
Regulation of Mitochondrial Permeability Transition Pore Opening by Monovalent Cations in Liver Mitochondria

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
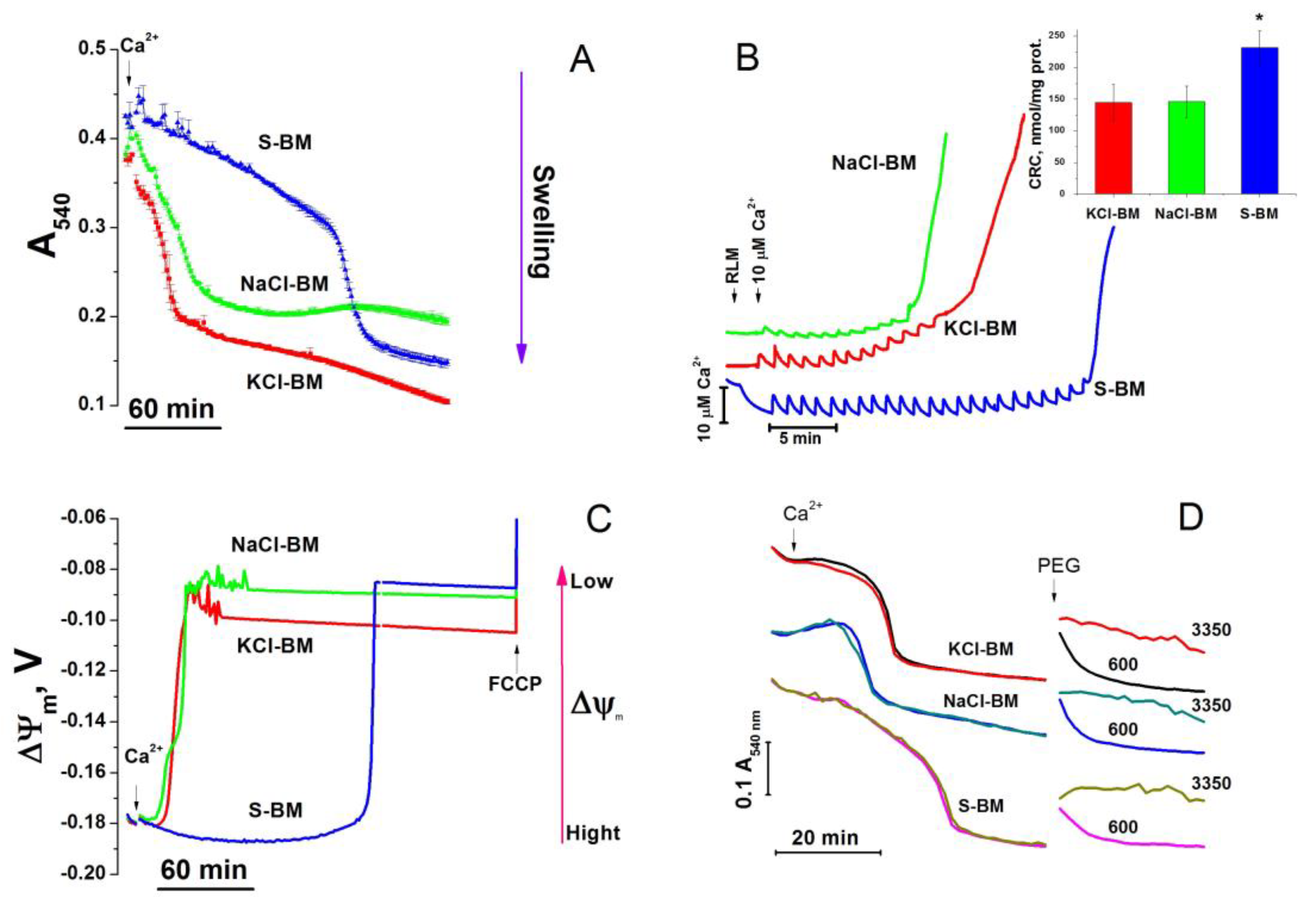
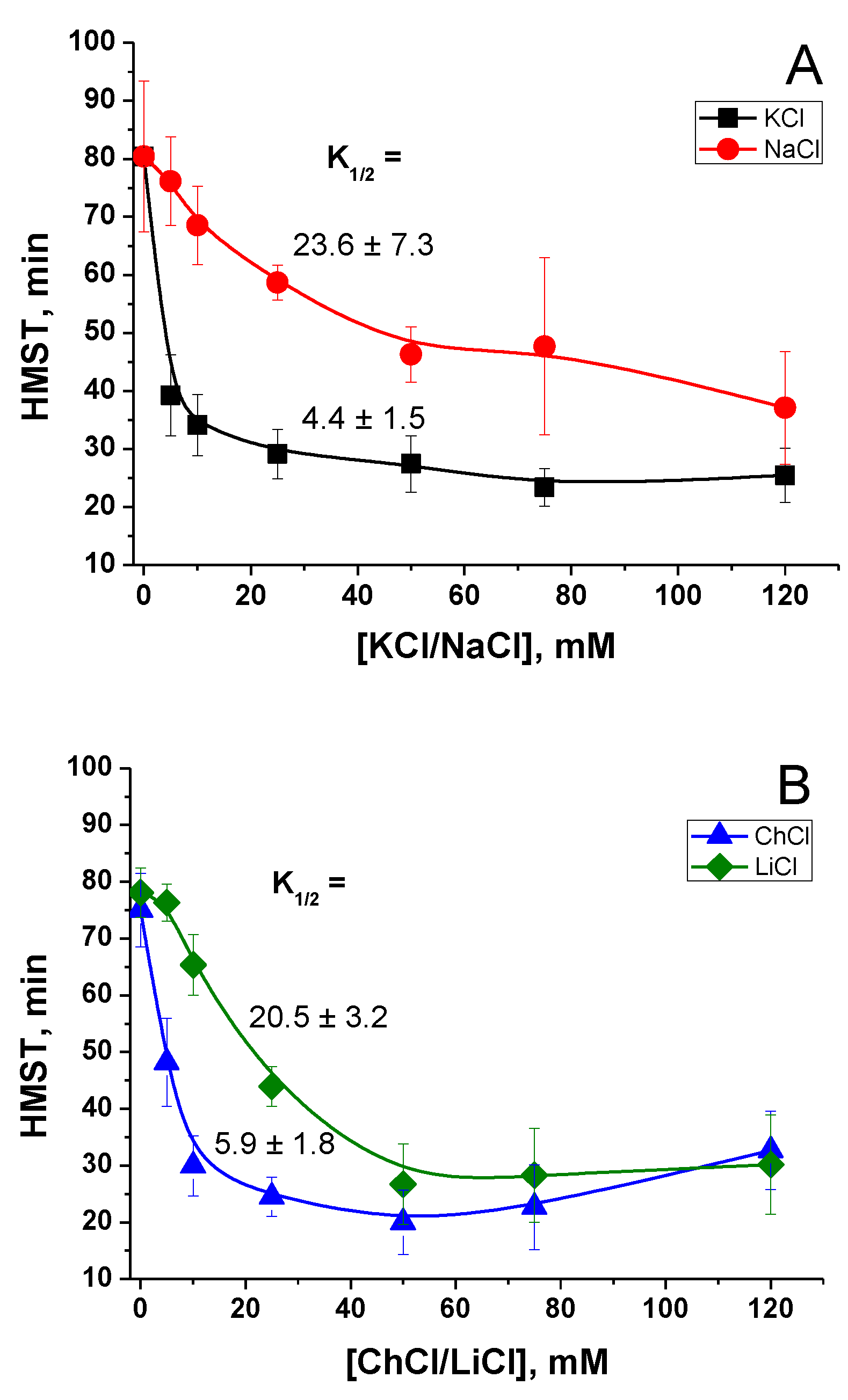
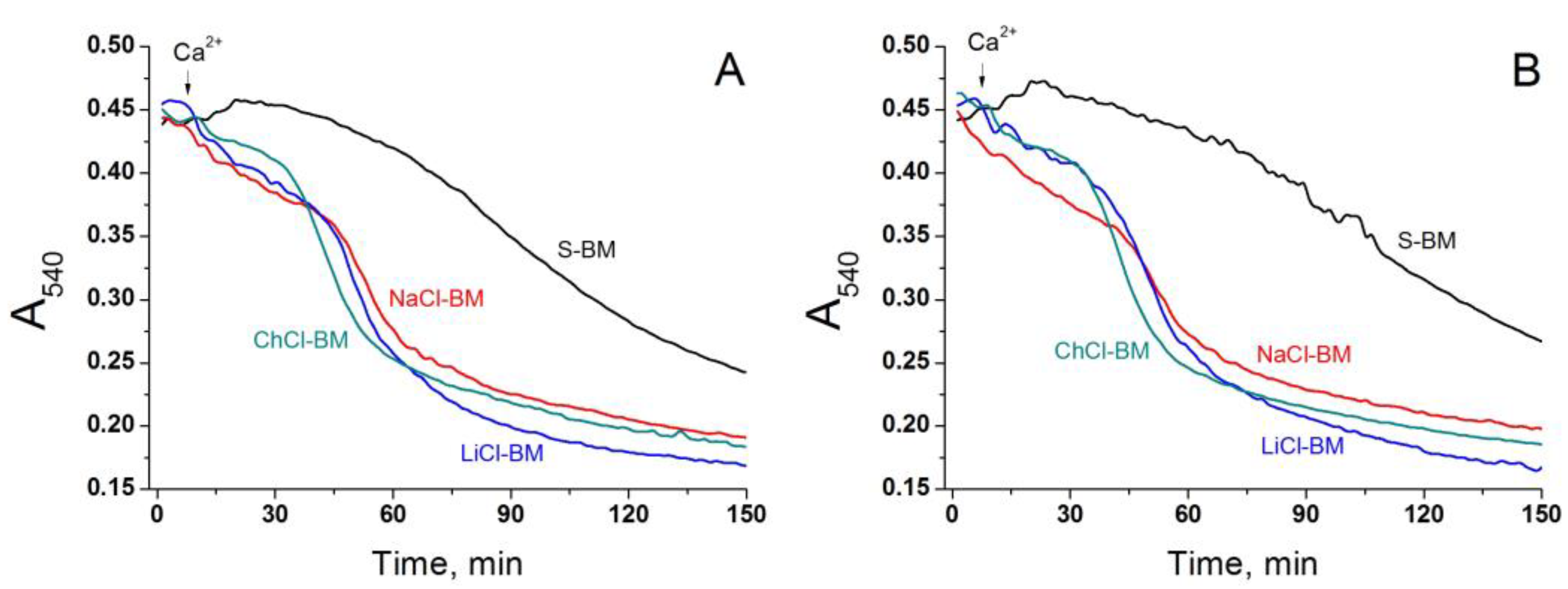
2.1. Effect of K+ and Na+ in the Incubation Medium on the Rate of PTP Opening
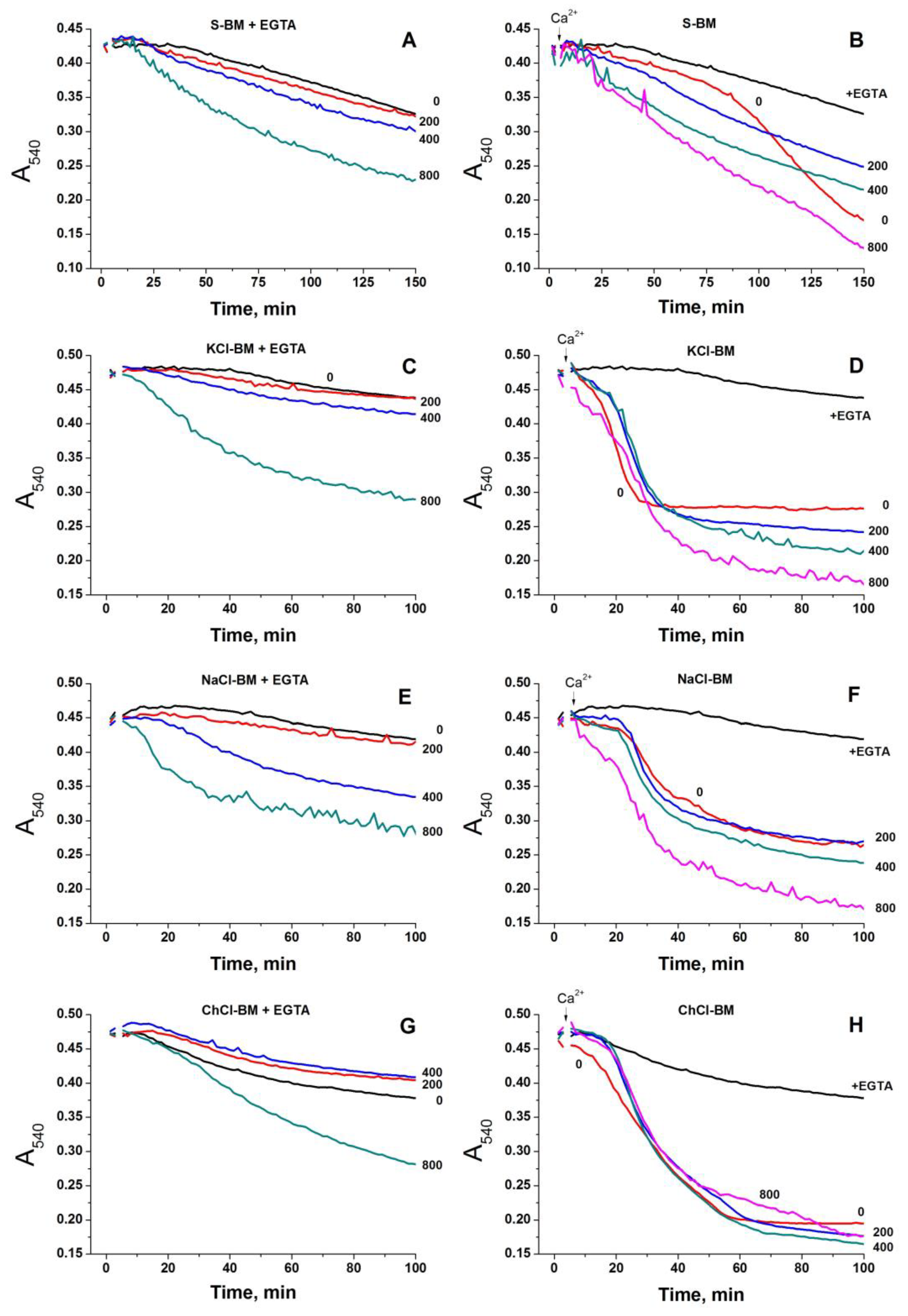
2.2. Effect of the Concentration of Salts in the Incubation Medium on the Rate of PTP Opening
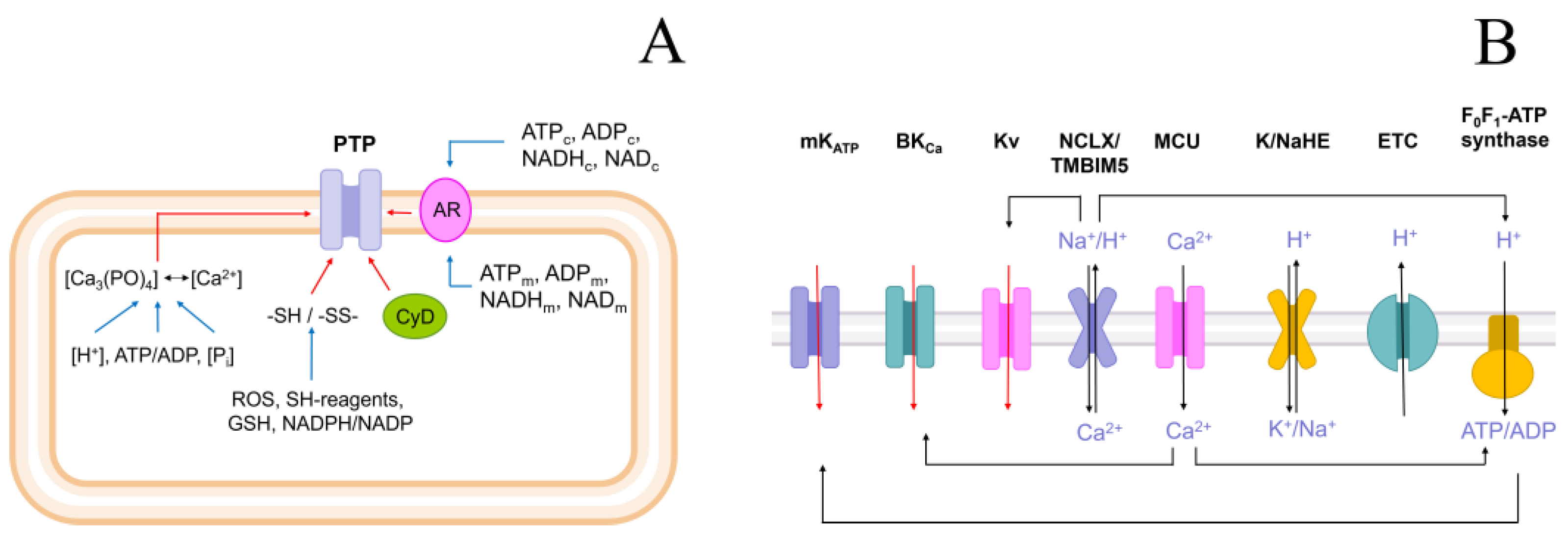
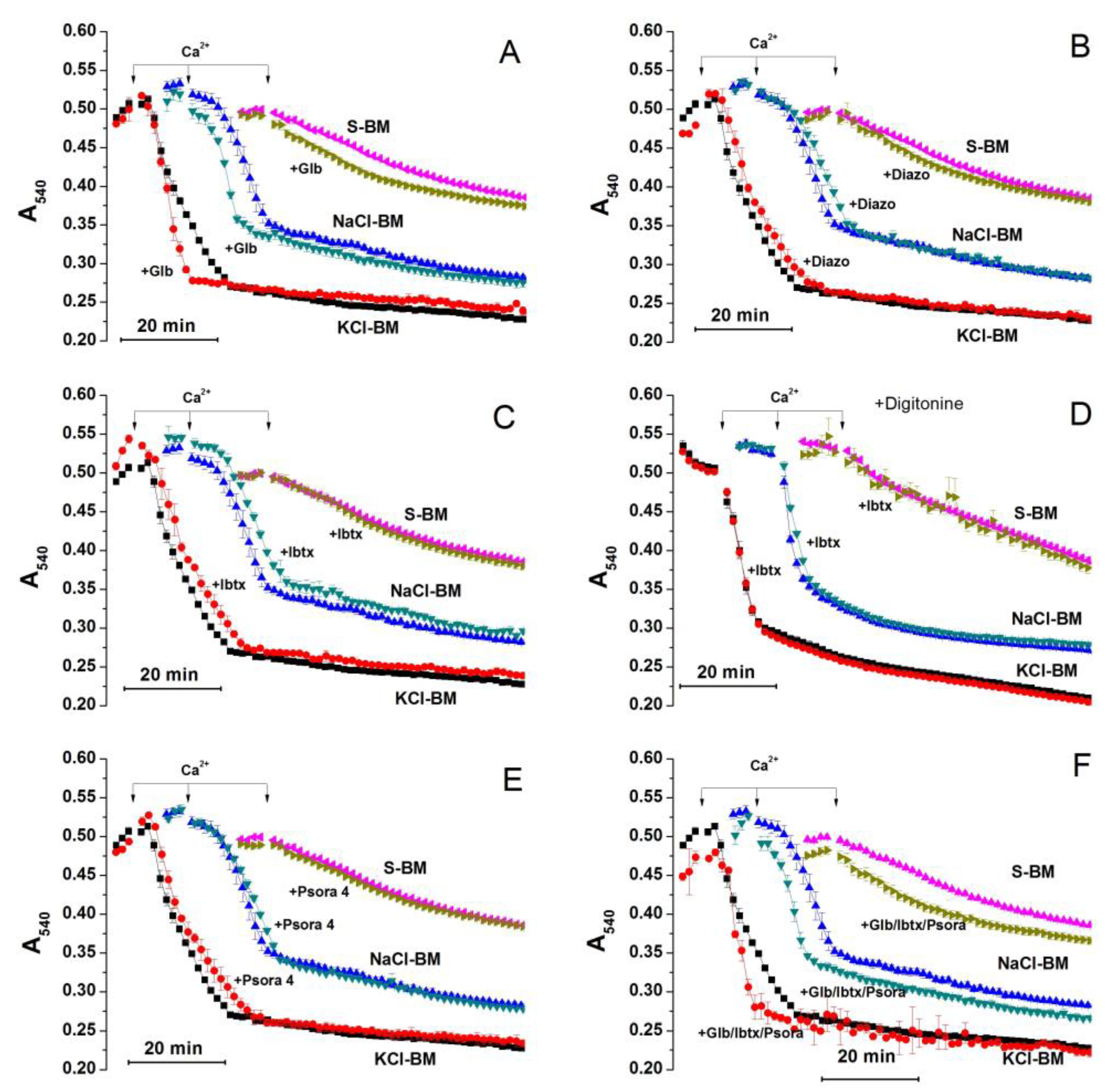
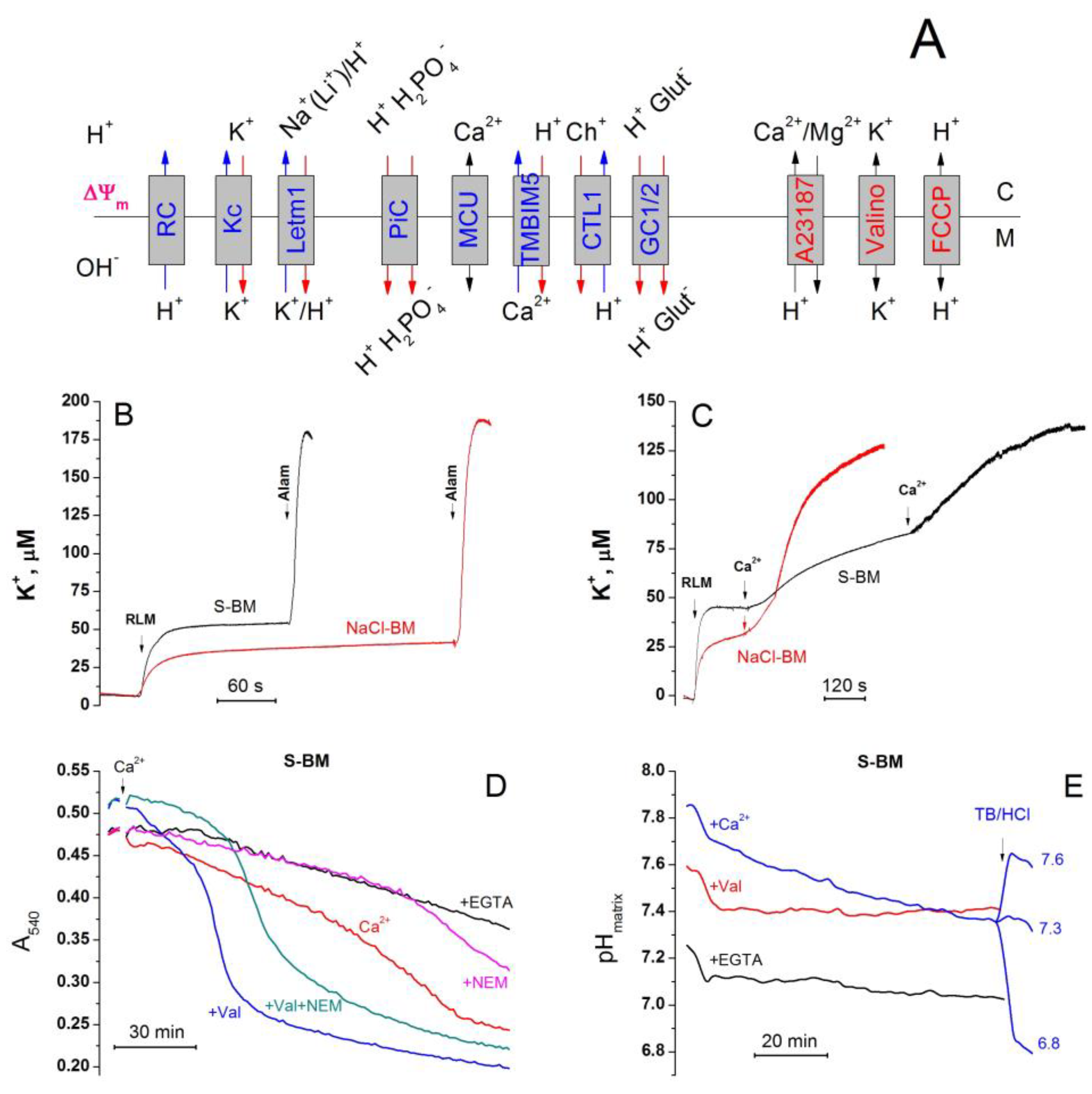
2.3. Role of K+ Channels in the Stimulation of PTP Opening
2.4. Role of Cations in the Transition of PTP from a Low- to a High-Conductance State
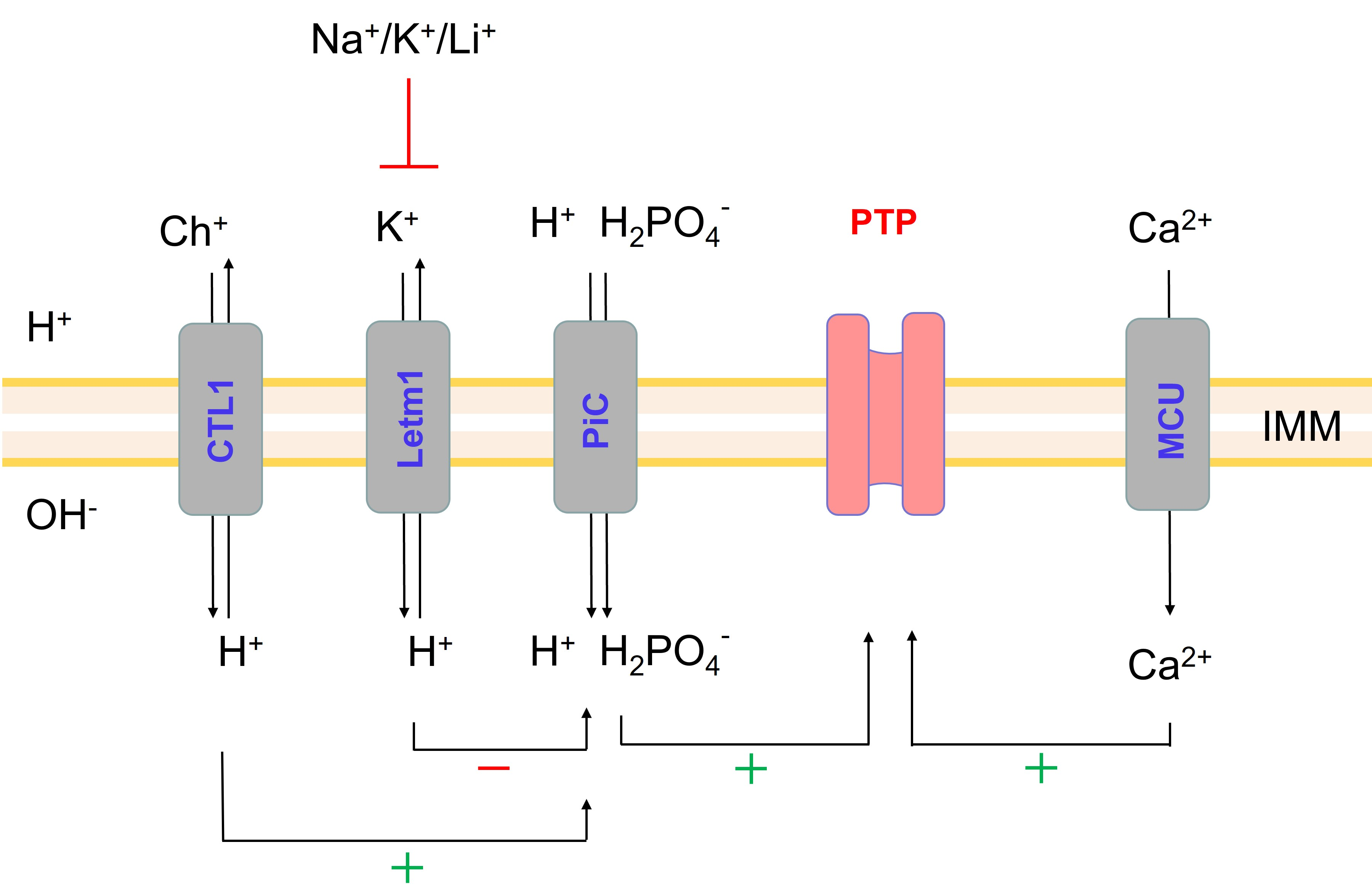
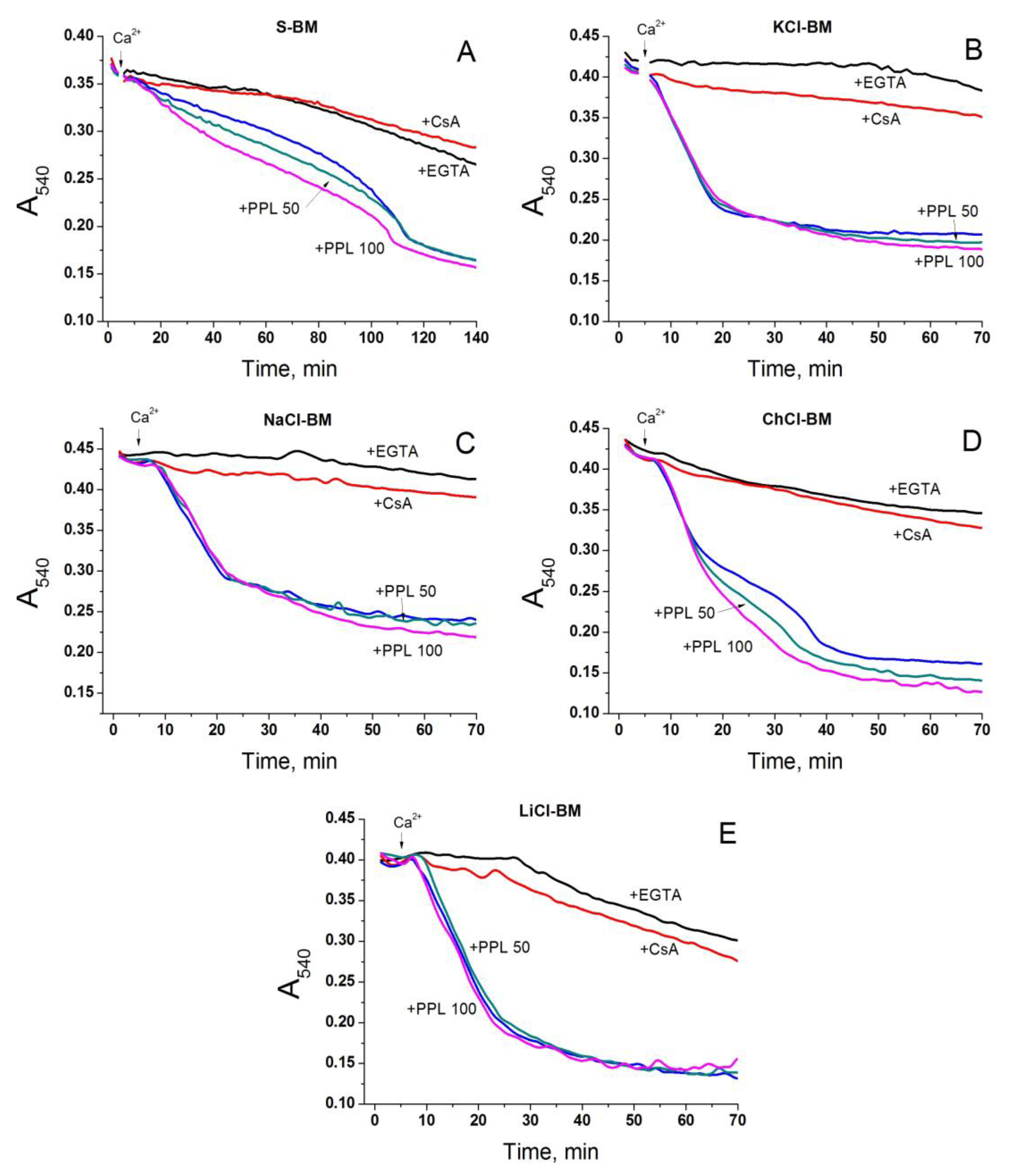
2.5. Role of Non-Selective Cation Channel in the PTP Triggering by Monovalent Cations
2.6. Role of IMAC in the Acceleration of PTP Opening in Salt-Containing Media
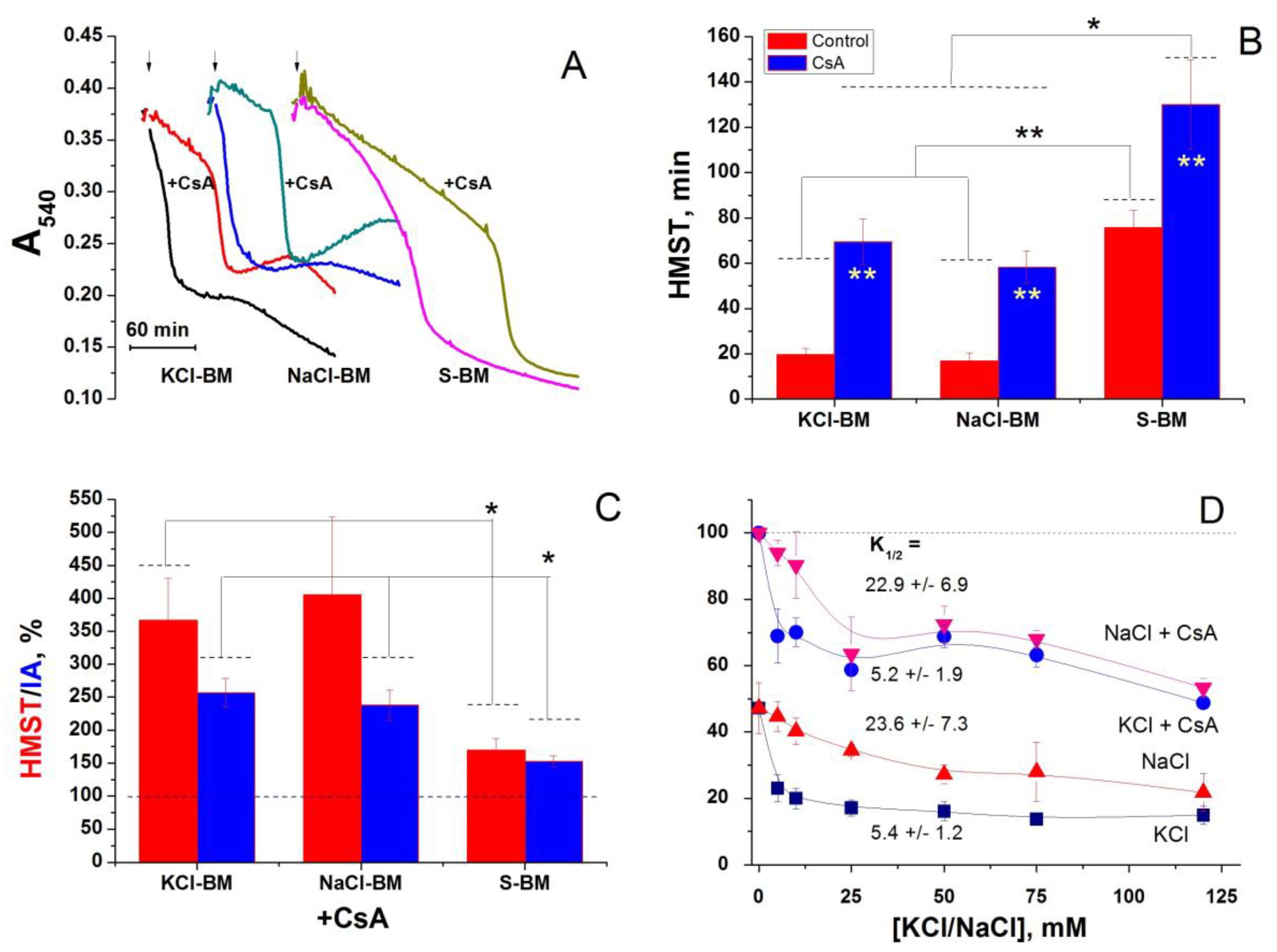
2.7. Effect of the Inhibition of K+(Na+, Li+)/H+ and Ch+/H+ Exchangers on the PTP Opening in Different Media
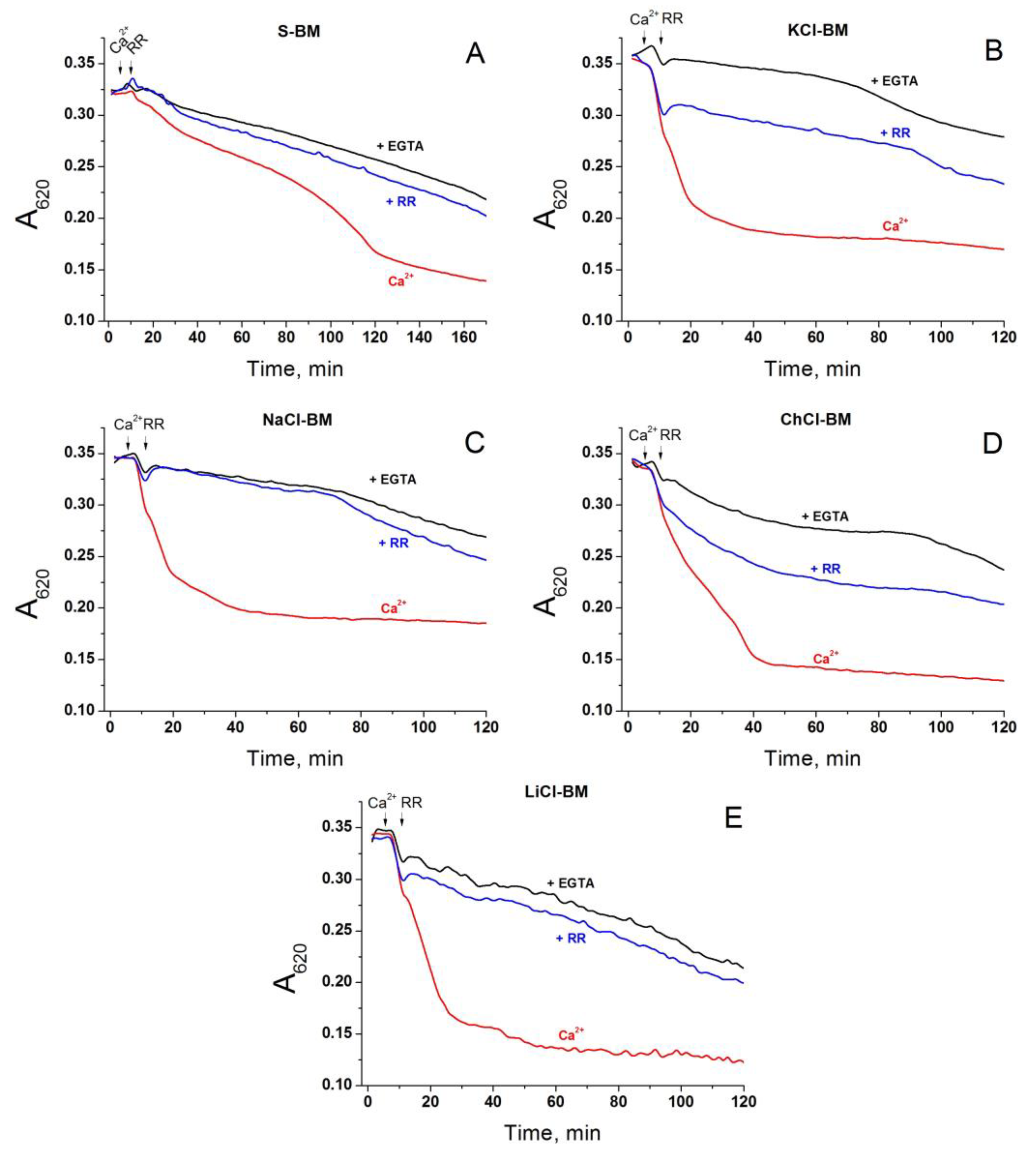
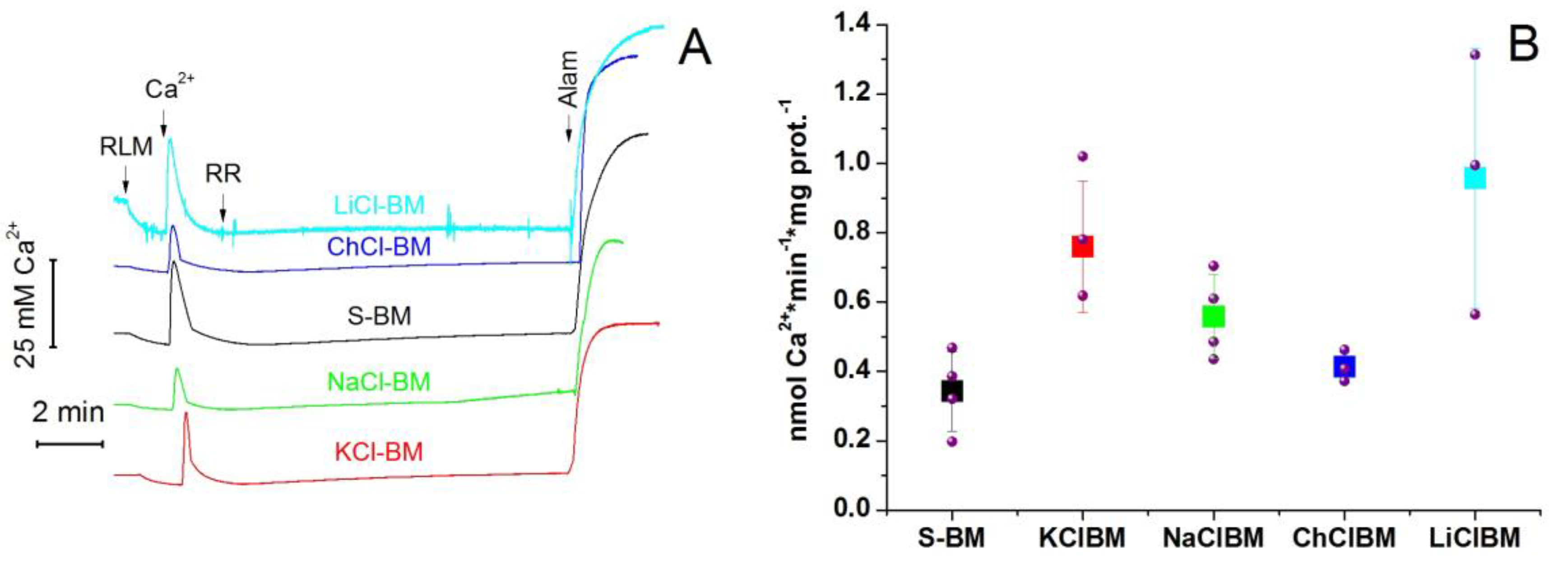
2.8. The Rate of Ca2+ Extrusion from Mitochondria in Different Incubation Media
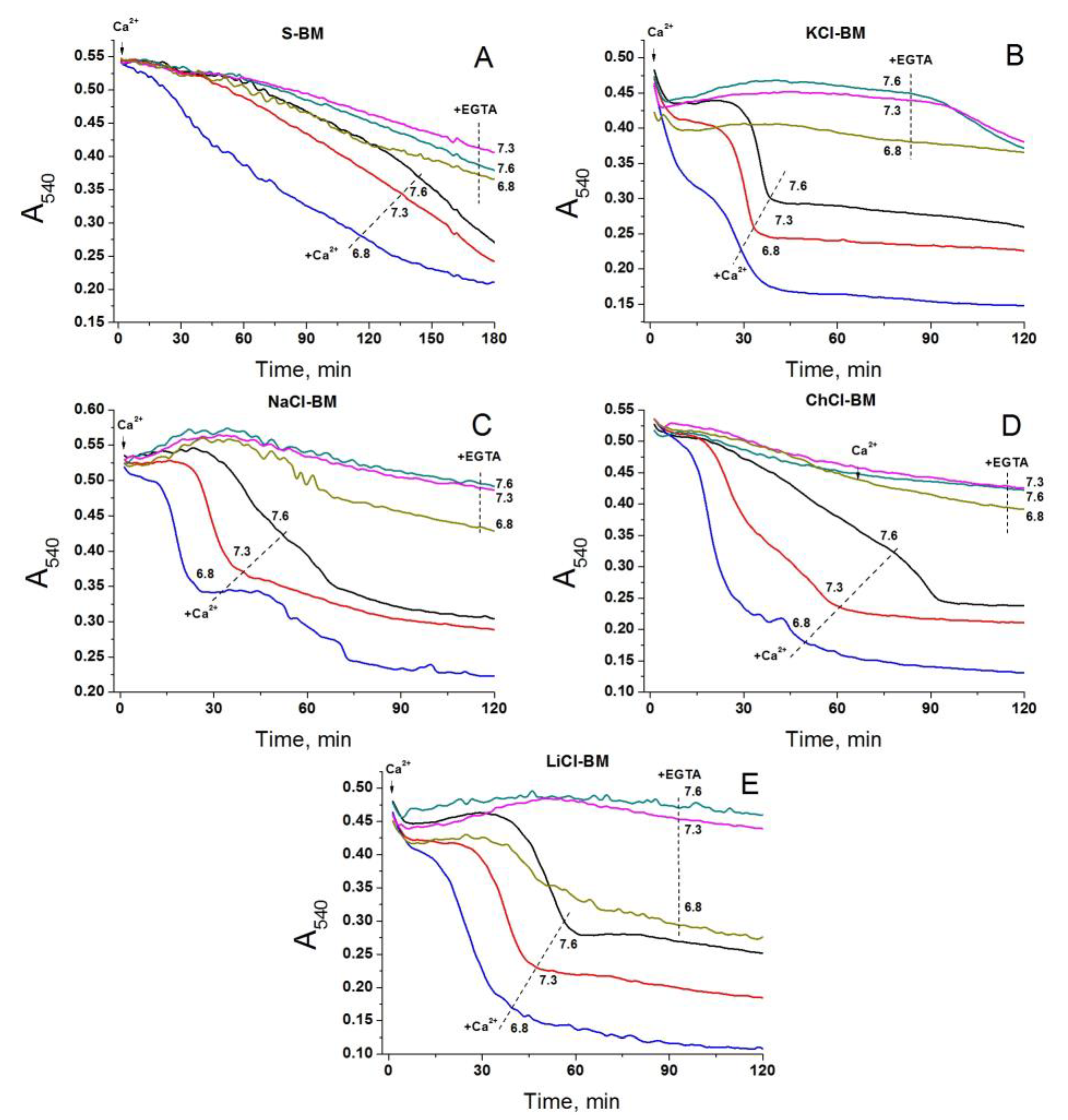
2.9. Effect of External pH on PTP Opening in Different Media
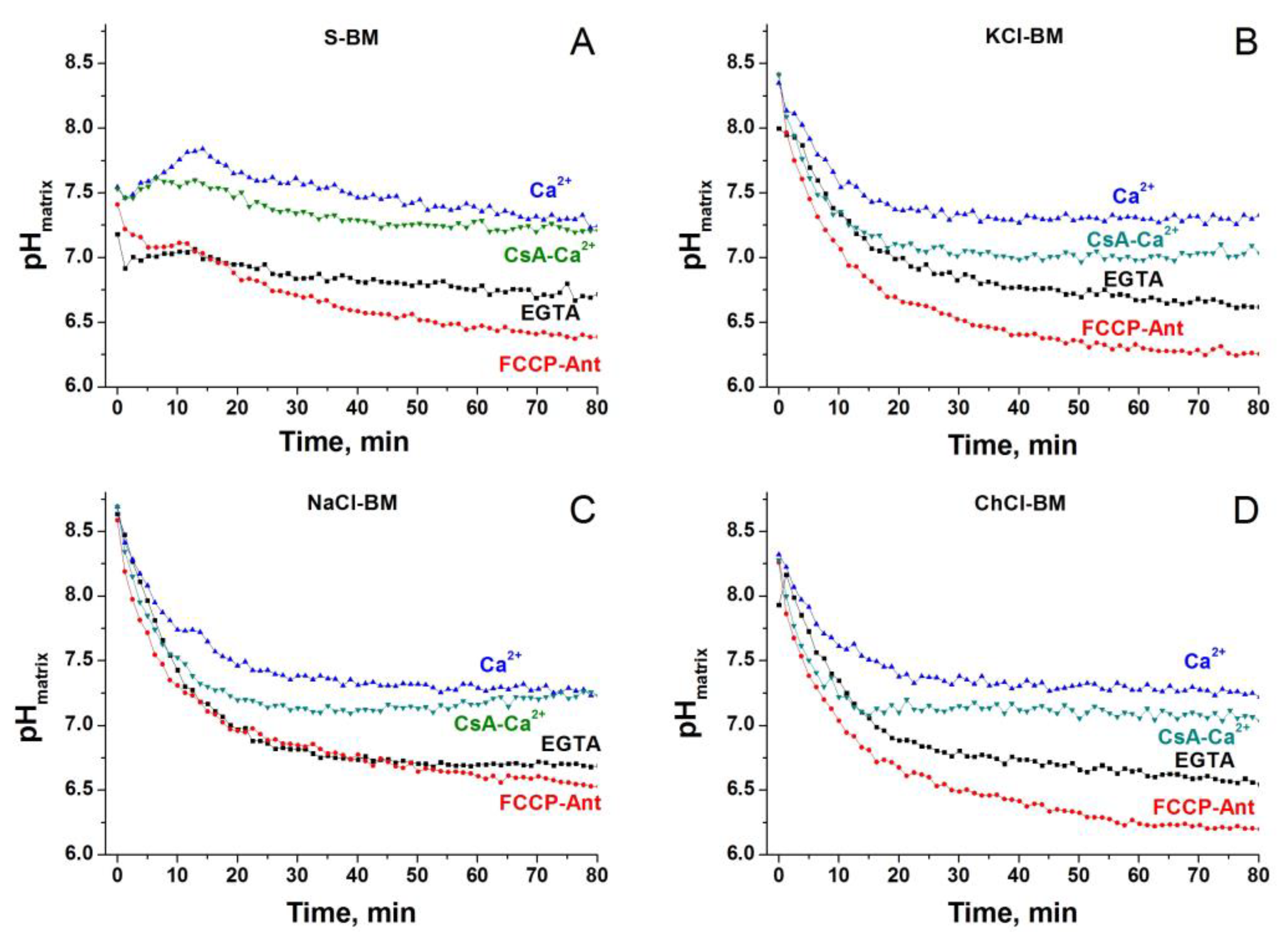
2.10. Dynamics of Matrix pH in Different Incubation Media
2.11. Cations Regulate the Activity of K+/H+ Exchanger, Matrix pH, and Pi Influx
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Isolation of Mitochondria from Rat Liver
4.3. Incubation Media
4.4. Induction and Registration of PTP Opening
4.5. Recording of the Mitochondrial Swelling
4.6. Measurements of ΔΨm in Isolated Mitochondria
4.7. Ca2+-Retention Capacity of Mitochondria
4.8. Measurements of the Oxygen Consumption Rate
4.9. Registration of the Release of Matrix K+ from Mitochondria
4.10. Determination of Matrix pH
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buja, L.M. Pathobiology of myocardial ischemia and reperfusion injury: Models, modes, molecular mechanisms, modulation and clinical applications. Cardiol. Rev. 2022. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Pan, M.; Ma, J.; Song, X.; Cao, W.; Zhang, P. Recent progress in the use of mitochondrial membrane permeability transition pore in mitochondrial dysfunction-related disease therapies. Mol. Cell. Biochem. 2021, 476, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, Y.; Wang, S.; McDonald, T.; Van Eyk, J.E.; Sidor, A.; O’Rourke, B. Cytoprotective role of Ca2+- activated K+ channels in the cardiac inner mitochondrial membrane. Science 2002, 298, 1029–1033. [Google Scholar] [CrossRef]
- Shen, F.; Wu, L.; Lu, Y.; Liang, H.; Bruce, I.; Xia, Q. Mitochondrial Permeability Transition Dynamics: An Indicator of Mitochondrial Potassium Channel Opener. In Proceedings of the 2005 IEEE Engineering in Medicine and Biology 27th Annual Conference, Shanghai, China, 17–18 January 2006. [Google Scholar] [CrossRef]
- Prendes, M.G.M.; Hermann, R.; Torresin, M.E.; Vélez, D.; Savino, E.A.; Varela, A. Role of mitochondrial permeability transition pore and mitochondrial ATP-sensitive potassium channels in the protective effects of ischemic preconditioning in isolated hearts from fed and fasted rats. J. Physiol. Biochem. 2014, 70, 791–800. [Google Scholar] [CrossRef]
- Santos, P.D.; Kowaltowski, A.J.; Laclau, M.N.; Seetharaman, S.; Paucek, P.; Boudina, S.; Thambo, J.B.; Tariosse, L.; Garlid, K.D. Mechanisms by which opening the mitochondrial ATP- sensitive K+ channel protects the ischemic heart. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H284–H295. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D.; Paucek, P.; Yarov-Yarovoy, V.; Murray, H.N.; Darbenzio, R.B.; D’Alonzo, A.J.; Lodge, N.J.; Smith, M.A.; Grover, G.J. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ. Res. 1997, 81, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Rahgozar, M.; Willgoss, D.A.; Gobé, G.C.; Endre, Z.H. ATP-dependent K+ channels in renal ischemia reperfusion injury. Ren. Fail. 2003, 25, 885–896. [Google Scholar] [CrossRef]
- Wang, S.; Cone, J.; Liu, Y. Dual roles of mitochondrial KATP channels in diazoxide-mediated protection in isolated rabbit hearts. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H246–H255. [Google Scholar] [CrossRef]
- Liu, D.; Lu, C.; Wan, R.; Auyeung, W.W.; Mattson, M.P. Activation of mitochondrial ATP-dependent potassium channels protects neurons against ischemia-induced death by a mechanism involving suppression of bax translocation and cytochrome c release. J. Cereb. Blood Flow Metab. 2002, 22, 431–443. [Google Scholar] [CrossRef]
- Holmuhamedov, E.L.; Wang, L.; Terzic, A. ATP-sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. J. Physiol. 1999, 519 Pt 2, 347–360. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Bednarczyk, P.; Kicinska, A.; Laskowski, M.; Kulawiak, B.; Kampa, R.; Walewska, A.; Krajewska, M.; Jarmuszkiewicz, W.; Szewczyk, A. Evidence for a mitochondrial ATP-regulated potassium channel in human dermal fibroblasts. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Pereira, O., Jr.; Kowaltowski. A.J. Mitochondrial K+ Transport: Modulation and Functional Consequences. Molecules 2021, 26, 2935. [Google Scholar] [CrossRef] [PubMed]
- Ferranti, R.; da Silva, M.M.; Kowaltowski, A.J. Mitochondrial ATP-sensitive K+ channel opening decreases reactive oxygen species generation. FEBS Lett. 2003, 536, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Facundo, H.T.F.; de Paula, J.G.; Kowaltowski, A.J. Mitochondrial ATP-Sensitive K+ Channels Prevent Oxidative Stress, Permeability Transition and Cell Death. J. Bioenerg. Biomembr. 2005, 37, 75–82. [Google Scholar] [CrossRef]
- Kulawiak, B.; Kudin, A.P.; Szewczyk, A.; Kunz, W.S. BK channel openers inhibit ROS production of isolated rat brain mitochondria. Exp. Neurol. 2008, 212, 543–547. [Google Scholar] [CrossRef]
- Dahlem, Y.A.; Wolf, G.; Siemen, D.; Horn, T.F. Combined modulation of the mitochondrial ATP-dependent potassium channel and the permeability transition pore causes prolongation of the biphasic calcium dynamics. Cell Calcium 2006, 39, 387–400. [Google Scholar] [CrossRef]
- Kupsch, K.; Parvez, S.; Siemen, D.; Wolf, G. Modulation of the Permeability Transition Pore by Inhibition of the Mitochondrial KATP Channel in Liver vs. Brain Mitochondria. J. Membr. Biol. 2007, 215, 69–74. [Google Scholar] [CrossRef]
- Cheng, Y.; Gu, X.Q.; Bednarczyk, P.; Wiedemann, F.R.; Haddad, G.G.; Siemen, D. Hypoxia increases activity of the BK-channel in the inner mitochondrial membrane and reduces activity of the permeability transition pore. Cell. Physiol. Biochem. 2008, 22, 127–136. [Google Scholar] [CrossRef]
- Cheng, Y.; Gulbins, E.; Siemen, D. Activation of the permeability transition pore by Bax via inhibition of the mitochondrial BK channel. Cell. Physiol. Biochem. 2011, 27, 191–200. [Google Scholar] [CrossRef]
- Korotkov, S.M.; Brailovskaya, I.V.; Shumakov, A.R.; Emelyanova, L.V. Closure of mitochondrial potassium channels favors opening of the Tl(+)-induced permeability transition pore in Ca(2+)-loaded rat liver mitochondria. J. Bioenerg. Biomembr. 2015, 47, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.D.T.; Jakob, R.; Costa, C.L.; Andrukhiv, K.; West, I.C.; Garlid, K.D. The Mechanism by Which the Mitochondrial ATP-sensitive K Channel Opening and H2O2 Inhibit the Mitochondrial Permeability Transition. J. Biol. Chem. 2006, 281, 20801–20808. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S.; Brandt, U.; Hanley, P.J. K+-independent actions of diazoxide question the role of inner membrane KATP channels in mitochondrial cytoprotective signaling. J. Biol. Chem. 2006, 281, 23733–23739. [Google Scholar] [CrossRef] [PubMed]
- Busija, D.W.; Katakam, P.; Rajapakse, N.C.; Kis, B.; Grover, G.; Domoki, F.; Bari, F. Effects of ATP-sensitive potassium channel activators diazoxide and BMS-191095 on membrane potential and reactive oxygen species production in isolated piglet mitochondria. Brain Res. Bull. 2005, 66, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Ziemys, A.; Toleikis, A.; Kopustinskiene, D.M. Molecular modelling of K(ATP) channel blockers-ADP/ATP carrier interactions. Syst. Biol. 2006, 153, 390–393. [Google Scholar] [CrossRef]
- Skalska, J.; Debska, G.; Kunz, W.S.; Szewczyk, A. Antidiabetic sulphonylureas activate mitochondrial permeability transition in rat skeletal muscle. Br. J. Pharmacol. 2005, 145, 785–791. [Google Scholar] [CrossRef]
- Hansson, M.J.; Morota, S.; Teilum, M.; Mattiasson, G.; Uchino, H.; Elmér, E. Increased potassium conductance of brain mitochondria induces resistance to permeability transition by enhancing matrix volume. J. Biol. Chem. 2010, 285, 741–750. [Google Scholar] [CrossRef]
- Traba, J.; del Arco, A.; Duchen, M.R.; Szabadkai, G.; Satrústegui, J. SCaMC-1 promotes cancer cell survival by desensitizing mitochondrial permeability transition via ATP/ADP-mediated matrix Ca(2+) buffering. Cell Death Differ. 2012, 19, 650–660. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Chalmers, S. The integration of mitochondrial calcium transport and storage. J. Bioenerg. Biomembr. 2004, 36, 277–281. [Google Scholar] [CrossRef]
- Solesio, M.E.; Demirkhanyan, L.; Zakharian, E.; Pavlov, E.V. Contribution of inorganic polyphosphate towards regulation of mitochondrial free calcium. Biochim. Biophys. Acta 2016, 1860, 1317–1325. [Google Scholar] [CrossRef]
- Kristian, T.; Pivovarova, N.B.; Fiskum, G.; Andrews, S.B. Calcium-induced precipitate formation in brain mitochondria: Composition, calcium capacity, and retention. J. Neurochem. 2007, 102, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Vajda, S.; Mándi, M.; Konràd, C.; Kiss, G.; Ambrus, A.; Adam-Vizi, V.; Chinopoulos, C. A re-evaluation of the role of matrix acidification in uncoupler-induced Ca2+ release from mitochondria. FEBS J. 2009, 276, 2713–2724. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C.; Adam-Vizi, V. Mitochondrial Ca2+ sequestration and precipitation revisited. FEBS J. 2010, 277, 3637–3651. [Google Scholar] [CrossRef] [PubMed]
- Linard, D.; Kandlbinder, A.; Degand, H.; Morsomme, P.; Dietz, K.J.; Knoops, B. Redox characterization of human cyclophilin D: Identification of a new mammalian mitochondrial redox sensor? Arch. Biochem. Biophys. 2009, 491, 39–45. [Google Scholar] [CrossRef]
- Majima, E.; Ikawa, K.; Takeda, M.; Hashimoto, M.; Shinohara, Y.; Terada, H. Translocation of loops regulates transport activity of mitochondrial ADP/ATP carrier deduced from formation of a specific intermolecular disulfide bridge catalyzed by copper-o-phenanthroline. J. Biol. Chem. 1995, 270, 29548–29554. [Google Scholar] [CrossRef]
- Costantini, P.; Belzacq, A.S.; Vieira, H.L.; Larochette, N.; de Pablo, M.A.; Zamzami, N.; Susin, S.A.; Brenner, C.; Kroemer, G. Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces Bcl-2-independent permeability transition pore opening and apoptosis. Oncogene 2000, 19, 307–314. [Google Scholar] [CrossRef]
- Woodfield, K.; Rück, A.; Brdiczka, D.; Halestrap, A.P. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem. J. 1998, 336 Pt 2, 287–290. [Google Scholar] [CrossRef]
- Kaludercic, N.; Giorgio, V. The Dual Function of Reactive Oxygen/Nitrogen Species in Bioenergetics and Cell Death: The Role of ATP Synthase. Oxid. Med. Cell. Longev. 2016, 2016, 3869610. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Dalle-Donne, I. Redox proteomics: From protein modifications to cellular dysfunction and disease. Mass Spectrom. Rev. 2014, 33, 1–6. [Google Scholar] [CrossRef]
- Petronilli, V.; Costantini, P.; Scorrano, L.; Colonna, R.; Passamonti, S.; Bernardi, P. The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation reduction state of vicinal thiols. Increase of the gating potential by oxidants and its reversal by reducing agents. J. Biol. Chem. 1994, 269, 16638–16642. [Google Scholar] [CrossRef]
- Costantini, P.; Chernyak, B.V.; Petronilli, V.; Bernardi, P. Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J. Biol. Chem. 1996, 271, 6746–6751. [Google Scholar] [CrossRef] [PubMed]
- McStay, G.P.; Clarke, S.J.; Halestrap, A.P. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem. J. 2002, 367 Pt 2, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Chernyak, B.V. Redox regulation of the mitochondrial permeability transition pore. Biosci. Rep. 1997, 17, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Rigobello, M.P.; Turcato, F.; Bindoli, A. Inhibition of rat liver mitochondrial permeability transition by respiratory substrates. Arch. Biochem. Biophys. 1995, 319, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Zago, E.B.; Castilho, R.F.; Vercesi, A.E. The redox state of endogenous pyridine nucleotides can determine both the degree of mitochondrial oxidative stress and the solute selectivity of the permeability transition pore. FEBS Lett. 2000, 478, 29–33. [Google Scholar] [CrossRef]
- Carrer, A.; Tommasin, L.; Šileikytė, J.; Ciscato, F.; Filadi, R.; Urbani, A.; Forte, M.l.; Rasola, A.; Szabò, I.; Carraro, M.; et al. Defining the molecular mechanisms of the mitochondrial permeability transition through genetic manipulation of F-ATP synthase. Nat. Commun. 2021, 12, 4835. [Google Scholar] [CrossRef]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep. 2019, 26, 11–17. [Google Scholar] [CrossRef]
- Galber, C.; Minervini, G.; Cannino, G.; Boldrin, F.; Petronilli, V.; Tosatto, S.; Lippe, G.; Giorgio, V. The f subunit of human ATP synthase is essential for normal mitochondrial morphology and permeability transition. Cell Rep. 2021, 35, 109111. [Google Scholar] [CrossRef]
- Carroll, J.; He, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the permeability transition pore in human mitochondria devoid of an assembled ATP synthase. Proc. Natl. Acad. Sci. USA 2019, 116, 12816–12821. [Google Scholar] [CrossRef]
- Brustovetsky, N. The Role of Adenine Nucleotide Translocase in the Mitochondrial Permeability Transition. Cells 2020, 9, 2686. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Brenner, C. The adenine nucleotide translocase: A central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 2003, 10, 1507–1525. [Google Scholar] [CrossRef] [PubMed]
- Haworth, R.A.; Hunter, D.R. Allosteric inhibition of the Ca2+-activated hydrophilic channel of the mitochondrial inner membrane by nucleotides. J. Membr. Biol. 1980, 54, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.R.; Haworth, R.A. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch. Biochem. Biophys. 1979, 195, 453–459. [Google Scholar] [CrossRef]
- Kharechkina, E.; Nikiforova, A.; Kruglov, A. NAD(H) Regulates the Permeability Transition Pore in Mitochondria through an External Site. Int. J. Mol. Sci. 2021, 22, 8560. [Google Scholar] [CrossRef] [PubMed]
- Kharechkina, E.S.; Nikiforova, A.B.; Teplova, V.V.; Odinokova, I.V.; Krestinina, O.V.; Baburina, Y.L.; Kruglova, S.A.; Kruglov, A.G. Regulation of permeability transition pore opening in mitochondria by external NAD(H). Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef]
- Giorgio, V.; Bisetto, E.; Soriano, M.E.; Dabbeni-Sala, F.; Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P.; Lippe, G. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem. 2009, 284, 33982–33988. [Google Scholar] [CrossRef]
- Bernardi, P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J. Biol. Chem. 1992, 267, 8834–8839. [Google Scholar] [CrossRef]
- Bernardi, P.; Veronese, P.; Petronilli, V. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore. I. Evidence for two separate Me2+ binding sites with opposing effects on the pore open probability. J. Biol. Chem. 1993, 268, 1005–1010. [Google Scholar] [CrossRef]
- Petronilli, V.; Cola, C.; Bernardi, P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore. II. The minimal requirements for pore induction underscore a key role for transmembrane electrical potential, matrix pH, and matrix Ca2+. J. Biol. Chem. 1993, 268, 1011–1016. [Google Scholar] [CrossRef]
- Petronilli, V.; Cola, C.; Massari, S.; Colonna, R.; Bernardi, P. Physiological effectors modify voltage sensing by the cyclosporin A-sensitive permeability transition pore of mitochondria. J. Biol. Chem. 1993, 268, 21939–21945. [Google Scholar] [CrossRef] [PubMed]
- Malas, K.M.; Lambert, D.S.; Heisner, J.S.; Camara, A.K.S.; Stowe, D.F. Time and charge/pH-dependent activation of K+ channel-mediated K+ influx and K+/H+ exchange in guinea pig heart isolated mitochondria; role in bioenergetic stability. Biochim. Biophys. Acta Bioenerg. 2022, 1863, 148908. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.D.T.; Quinlan, C.L.; Andrukhiv, A.; West, I.C.; Jabůrek, M.; Garlid, K.D. The direct physiological effects of mitoK(ATP) opening on heart mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H406–H415. [Google Scholar] [CrossRef]
- Czyz, A.; Szewczyk, A.; Nalecz, M.J.; Wojtczak, L. The Role of Mitochondrial Potassium Fluxes in Controlling the Protonmotive Force in Energized Mitochondria. Biochem. Biophys. Res. Commun. 1995, 210, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; Seetharaman, S.; Paucek, P.; Garlid, K.D. Bioenergetic consequences of opening the ATP-sensitive K+ channel of heart mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H649–H657. [Google Scholar] [CrossRef]
- Morota, S.; Piel, S.; Hansson, M.J. Respiratory uncoupling by increased H+ or K+ flux is beneficial for heart mitochondrial turnover of reactive oxygen species but not for permeability transition. BMC Cell Biol. 2013, 14, 40. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Schulz, R.; Girao, H.; Kwak, B.R.; de Stefani, D.; Rizzuto, R.; Bernardi, P.; Di Lisa, F. Mitochondrial ion channels as targets for cardioprotection. J. Cell. Mol. Med. 2020, 24, 7102–7114. [Google Scholar] [CrossRef]
- Kulawiak, B.; Bednarczyk, P.; Szewczyk, A. Multidimensional Regulation of Cardiac Mitochondrial Potassium Channels. Cells 2021, 10, 1554. [Google Scholar] [CrossRef]
- Kulawiak, B.; Szewczyk, A. Current Challenges of Mitochondrial Potassium Channel Research. Front. Physiol. 2022, 13, 907015. [Google Scholar] [CrossRef]
- Jabůrek, M.; Yarov-Yarovoy, V.; Paucek, P.; Garlid, K.D. State-dependent inhibition of the mitochondrial KATP channel by glyburide and 5-hydroxydecanoate. J. Biol. Chem. 1998, 273, 13578–13582. [Google Scholar] [CrossRef] [PubMed]
- Vennekamp, J.; Wulff, H.; Beeton, C.; Calabresi, P.A.; Grissmer, S.; Hänsel, W.; Chandy, K.G. Kv1.3-blocking 5-phenylalkoxypsoralens: A new class of immunomodulators. Mol. Pharmacol. 2004, 65, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Kruglov, A.G.; Kharechkina, E.S.; Nikiforova, A.B.; Odinokova, I.V.; Kruglova, S.A. Dynamics of the permeability transition pore size in isolated mitochondria and mitoplasts. FASEB J. 2021, 35, e21764. [Google Scholar] [CrossRef]
- Novgorodov, S.A.; Gudz, T.I.; Milgrom, Y.M.; Brierley, G.P. The permeability transition in heart mitochondria is regulated synergistically by ADP and cyclosporin A. J. Biol. Chem. 1992, 267, 16274–16282. [Google Scholar] [CrossRef] [PubMed]
- Petronilli, V.; Miotto, G.; Canton, M.; Brini, M.; Colonna, R.; Bernardi, P.; Di Lisa, F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys. J. 1999, 76, 725–734. [Google Scholar] [CrossRef]
- Zorov, D.B.; Kinnally, K.W.; Perini, S.; Tedeschi, H. Multiple conductance levels in rat heart inner mitochondrial membranes studied by patch clamping. Biochim. Biophys. Acta 1992, 1105, 263–270. [Google Scholar] [CrossRef]
- Kruglov, A.G.; Subbotina, K.B.; Saris, N.L. Redox-cycling compounds can cause the permeabilization of mitochondrial membranes by mechanisms other than ROS production. Free Radic. Biol. Med. 2008, 44, 646–656. [Google Scholar] [CrossRef]
- Kapus, A.; Szászi, K.; Káldi, K.; Ligeti, E.; Fonyó, A. Ruthenium red inhibits mitochondrial Na+ and K+ uniports induced by magnesium removal. J. Biol. Chem. 1990, 265, 18063–18066. [Google Scholar] [CrossRef]
- Beavis, A.D. Properties of the inner membrane anion channel in intact mitochondria. J. Bioenerg. Biomembr. 1992, 24, 77–90. [Google Scholar] [CrossRef]
- Bernardi, P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef]
- Michel, V.; Bakovic, M. The solute carrier 44A1 is a mitochondrial protein and mediates choline transport. FASEB J. 2009, 23, 2749–2758. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.; Mekis, R.; Mohammed, S.E.M.; Scalise, M.; Wang, W.A.; Galluccio, M.; Pfeiffer, C.; Borovec, T.; Parapatics, K.; Vitko, D.; et al. TMBIM5 is the Ca2+/H+ antiporter of mammalian mitochondria. EMBO Rep. 2022, 23, e54978. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Vassanelli, S.; Veronese, P.; Colonna, R.; Szabó, I.; Zoratti, M. Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J. Biol. Chem. 1992, 267, 2934–2939. [Google Scholar] [CrossRef]
- Diwan, J.J. Effect of quinine on mitochondrial K+ and Mg++ flux. Biochem. Biophys. Res. Commun. 1986, 135, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, R.A.; Garlid, K.D. Quinine inhibition of Na+ and K+ transport provides evidence for two cation/H+ exchangers in rat liver mitochondria. J. Biol. Chem. 1982, 257, 9252–9254. [Google Scholar] [CrossRef]
- Taylor, A.; Grapentine, S.; Ichhpuniani, J.; Bakovic, M. Choline transporter-like proteins 1 and 2 are newly identified plasma membrane and mitochondrial ethanolamine transporters. J. Biol. Chem. 2021, 296, 100604. [Google Scholar] [CrossRef]
- Inazu, M.; Takeda, H.; Matsumiya, T. Molecular and functional characterization of an Na+-independent choline transporter in rat astrocytes. J. Neurochem. 2005, 94, 1427–1437. [Google Scholar] [CrossRef]
- Brierley, G.P.; Panzeter, E.S.; Jung, D.W. Regulation of mitochondrial K+/H+ antiport activity by hydrogen ions. Arch. Biochem. Biophys. 1991, 288, 358–367. [Google Scholar] [CrossRef]
- Haumann, J.; Camara, A.K.S.; Gadicherla, A.K.; Navarro, C.D.; Boelens, A.D.; Blomeyer, C.A.; Dash, R.K.; Boswell, M.R.; Kwok, W.M.; Stowe, D.F. Slow Ca2+ Efflux by Ca2+/H+ Exchange in Cardiac Mitochondria Is Modulated by Ca2+ Re-uptake via MCU, Extra-Mitochondrial pH, and H+ Pumping by FOF1-ATPase. Front. Physiol. 2019, 9, 1914. [Google Scholar] [CrossRef]
- Kristian, T.; Bernardi, P.; Siesjö, B.K. Acidosis promotes the permeability transition in energized mitochondria: Implications for reperfusion injury. J. Neurotrauma 2001, 18, 1059–1074. [Google Scholar] [CrossRef] [PubMed]
- Nicolli, A.; Petronilli, V.; Bernardi, P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by matrix pH. Evidence that the pore open-closed probability is regulated by reversible histidine protonation. Biochemistry 1993, 32, 4461–4465. [Google Scholar] [CrossRef] [PubMed]
- Wrzosek, A.; Augustynek, B.; Żochowska, M.; Szewczyk, A. Mitochondrial Potassium Channels as Druggable Targets. Biomolecules 2020, 10, 1200. [Google Scholar] [CrossRef]
- Al-Nasser, I.; Crompton, M. The reversible Ca2+-induced permeabilization of rat liver mitochondria. Biochem. J. 1986, 239, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Broekemeier, K.M.; Schmid, P.C.; Schmid, H.H.; Pfeiffer, D.R. Effects of phospholipase A2 inhibitors on ruthenium red-induced Ca2+ release from mitochondria. J. Biol. Chem. 1985, 260, 105–113. [Google Scholar] [CrossRef]
- Broekemeier, K.M.; Klocek, C.K.; Pfeiffer, D.R. Proton selective substate of the mitochondrial permeability transition pore: Regulation by the redox state of the electron transport chain. Biochemistry 1998, 37, 13059–13065. [Google Scholar] [CrossRef]
- Fernandes, M.A.; Santos, M.S.; Moreno, A.J.; Duburs, G.; Oliveira, C.R.; Vicente, J.A. Glibenclamide interferes with mitochondrial bioenergetics by inducing changes on membrane ion permeability. J. Biochem. Mol. Toxicol. 2004, 18, 162–169. [Google Scholar] [CrossRef]
- Ponnalagu, D.; Singh, H. Anion Channels of Mitochondria. Handb. Exp. Pharmacol. 2017, 240, 71–101. [Google Scholar] [CrossRef]
- Rysted, J.E.; Lin, Z.; Walters, G.C.; Rauckhorst, A.J.; Noterman, M.; Liu, G.; Taylor, E.B.; Strack, S.; Usachev, Y.M. Distinct Properties of Ca2+ Efflux from Brain, Heart and Liver Mitochondria: The Effects of Na+, Li+ and the Mitochondrial Na+/Ca2+ Exchange Inhibitor CGP37157. Cell Calcium 2021, 96, 102382. [Google Scholar] [CrossRef]
- Johnson, D.; Lardy, H. Isolation of liver or kidney mitochondria. Methods Enzymol. 1967, 10, 94–96. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilising the principle of protein dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Chalmers, S.; Nicholls, D.G. The Relationship between Free and Total Calcium Concentrations in the Matrix of Liver and Brain Mitochondria. J. Biol. Chem. 2003, 278, 19062–19070. [Google Scholar] [CrossRef]
- Csordás, G.; Várnai, P.; Golenár, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnóczky, G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; Guo, L.; Bassot, C.; Petronilli, V.; Bernardi, P. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium 2018, 70, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Kharechkina, E.S.; Nikiforova, A.B.; Belosludtsev, K.N.; Rokitskaya, T.I.; Antonenko, Y.N.; Kruglov, A.G. Pioglitazone Is a Mild Carrier-Dependent Uncoupler of Oxidative Phosphorylation and a Modulator of Mitochondrial Permeability Transition. Pharmaceuticals 2021, 14, 1045. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C.; Vajda, S.; Csanády, L.; Mándi, M.; Mathe, K.; Adam-Vizi, V. A novel kinetic assay of mitochondrial ATP-ADP exchange rate mediated by the ANT. Biophys. J. 2009, 96, 2490–2504. [Google Scholar] [CrossRef]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kharechkina, E.S.; Nikiforova, A.B.; Kruglov, A.G. Regulation of Mitochondrial Permeability Transition Pore Opening by Monovalent Cations in Liver Mitochondria. Int. J. Mol. Sci. 2023, 24, 9237. https://doi.org/10.3390/ijms24119237
Kharechkina ES, Nikiforova AB, Kruglov AG. Regulation of Mitochondrial Permeability Transition Pore Opening by Monovalent Cations in Liver Mitochondria. International Journal of Molecular Sciences. 2023; 24(11):9237. https://doi.org/10.3390/ijms24119237
Chicago/Turabian StyleKharechkina, Ekaterina S., Anna B. Nikiforova, and Alexey G. Kruglov. 2023. "Regulation of Mitochondrial Permeability Transition Pore Opening by Monovalent Cations in Liver Mitochondria" International Journal of Molecular Sciences 24, no. 11: 9237. https://doi.org/10.3390/ijms24119237