
Alu-Mediated Insertions in the DMD Gene: A Difficult Puzzle to Interpret Clinically

 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
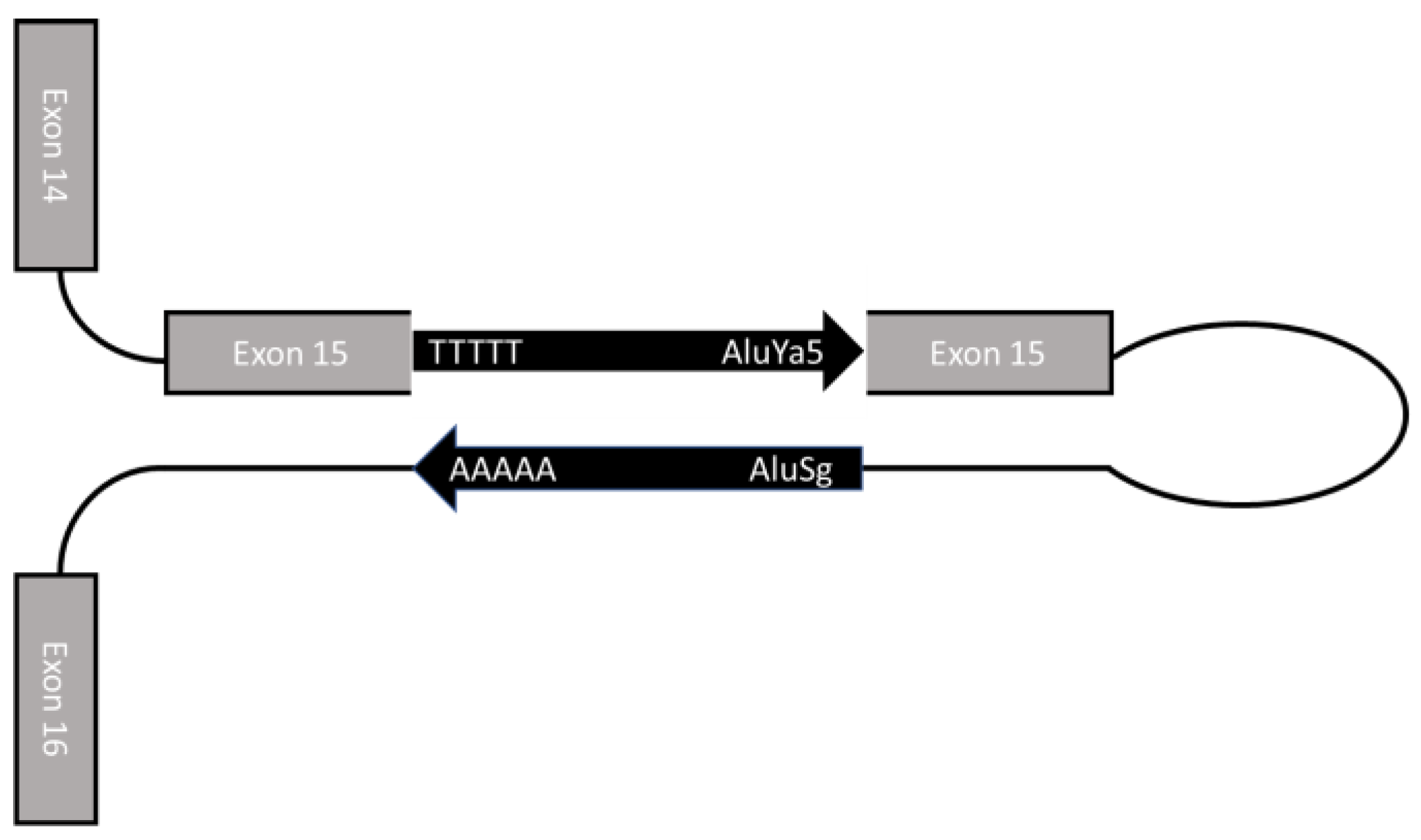
2.1. Case Presentation
2.2. Molecular Diagnosis
3. Discussion
4. Materials and Methods
4.1. Multiplex Ligation-Dependent Probe Amplification
4.2. Log-PCR
4.3. Polymerase Chain Reaction and Sanger Sequencing
4.4. RT-PCR
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nigro, V.; Piluso, G. Spectrum of muscular dystrophies associated with sarcolemmal-protein genetic defects. Biochim. Biophys. Acta 2015, 1852, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M. Duchenne and Becker muscular dystrophies. Neurol. Clin. 2014, 32, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Neri, M.; Rossi, R.; Trabanelli, C.; Mauro, A.; Selvatici, R.; Falzarano, M.S.; Spedicato, N.; Margutti, A.; Rimessi, P.; Fortunato, F.; et al. The Genetic Landscape of Dystrophin Mutations in Italy: A Nationwide Study. Front. Genet. 2020, 11, 131. [Google Scholar] [CrossRef] [PubMed]
- Torella, A.; Zanobio, M.; Zeuli, R.; Del Vecchio Blanco, F.; Savarese, M.; Giugliano, T.; Garofalo, A.; Piluso, G.; Politano, L.; Nigro, V. The position of nonsense mutations can predict the phenotype severity: A survey on the DMD gene. PLoS ONE 2020, 15, e0237803. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C.; Pritham, E.J. DNA transposons and the evolution of eukaryotic genomes. Annu. Rev. Genet. 2007, 41, 331–368. [Google Scholar] [CrossRef] [PubMed]
- Batzer, M.A.; Deininger, P.L. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002, 3, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Nakamura, A.; Yazaki, M.; Ikeda, S.; Takeda, S. Insertional mutation by transposable element, L1, in the DMD gene results in X-linked dilated cardiomyopathy. Hum. Mol. Genet. 1998, 7, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Ferlini, A.; Galie, N.; Merlini, L.; Sewry, C.; Branzi, A.; Muntoni, F. A novel Alu-like element rearranged in the dystrophin gene causes a splicing mutation in a family with X-linked dilated cardiomyopathy. Am. J. Hum. Genet. 1998, 63, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Sun, C.; Liu, Y.; Yu, M.; Zheng, Y.; Meng, L.; Wang, G.; Cornejo-Sanchez, D.M.; Bharadwaj, T.; Yan, J.; et al. Practical approach to the genetic diagnosis of unsolved dystrophinopathies: A stepwise strategy in the genomic era. J. Med. Genet. 2021, 58, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Sedlackova, J.; Vondracek, P.; Hermanova, M.; Zamecnik, J.; Hruba, Z.; Haberlova, J.; Kraus, J.; Marikova, T.; Hedvicakova, P.; Vohanka, S.; et al. Point mutations in Czech DMD/BMD patients and their phenotypic outcome. Neuromuscul. Disord. 2009, 19, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Trimarco, A.; Torella, A.; Piluso, G.; Maria Ventriglia, V.; Politano, L.; Nigro, V. Log-PCR: A new tool for immediate and cost-effective diagnosis of up to 85% of dystrophin gene mutations. Clin. Chem. 2008, 54, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Viggiano, E.; Picillo, E.; Passamano, L.; Onore, M.E.; Piluso, G.; Scutifero, M.; Torella, A.; Nigro, V.; Politano, L. Spectrum of Genetic Variants in the Dystrophin Gene: A Single Centre Retrospective Analysis of 750 Duchenne and Becker Patients from Southern Italy. Genes 2023, 14, 214. [Google Scholar] [CrossRef] [PubMed]
- Savarese, M.; Qureshi, T.; Torella, A.; Laine, P.; Giugliano, T.; Jonson, P.H.; Johari, M.; Paulin, L.; Piluso, G.; Auvinen, P.; et al. Identification and Characterization of Splicing Defects by Single-Molecule Real-Time Sequencing Technology (PacBio). J. Neuromuscul. Dis. 2020, 7, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gao, M.; Zhang, K.; Jin, R.; Lv, Y.; Liu, Y.; Ma, J.; Wan, Y.; Gai, Z.; Liu, Y. Molecular Genetics Analysis of 70 Chinese Families With Muscular Dystrophy Using Multiplex Ligation-Dependent Probe Amplification and Next-Generation Sequencing. Front. Pharmacol. 2019, 10, 814. [Google Scholar] [CrossRef] [PubMed]
- Lerario, A.; Colombo, I.; Milani, D.; Peverelli, L.; Villa, L.; Del Bo, R.; Sciacco, M.; Comi, G.P.; Esposito, S.; Moggio, M. A case report with the peculiar concomitance of 2 different genetic syndromes. Medicine 2016, 95, e5567. [Google Scholar] [CrossRef] [PubMed]
- Nakama, M.; Otsuka, H.; Ago, Y.; Sasai, H.; Abdelkreem, E.; Aoyama, Y.; Fukao, T. Intronic antisense Alu elements have a negative splicing effect on the inclusion of adjacent downstream exons. Gene 2018, 664, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Nakama, M.; Otsuka, H.; Sasai, H.; Ohnishi, H.; Morishige, K.-I. A short sequence within AluSx induces downstream exon skipping in an ACAT1 minigene model. All Life 2021, 14, 869–873. [Google Scholar] [CrossRef]
- Lev-Maor, G.; Ram, O.; Kim, E.; Sela, N.; Goren, A.; Levanon, E.Y.; Ast, G. Intronic Alus influence alternative splicing. PLoS Genet. 2008, 4, e1000204. [Google Scholar] [CrossRef] [PubMed]
- Santoro, L.; Nolano, M.; Faraso, S.; Fiorillo, C.; Vitiello, C.; Provitera, V.; Aurino, S.; Nigro, V. Perioral skin biopsy to study skeletal muscle protein expression. Muscle Nerve 2010, 41, 392–398. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torella, A.; Budillon, A.; Zanobio, M.; Del Vecchio Blanco, F.; Picillo, E.; Politano, L.; Nigro, V.; Piluso, G. Alu-Mediated Insertions in the DMD Gene: A Difficult Puzzle to Interpret Clinically. Int. J. Mol. Sci. 2023, 24, 9241. https://doi.org/10.3390/ijms24119241
Torella A, Budillon A, Zanobio M, Del Vecchio Blanco F, Picillo E, Politano L, Nigro V, Piluso G. Alu-Mediated Insertions in the DMD Gene: A Difficult Puzzle to Interpret Clinically. International Journal of Molecular Sciences. 2023; 24(11):9241. https://doi.org/10.3390/ijms24119241
Chicago/Turabian StyleTorella, Annalaura, Alberto Budillon, Mariateresa Zanobio, Francesca Del Vecchio Blanco, Esther Picillo, Luisa Politano, Vincenzo Nigro, and Giulio Piluso. 2023. "Alu-Mediated Insertions in the DMD Gene: A Difficult Puzzle to Interpret Clinically" International Journal of Molecular Sciences 24, no. 11: 9241. https://doi.org/10.3390/ijms24119241