induced obesity, fat absorption, and feeding behavior in CB(1) cannabinoid receptor-deficient mice. FASEB J. 2019, 33, 2484–2497. [Google Scholar] [CrossRef] [PubMed]
- Chon, S.H.; Douglass, J.D.; Zhou, Y.X.; Malik, N.; Dixon, J.L.; Brinker, A.; Quadro, L.; Storch, J. Over-expression of monoacylglycerol lipase (MGL) in small intestine alters endocannabinoid levels and whole body energy balance, resulting in obesity. PLoS ONE 2012, 7, e43962. [Google Scholar] [CrossRef]
- Ledent, C.; Valverde, O.; Cossu, G.; Petitet, F.; Aubert, J.F.; Beslot, F.; Bohme, G.A.; Imperato, A.; Pedrazzini, T.; Roques, B.P.; et al. Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science 1999, 283, 401–404. [Google Scholar] [CrossRef]
- Zimmer, A.; Zimmer, A.M.; Hohmann, A.G.; Herkenham, M.; Bonner, T.I. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 5780–5785. [Google Scholar] [CrossRef]
- Varvel, S.A.; Lichtman, A.H. Evaluation of CB1 receptor knockout mice in the Morris water maze. J. Pharmacol. Exp. Ther. 2002, 301, 915–924. [Google Scholar] [CrossRef]
- Di Marzo, V.; Goparaju, S.K.; Wang, L.; Liu, J.; Batkai, S.; Jarai, Z.; Fezza, F.; Miura, G.I.; Palmiter, R.D.; Sugiura, T.; et al. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 2001, 410, 822–825. [Google Scholar] [CrossRef]
- Cota, D.; Marsicano, G.; Tschop, M.; Grubler, Y.; Flachskamm, C.; Schubert, M.; Auer, D.; Yassouridis, A.; Thone-Reineke, C.; Ortmann, S.; et al. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J. Clin. Investig. 2003, 112, 423–431. [Google Scholar] [CrossRef]
- Song, D.; Bandsma, R.H.; Xiao, C.; Xi, L.; Shao, W.; Jin, T.; Lewis, G.F. Acute cannabinoid receptor type 1 (CB1R) modulation influences insulin sensitivity by an effect outside the central nervous system in mice. Diabetologia 2011, 54, 1181–1189. [Google Scholar] [CrossRef]
- Bellocchio, L.; Lafenetre, P.; Cannich, A.; Cota, D.; Puente, N.; Grandes, P.; Chaouloff, F.; Piazza, P.V.; Marsicano, G. Bimodal control of stimulated food intake by the endocannabinoid system. Nat. Neurosci. 2010, 13, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Bellocchio, L.; Soria-Gomez, E.; Quarta, C.; Metna-Laurent, M.; Cardinal, P.; Binder, E.; Cannich, A.; Delamarre, A.; Haring, M.; Martin-Fontecha, M.; et al. Activation of the sympathetic nervous system mediates hypophagic and anxiety-like effects of CB(1) receptor blockade. Proc. Natl. Acad. Sci. USA 2013, 110, 4786–4791. [Google Scholar] [CrossRef]
- Massa, F.; Mancini, G.; Schmidt, H.; Steindel, F.; Mackie, K.; Angioni, C.; Oliet, S.H.; Geisslinger, G.; Lutz, B. Alterations in the hippocampal endocannabinoid system in diet-induced obese mice. J. Neurosci. 2010, 30, 6273–6281. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, L.; Xiong, K.; Godlewski, G.; Mukhopadhyay, B.; Tam, J.; Yin, S.; Gao, P.; Shan, X.; Pickel, J.; et al. Hepatic cannabinoid receptor-1 mediates diet-induced insulin resistance via inhibition of insulin signaling and clearance in mice. Gastroenterology 2012, 142, 1218–1228.e1. [Google Scholar] [CrossRef] [PubMed]
- Osei-Hyiaman, D.; DePetrillo, M.; Pacher, P.; Liu, J.; Radaeva, S.; Batkai, S.; Harvey-White, J.; Mackie, K.; Offertaler, L.; Wang, L.; et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J. Clin. Investig. 2005, 115, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Osei-Hyiaman, D.; Liu, J.; Zhou, L.; Godlewski, G.; Harvey-White, J.; Jeong, W.I.; Batkai, S.; Marsicano, G.; Lutz, B.; Buettner, C.; et al. Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J. Clin. Investig. 2008, 118, 3160–3169. [Google Scholar] [CrossRef]
- Soria-Gomez, E.; Bellocchio, L.; Reguero, L.; Lepousez, G.; Martin, C.; Bendahmane, M.; Ruehle, S.; Remmers, F.; Desprez, T.; Matias, I.; et al. The endocannabinoid system controls food intake via olfactory processes. Nat. Neurosci. 2014, 17, 407–415. [Google Scholar] [CrossRef]
- Ravinet Trillou, C.; Arnone, M.; Delgorge, C.; Gonalons, N.; Keane, P.; Maffrand, J.P.; Soubrie, P. Anti-obesity effect of SR141716, a CB1 receptor antagonist, in diet-induced obese mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R345–R353. [Google Scholar] [CrossRef]
- Ravinet Trillou, C.; Delgorge, C.; Menet, C.; Arnone, M.; Soubrie, P. CB1 cannabinoid receptor knockout in mice leads to leanness, resistance to diet-induced obesity and enhanced leptin sensitivity. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 640–648. [Google Scholar] [CrossRef]
- Bura, S.A.; Burokas, A.; Martin-Garcia, E.; Maldonado, R. Effects of chronic nicotine on food intake and anxiety-like behaviour in CB(1) knockout mice. Eur. Neuropsychopharmacol. 2010, 20, 369–378. [Google Scholar] [CrossRef]
- Brommage, R.; Desai, U.; Revelli, J.P.; Donoviel, D.B.; Fontenot, G.K.; Dacosta, C.M.; Smith, D.D.; Kirkpatrick, L.L.; Coker, K.J.; Donoviel, M.S.; et al. High-throughput screening of mouse knockout lines identifies true lean and obese phenotypes. Obesity 2008, 16, 2362–2367. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.R.; Gay, J.P.; Wilganowski, N.; Doree, D.; Savelieva, K.V.; Lanthorn, T.H.; Read, R.; Vogel, P.; Hansen, G.M.; Brommage, R.; et al. Diacylglycerol Lipase alpha Knockout Mice Demonstrate Metabolic and Behavioral Phenotypes Similar to Those of Cannabinoid Receptor 1 Knockout Mice. Front. Endocrinol. 2015, 6, 86. [Google Scholar] [CrossRef]
- Quarta, C.; Bellocchio, L.; Mancini, G.; Mazza, R.; Cervino, C.; Braulke, L.J.; Fekete, C.; Latorre, R.; Nanni, C.; Bucci, M.; et al. CB(1) signaling in forebrain and sympathetic neurons is a key determinant of endocannabinoid actions on energy balance. Cell Metab. 2010, 11, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Poncelet, M.; Maruani, J.; Calassi, R.; Soubrie, P. Overeating, alcohol and sucrose consumption decrease in CB1 receptor deleted mice. Neurosci. Lett. 2003, 343, 216–218. [Google Scholar] [CrossRef]
- Sanchis-Segura, C.; Cline, B.H.; Marsicano, G.; Lutz, B.; Spanagel, R. Reduced sensitivity to reward in CB1 knockout mice. Psychopharmacology 2004, 176, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.J.; Dykstra, L.A. The role of CB1 receptors in sweet versus fat reinforcement: Effect of CB1 receptor deletion, CB1 receptor antagonism (SR141716A) and CB1 receptor agonism (CP-55940). Behav. Pharmacol. 2005, 16, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Guegan, T.; Cutando, L.; Ayuso, E.; Santini, E.; Fisone, G.; Bosch, F.; Martinez, A.; Valjent, E.; Maldonado, R.; Martin, M. Operant behavior to obtain palatable food modifies neuronal plasticity in the brain reward circuit. Eur. Neuropsychopharmacol. 2013, 23, 146–159. [Google Scholar] [CrossRef]
- Mancino, S.; Burokas, A.; Gutierrez-Cuesta, J.; Gutierrez-Martos, M.; Martin-Garcia, E.; Pucci, M.; Falconi, A.; D’Addario, C.; Maccarrone, M.; Maldonado, R. Epigenetic and Proteomic Expression Changes Promoted by Eating Addictive-Like Behavior. Neuropsychopharmacology 2015, 40, 2788–2800. [Google Scholar] [CrossRef]
- Lafenetre, P.; Chaouloff, F.; Marsicano, G. Bidirectional regulation of novelty-induced behavioral inhibition by the endocannabinoid system. Neuropharmacology 2009, 57, 715–721. [Google Scholar] [CrossRef]
- Domingo-Rodriguez, L.; Ruiz de Azua, I.; Dominguez, E.; Senabre, E.; Serra, I.; Kummer, S.; Navandar, M.; Baddenhausen, S.; Hofmann, C.; Andero, R.; et al. A specific prelimbic-nucleus accumbens pathway controls resilience versus vulnerability to food addiction. Nat. Commun. 2020, 11, 782. [Google Scholar] [CrossRef]
- Ruiz de Azua, I.; Martin-Garcia, E.; Domingo-Rodriguez, L.; Aparisi Rey, A.; Pascual Cuadrado, D.; Islami, L.; Turunen, P.; Remmers, F.; Lutz, B.; Maldonado, R. Cannabinoid CB1 receptor in dorsal telencephalic glutamatergic neurons drives overconsumption of palatable food and obesity. Neuropsychopharmacology 2021, 46, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Cardinal, P.; Andre, C.; Quarta, C.; Bellocchio, L.; Clark, S.; Elie, M.; Leste-Lasserre, T.; Maitre, M.; Gonzales, D.; Cannich, A.; et al. CB1 cannabinoid receptor in SF1-expressing neurons of the ventromedial hypothalamus determines metabolic responses to diet and leptin. Mol. Metab. 2014, 3, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Cardinal, P.; Bellocchio, L.; Guzman-Quevedo, O.; Andre, C.; Clark, S.; Elie, M.; Leste-Lasserre, T.; Gonzales, D.; Cannich, A.; Marsicano, G.; et al. Cannabinoid type 1 (CB1) receptors on Sim1-expressing neurons regulate energy expenditure in male mice. Endocrinology 2015, 156, 411–418. [Google Scholar] [CrossRef]
- Cardinal, P.; Bellocchio, L.; Clark, S.; Cannich, A.; Klugmann, M.; Lutz, B.; Marsicano, G.; Cota, D. Hypothalamic CB1 cannabinoid receptors regulate energy balance in mice. Endocrinology 2012, 153, 4136–4143. [Google Scholar] [CrossRef]
- Pang, Z.; Wu, N.N.; Zhao, W.; Chain, D.C.; Schaffer, E.; Zhang, X.; Yamdagni, P.; Palejwala, V.A.; Fan, C.; Favara, S.G.; et al. The central cannabinoid CB1 receptor is required for diet-induced obesity and rimonabant’s antiobesity effects in mice. Obesity 2011, 19, 1923–1934. [Google Scholar] [CrossRef] [PubMed]
- Avalos, B.; Argueta, D.A.; Perez, P.A.; Wiley, M.; Wood, C.; DiPatrizio, N.V. Cannabinoid CB(1) Receptors in the Intestinal Epithelium Are Required for Acute Western-Diet Preferences in Mice. Nutrients 2020, 12, 2874. [Google Scholar] [CrossRef]
- Agudo, J.; Martin, M.; Roca, C.; Molas, M.; Bura, A.S.; Zimmer, A.; Bosch, F.; Maldonado, R. Deficiency of CB2 cannabinoid receptor in mice improves insulin sensitivity but increases food intake and obesity with age. Diabetologia 2010, 53, 2629–2640. [Google Scholar] [CrossRef]
- Deveaux, V.; Cadoudal, T.; Ichigotani, Y.; Teixeira-Clerc, F.; Louvet, A.; Manin, S.; Nhieu, J.T.; Belot, M.P.; Zimmer, A.; Even, P.; et al. Cannabinoid CB2 receptor potentiates obesity-associated inflammation, insulin resistance and hepatic steatosis. PLoS ONE 2009, 4, e5844. [Google Scholar] [CrossRef]
- Flake, N.M.; Zweifel, L.S. Behavioral effects of pulp exposure in mice lacking cannabinoid receptor 2. J. Endod. 2012, 38, 86–90. [Google Scholar] [CrossRef]
- Pradier, B.; Erxlebe, E.; Markert, A.; Racz, I. Interaction of cannabinoid receptor 2 and social environment modulates chronic alcohol consumption. Behav. Brain. Res. 2015, 287, 163–171. [Google Scholar] [CrossRef]
- Garcia-Blanco, A.; Ramirez-Lopez, A.; Navarrete, F.; Garcia-Gutierrez, M.S.; Manzanares, J.; Martin-Garcia, E.; Maldonado, R. Role of CB2 cannabinoid receptor in the development of food addiction in male mice. Neurobiol. Dis. 2023, 179, 106034. [Google Scholar] [CrossRef]
- Romero-Zerbo, S.Y.; Garcia-Gutierrez, M.S.; Suarez, J.; Rivera, P.; Ruz-Maldonado, I.; Vida, M.; Rodriguez de Fonseca, F.; Manzanares, J.; Bermudez-Silva, F.J. Overexpression of cannabinoid CB2 receptor in the brain induces hyperglycaemia and a lean phenotype in adult mice. J. Neuroendocrinol. 2012, 24, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Vasilyev, D.V.; Goncalves, M.B.; Howell, F.V.; Hobbs, C.; Reisenberg, M.; Shen, R.; Zhang, M.Y.; Strassle, B.W.; Lu, P.; et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J. Neurosci. 2010, 30, 2017–2024. [Google Scholar] [CrossRef] [PubMed]
- Cravatt, B.F.; Demarest, K.; Patricelli, M.P.; Bracey, M.H.; Giang, D.K.; Martin, B.R.; Lichtman, A.H. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2001, 98, 9371–9376. [Google Scholar] [CrossRef] [PubMed]
- Tourino, C.; Oveisi, F.; Lockney, J.; Piomelli, D.; Maldonado, R. FAAH deficiency promotes energy storage and enhances the motivation for food. Int. J. Obes. 2010, 34, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Bello, N.T.; Coughlin, J.W.; Redgrave, G.W.; Ladenheim, E.E.; Moran, T.H.; Guarda, A.S. Dietary conditions and highly palatable food access alter rat cannabinoid receptor expression and binding density. Physiol. Behav. 2012, 105, 720–726. [Google Scholar] [CrossRef]
- Blanco-Gandia, M.C.; Cantacorps, L.; Aracil-Fernandez, A.; Montagud-Romero, S.; Aguilar, M.A.; Manzanares, J.; Valverde, O.; Minarro, J.; Rodriguez-Arias, M. Effects of bingeing on fat during adolescence on the reinforcing effects of cocaine in adult male mice. Neuropharmacology 2017, 113, 31–44. [Google Scholar] [CrossRef]
- Wong, K.J.; Wojnicki, F.H.; Corwin, R.L. Baclofen, raclopride, and naltrexone differentially affect intake of fat/sucrose mixtures under limited access conditions. Pharmacol. Biochem. Behav. 2009, 92, 528–536. [Google Scholar] [CrossRef]
- Tam, J.; Vemuri, V.K.; Liu, J.; Batkai, S.; Mukhopadhyay, B.; Godlewski, G.; Osei-Hyiaman, D.; Ohnuma, S.; Ambudkar, S.V.; Pickel, J.; et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J. Clin. Investig. 2010, 120, 2953–2966. [Google Scholar] [CrossRef]
- Jamshidi, N.; Taylor, D.A. Anandamide administration into the ventromedial hypothalamus stimulates appetite in rats. Br. J. Pharmacol. 2001, 134, 1151–1154. [Google Scholar] [CrossRef]
- Williams, C.M.; Kirkham, T.C. Anandamide induces overeating: Mediation by central cannabinoid (CB1) receptors. Psychopharmacology 1999, 143, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Salaya-Velazquez, N.F.; Lopez-Mucino, L.A.; Mejia-Chavez, S.; Sanchez-Aparicio, P.; Dominguez-Guadarrama, A.A.; Venebra-Munoz, A. Anandamide and sucralose change DeltaFosB expression in the reward system. Neuroreport 2020, 31, 240–244. [Google Scholar] [CrossRef] [PubMed]
- De Luca, M.A.; Valentini, V.; Bimpisidis, Z.; Cacciapaglia, F.; Caboni, P.; Di Chiara, G. Endocannabinoid 2-Arachidonoylglycerol Self-Administration by Sprague-Dawley Rats and Stimulation of in vivo Dopamine Transmission in the Nucleus Accumbens Shell. Front. Psychiatry 2014, 5, 140. [Google Scholar] [CrossRef] [PubMed]
- Solinas, M.; Justinova, Z.; Goldberg, S.R.; Tanda, G. Anandamide administration alone and after inhibition of fatty acid amide hydrolase (FAAH) increases dopamine levels in the nucleus accumbens shell in rats. J. Neurochem. 2006, 98, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, P.; Maj, M. Dysfunctions of leptin, ghrelin, BDNF and endocannabinoids in eating disorders: Beyond the homeostatic control of food intake. Psychoneuroendocrinology 2013, 38, 312–330. [Google Scholar] [CrossRef]
- Melis, M.; Perra, S.; Muntoni, A.L.; Pillolla, G.; Lutz, B.; Marsicano, G.; Di Marzo, V.; Gessa, G.L.; Pistis, M. Prefrontal cortex stimulation induces 2-arachidonoyl-glycerol-mediated suppression of excitation in dopamine neurons. J. Neurosci. 2004, 24, 10707–10715. [Google Scholar] [CrossRef]
- Bossong, M.G.; van Berckel, B.N.; Boellaard, R.; Zuurman, L.; Schuit, R.C.; Windhorst, A.D.; van Gerven, J.M.; Ramsey, N.F.; Lammertsma, A.A.; Kahn, R.S. Delta 9-tetrahydrocannabinol induces dopamine release in the human striatum. Neuropsychopharmacology 2009, 34, 759–766. [Google Scholar] [CrossRef]
- DiPatrizio, N.V. Is fat taste ready for primetime? Physiol. Behav. 2014, 136, 145–154. [Google Scholar] [CrossRef]
- Tanda, G.; Goldberg, S.R. Cannabinoids: Reward, dependence, and underlying neurochemical mechanisms—A review of recent preclinical data. Psychopharmacology 2003, 169, 115–134. [Google Scholar] [CrossRef]
- Mahler, S.V.; Smith, K.S.; Berridge, K.C. Endocannabinoid hedonic hotspot for sensory pleasure: Anandamide in nucleus accumbens shell enhances ‘liking’ of a sweet reward. Neuropsychopharmacology 2007, 32, 2267–2278. [Google Scholar] [CrossRef]
- Barros, D.M.; Carlis, V.; Maidana, M.; Silva, E.S.; Baisch, A.L.; Ramirez, M.R.; Izquierdo, I. Interactions between anandamide-induced anterograde amnesia and post-training memory modulatory systems. Brain Res. 2004, 1016, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Robinson, L.; McKillop-Smith, S.; Ross, N.L.; Pertwee, R.G.; Hampson, R.E.; Platt, B.; Riedel, G. Hippocampal endocannabinoids inhibit spatial learning and limit spatial memory in rats. Psychopharmacology 2008, 198, 551–563. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Alvares, L.; Genro, B.P.; Diehl, F.; Quillfeldt, J.A. Differential role of the hippocampal endocannabinoid system in the memory consolidation and retrieval mechanisms. Neurobiol. Learn. Mem. 2008, 90, 1–9. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Alvares, L.; de Oliveira, L.F.; Camboim, C.; Diehl, F.; Genro, B.P.; Lanziotti, V.B.; Quillfeldt, J.A. Amnestic effect of intrahippocampal AM251, a CB1-selective blocker, in the inhibitory avoidance, but not in the open field habituation task, in rats. Neurobiol. Learn. Mem. 2005, 83, 119–124. [Google Scholar] [CrossRef]
- Bucherelli, C.; Baldi, E.; Mariottini, C.; Passani, M.B.; Blandina, P. Aversive memory reactivation engages in the amygdala only some neurotransmitters involved in consolidation. Learn. Mem. 2006, 13, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Campolongo, P.; Roozendaal, B.; Trezza, V.; Hauer, D.; Schelling, G.; McGaugh, J.L.; Cuomo, V. Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proc. Natl. Acad. Sci. USA 2009, 106, 4888–4893. [Google Scholar] [CrossRef]
- McGaugh, J.L. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu. Rev. Neurosci. 2004, 27, 1–28. [Google Scholar] [CrossRef]
- Soria-Gomez, E.; Massa, F.; Bellocchio, L.; Rueda-Orozco, P.E.; Ciofi, P.; Cota, D.; Oliet, S.H.; Prospero-Garcia, O.; Marsicano, G. Cannabinoid type-1 receptors in the paraventricular nucleus of the hypothalamus inhibit stimulated food intake. Neuroscience 2014, 263, 46–53. [Google Scholar] [CrossRef]
- De Ceglia, M.; Decara, J.; Gaetani, S.; Rodriguez de Fonseca, F. Obesity as a Condition Determined by Food Addiction: Should Brain Endocannabinoid System Alterations Be the Cause and Its Modulation the Solution? Pharmaceuticals 2021, 14, 1002. [Google Scholar] [CrossRef]
- Morales, I.; Berridge, K.C. ‘Liking’ and ‘wanting’ in eating and food reward: Brain mechanisms and clinical implications. Physiol. Behav. 2020, 227, 113152. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourdy, R.; Befort, K. The Role of the Endocannabinoid System in Binge Eating Disorder. Int. J. Mol. Sci. 2023, 24, 9574. https://doi.org/10.3390/ijms24119574
Bourdy R, Befort K. The Role of the Endocannabinoid System in Binge Eating Disorder. International Journal of Molecular Sciences. 2023; 24(11):9574. https://doi.org/10.3390/ijms24119574
Chicago/Turabian StyleBourdy, Romain, and Katia Befort. 2023. "The Role of the Endocannabinoid System in Binge Eating Disorder" International Journal of Molecular Sciences 24, no. 11: 9574. https://doi.org/10.3390/ijms24119574