
α-Tocopherol-13′-Carboxychromanol Induces Cell Cycle Arrest and Cell Death by Inhibiting the SREBP1-SCD1 Axis and Causing Imbalance in Lipid Desaturation
 , , , , , , and
, , , , , , and
Abstract
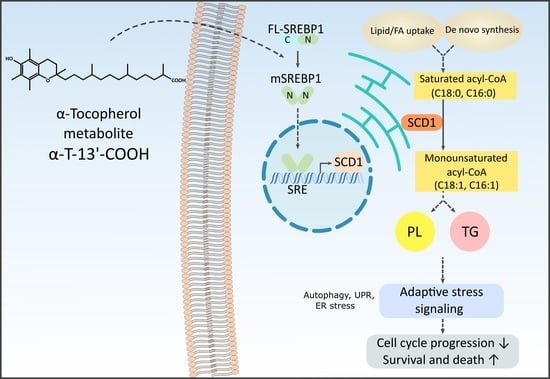
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
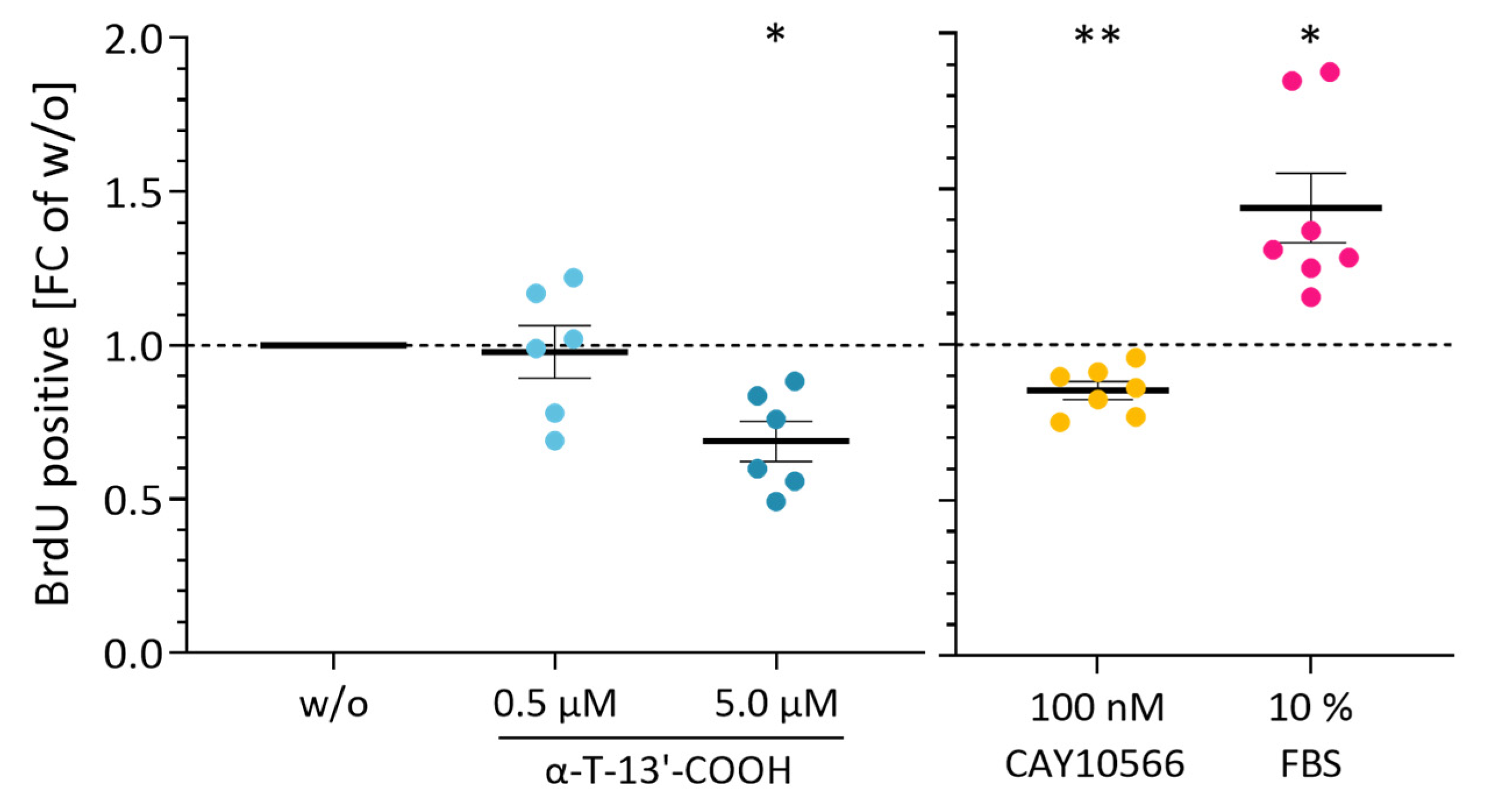
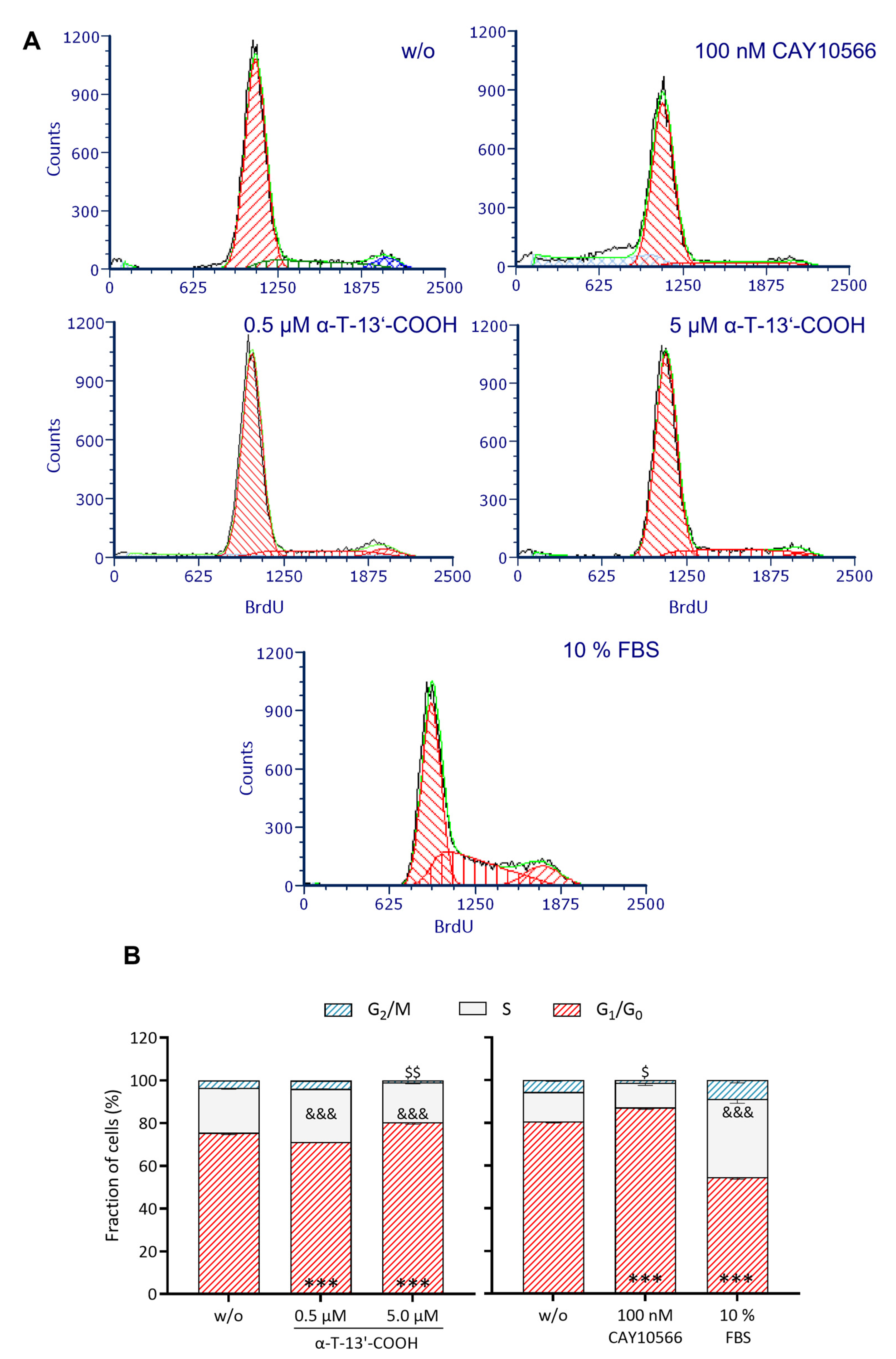
2.1. α-T-13′-COOH Suppresses Cell Proliferation following G0/G1 Cell Cycle Arrest
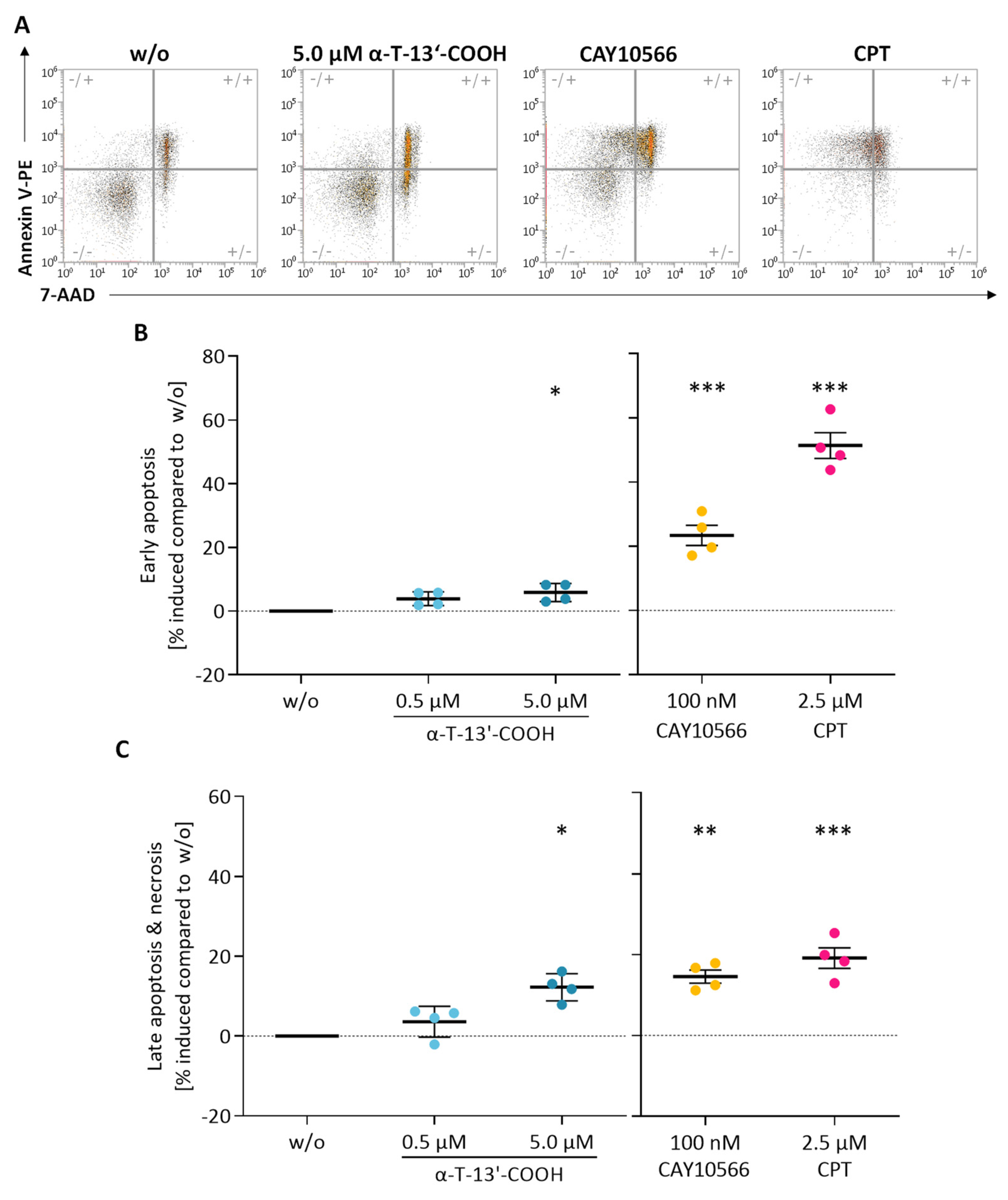
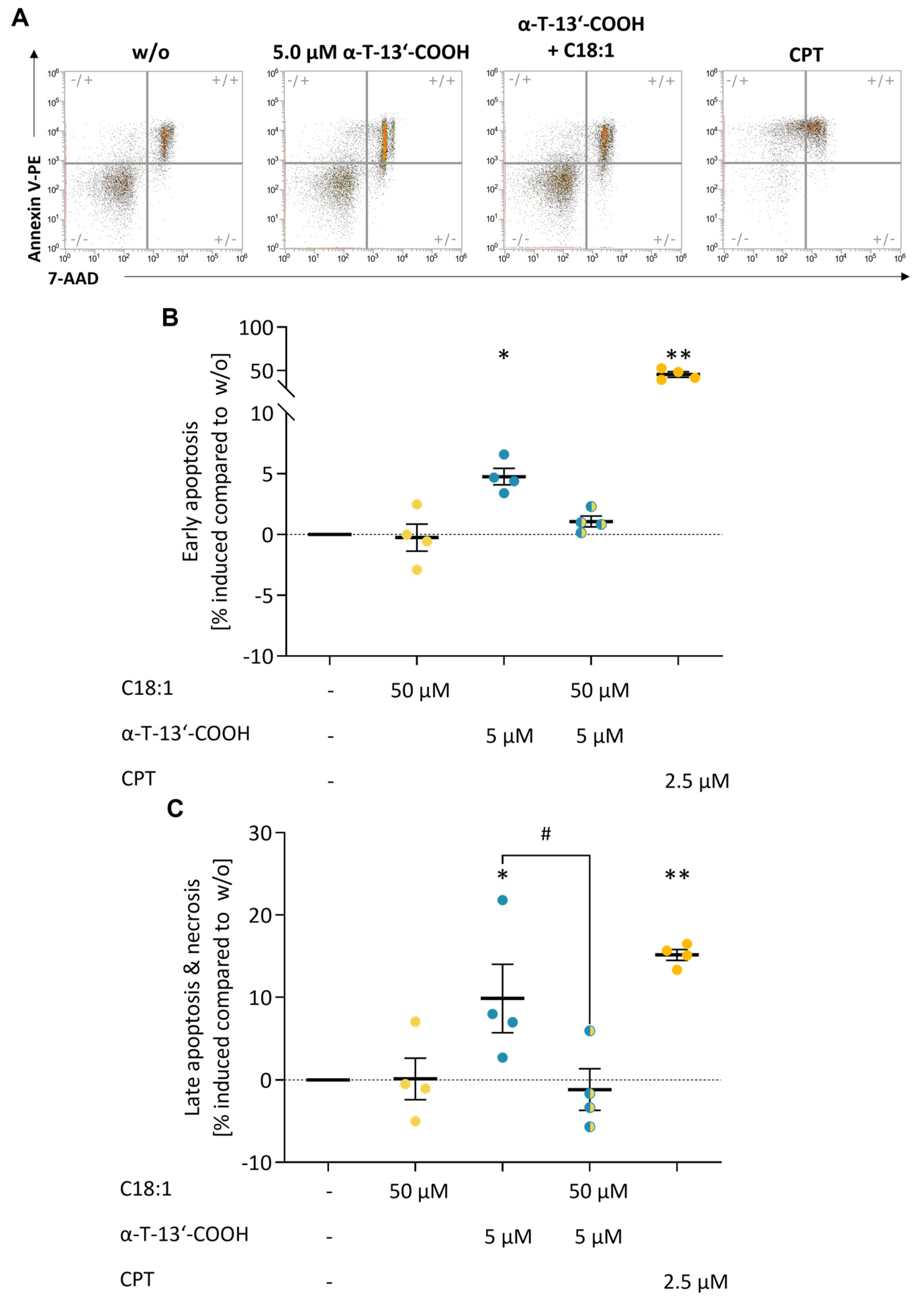
2.2. α-T-13′-COOH Induces Apoptosis
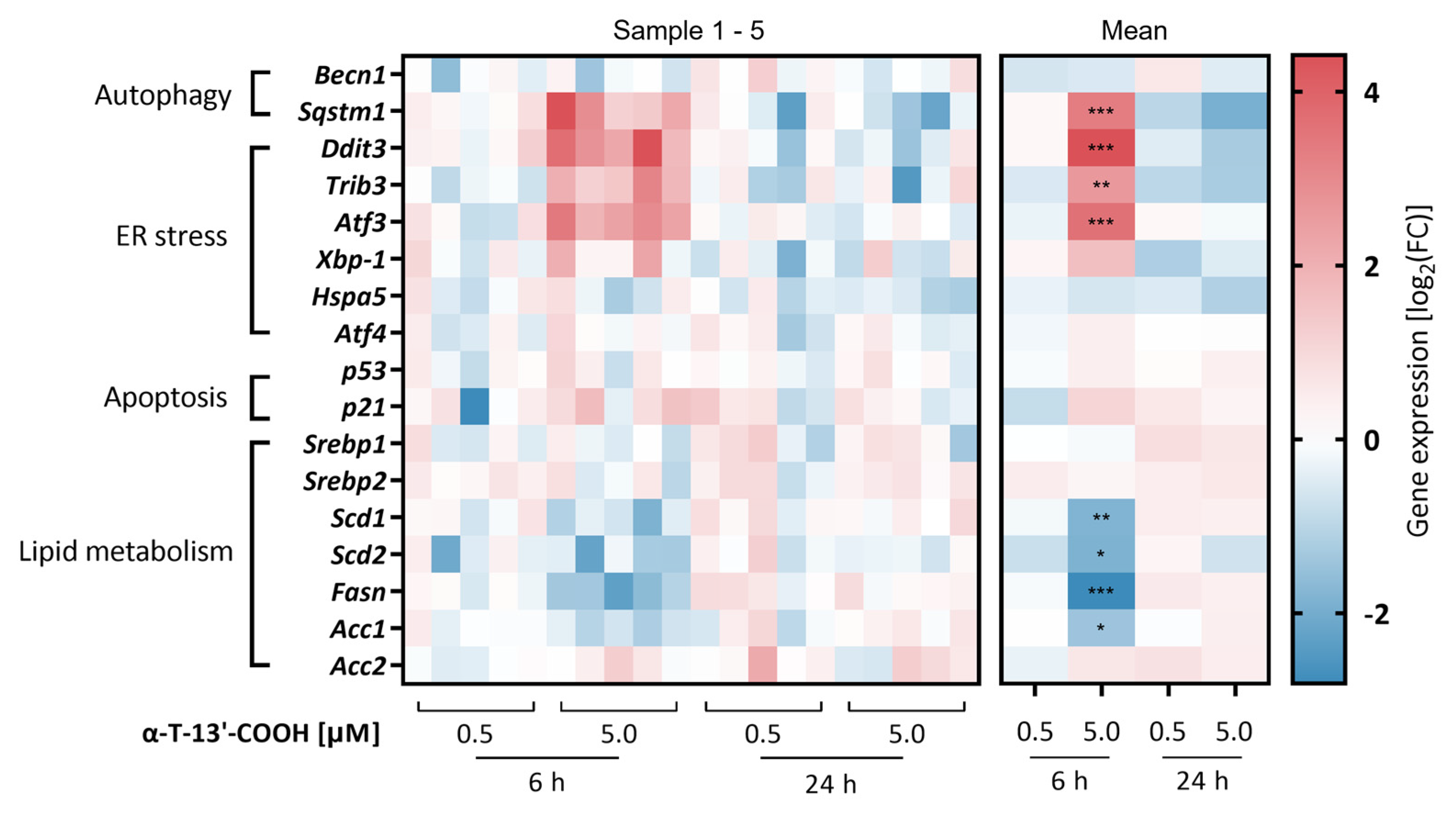
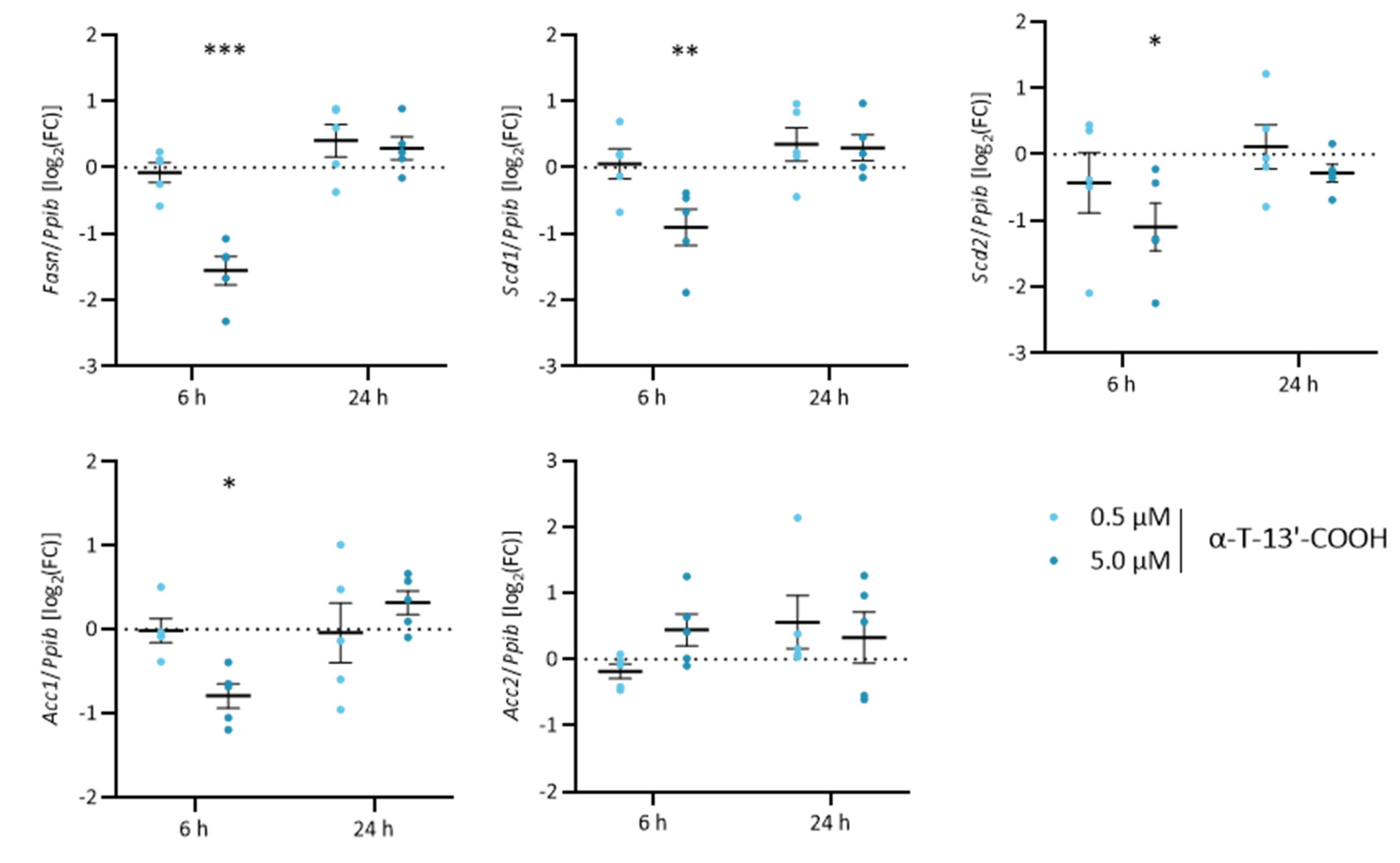
2.3. α-T-13′-COOH Modulates Expression of Stress-Related Genes
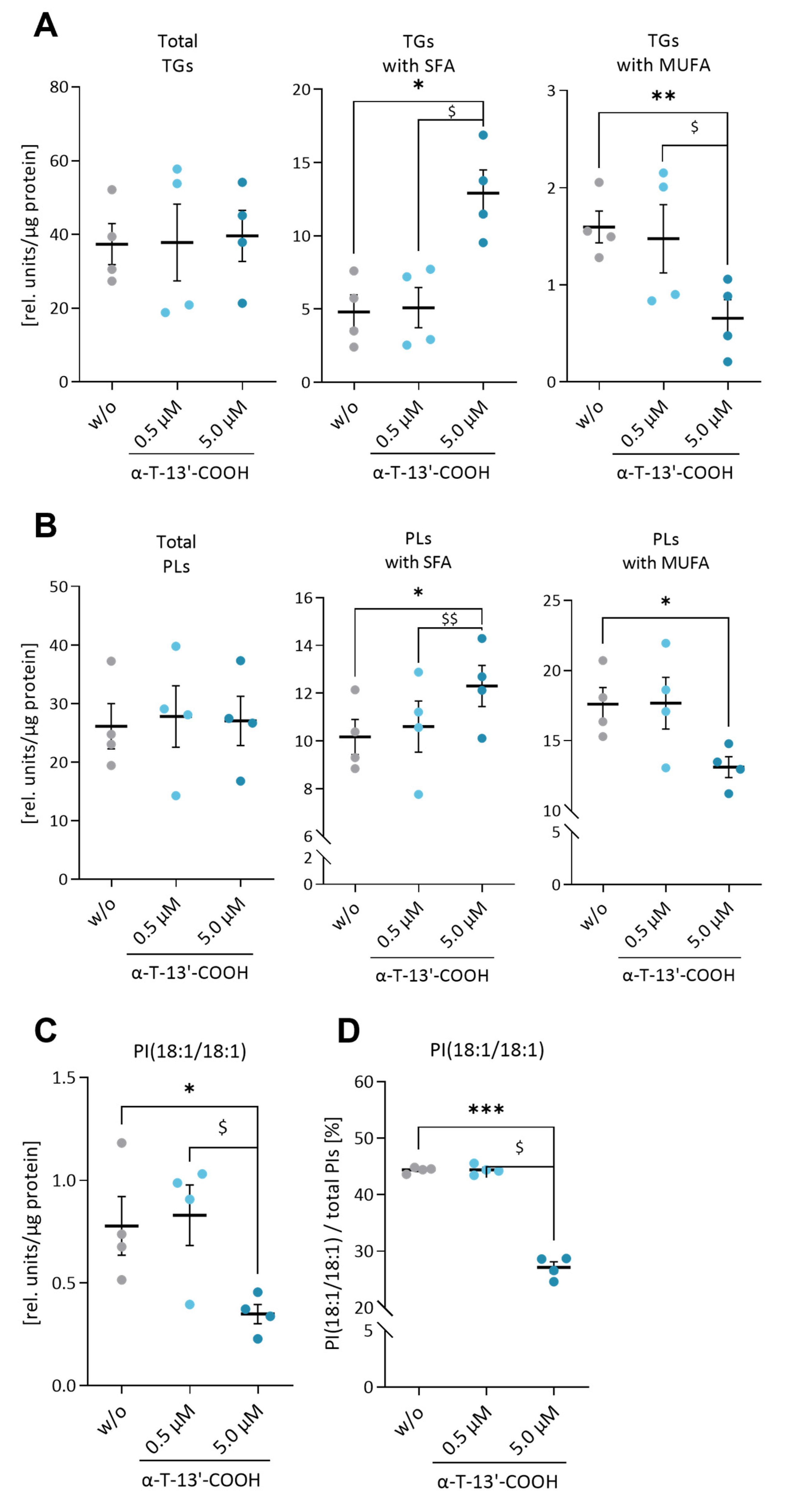
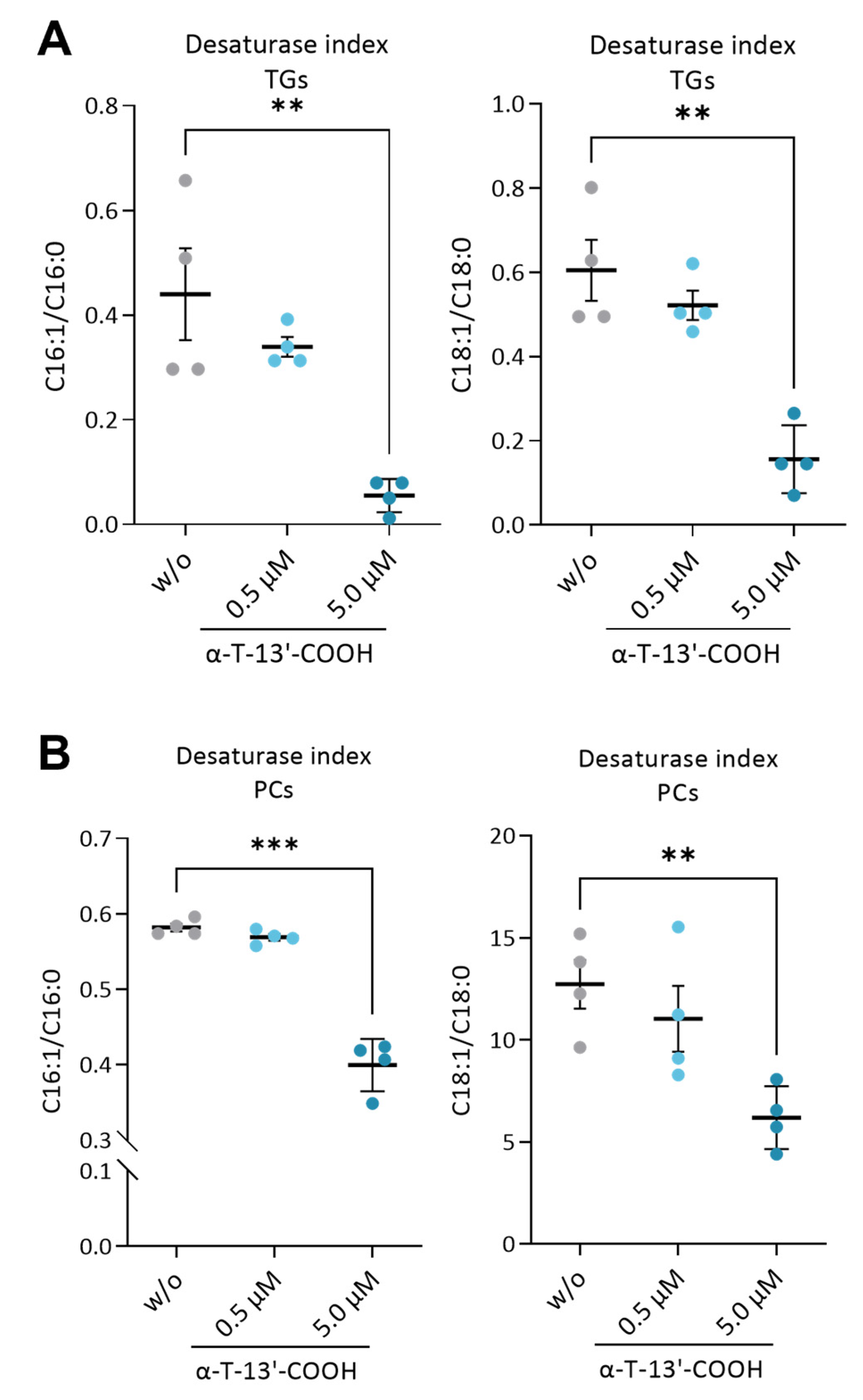
2.4. α-T-13′-COOH Decreases the MUFA/SFA Ratio in Triglycerides and Phospholipids
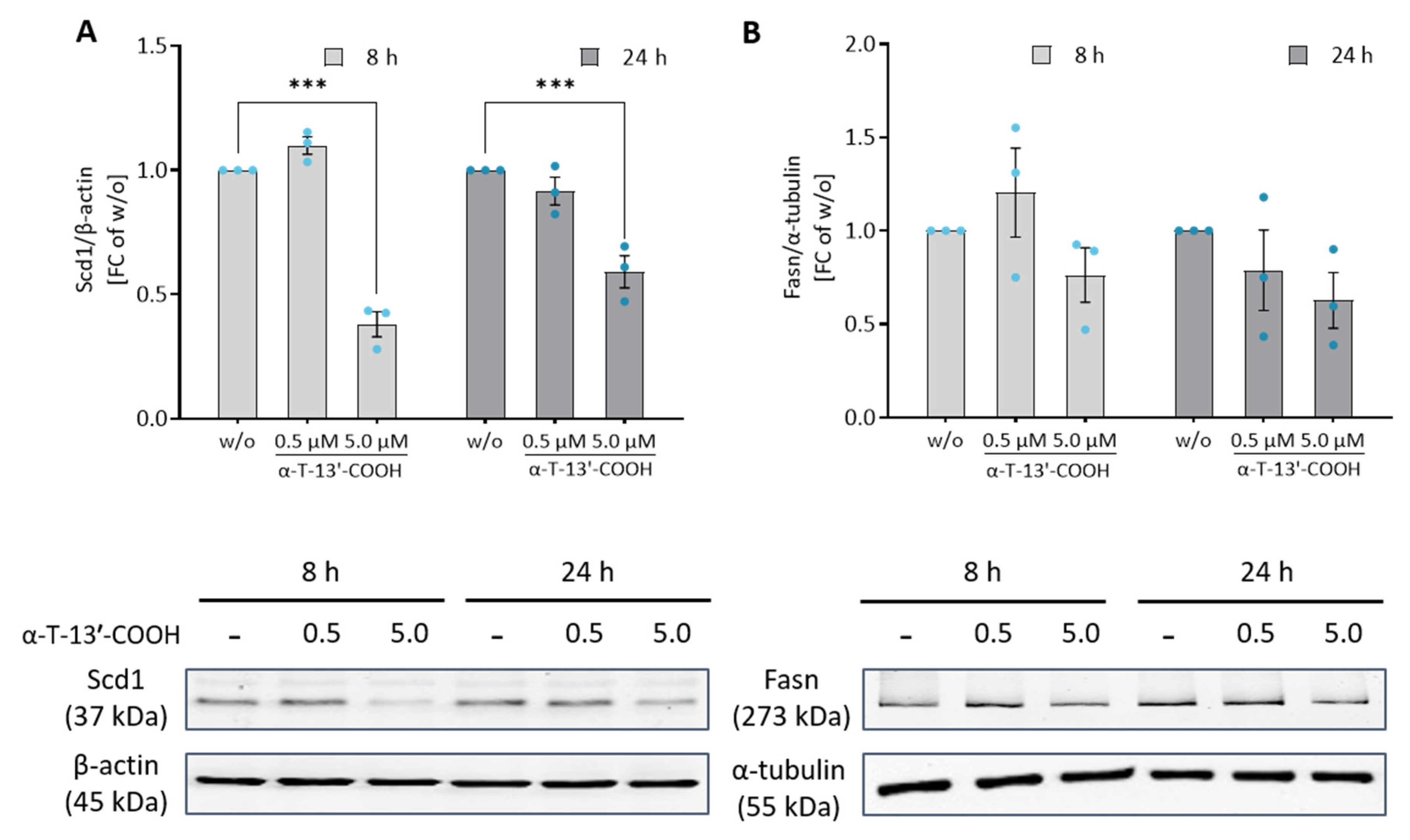
2.5. α-T-13′-COOH Modulates Expression of Scd1 and Other Genes Encoding for Enzymes in De Novo Fatty Acid Biosynthesis
2.6. SCD1 Repression Contributes to α-T-13′-COOH-Induced Cell Cycle Arrest and Apoptosis
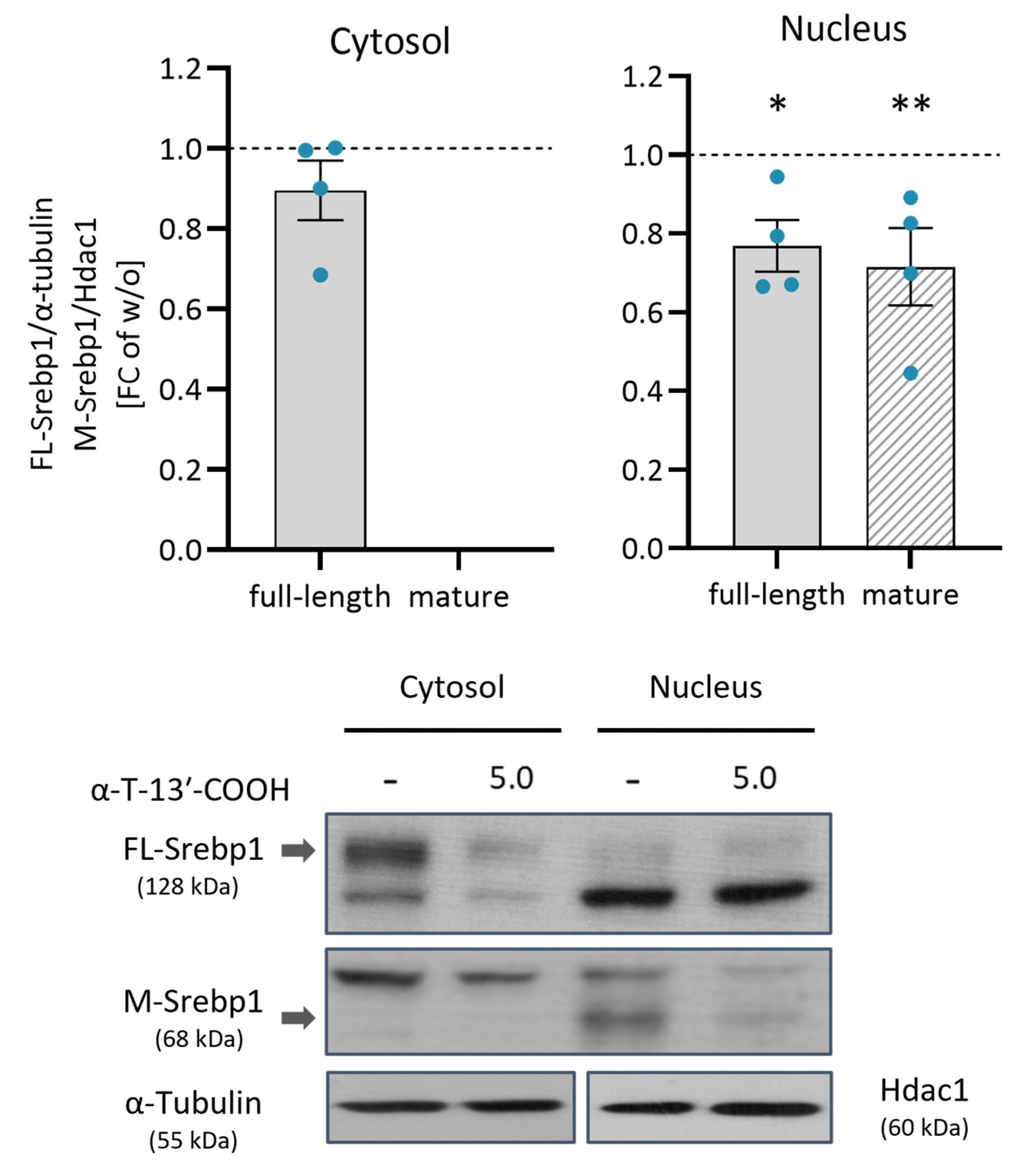
2.7. α-T-13′-COOH Inhibits the Proteolytic Activation of SREBP1
3. Discussion
4. Materials and Methods
4.1. Materials
4.1.1. Chemicals
4.1.2. Semi-Synthesis of Vitamin E Metabolites and Concentration Determination
4.2. Cell Culture
4.3. Colorimetric Cell Proliferation Assay
4.4. Flow Cytometric Analysis of Cell Cycle
4.5. Flow Cytometric Analysis of Apoptosis and Necrosis
4.6. Analysis of mRNA Expression
4.6.1. RNA Isolation and cDNA Synthesis
4.6.2. Quantitative Real-Time Reverse Transcription PCR (RT-qPCR)
4.7. Lipidomics Analysis
4.7.1. Lipid Extraction
4.7.2. Analysis of Phospholipid and TG Profiles by UPLC-MS/MS
4.7.3. Determination of the TG and PC Desaturase Index
4.8. Immunoblotting
4.8.1. Sample Preparation
4.8.2. SDS-PAGE and Western Blot Analysis
4.9. Data Presentation and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wallert, M.; Schmölz, L.; Galli, F.; Birringer, M.; Lorkowski, S. Regulatory Metabolites of Vitamin E and Their Putative Relevance for Atherogenesis. Redox Biol. 2014, 2, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Azzi, A.; Birringer, M.; Cook-Mills, J.M.; Eggersdorfer, M.; Frank, J.; Cruciani, G.; Lorkowski, S.; Özer, N.K. Vitamin E: Emerging Aspects and New Directions. Free Radic. Biol. Med. 2017, 102, 16–36. [Google Scholar] [CrossRef]
- Schubert, M.; Kluge, S.; Schmölz, L.; Wallert, M.; Galli, F.; Birringer, M.; Lorkowski, S. Long-Chain Metabolites of Vitamin E: Metabolic Activation as a General Concept for Lipid-Soluble Vitamins? Antioxidants 2018, 7, 10. [Google Scholar] [CrossRef]
- Wallert, M.; Mosig, S.; Rennert, K.; Funke, H.; Ristow, M.; Pellegrino, R.M.; Cruciani, G.; Galli, F.; Lorkowski, S.; Birringer, M. Long-Chain Metabolites of α-Tocopherol Occur in Human Serum and Inhibit Macrophage Foam Cell Formation In Vitro. Free Radic. Biol. Med. 2014, 68, 43–51. [Google Scholar] [CrossRef]
- Pein, H.; Ville, A.; Pace, S.; Temml, V.; Garscha, U.; Raasch, M.; Alsabil, K.; Viault, G.; Dinh, C.-P.; Guilet, D.; et al. Endogenous Metabolites of Vitamin E Limit Inflammation by Targeting 5-Lipoxygenase. Nat. Commun. 2018, 9, 3834. [Google Scholar] [CrossRef] [PubMed]
- Kluge, S.; Schubert, M.; Börmel, L.; Lorkowski, S. The Vitamin E Long-Chain Metabolite α-13′-COOH Affects Macrophage Foam Cell Formation via Modulation of the Lipoprotein Lipase System. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158875. [Google Scholar] [CrossRef]
- Schmölz, L. Complexity of Vitamin E Metabolism. World J. Biol. Chem. 2016, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Ciffolilli, S.; Wallert, M.; Bartolini, D.; Krauth, V.; Werz, O.; Piroddi, M.; Sebastiani, B.; Torquato, P.; Lorkowski, S.; Birringer, M.; et al. Human Serum Determination and in Vitro Anti-Inflammatory Activity of the Vitamin E Metabolite α-(13′-Hydroxy)-6-Hydroxychroman. Free Radic. Biol. Med. 2015, 89, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Schmölz, L.; Schubert, M.; Kirschner, J.; Kluge, S.; Galli, F.; Birringer, M.; Wallert, M.; Lorkowski, S. Long-Chain Metabolites of Vitamin E: Interference with Lipotoxicity via Lipid Droplet Associated Protein PLIN2. Biochim. Biophys. Acta 2018, 1863, 919–927. [Google Scholar] [CrossRef]
- Wallert, M.; Schmölz, L.; Koeberle, A.; Krauth, V.; Glei, M.; Galli, F.; Werz, O.; Birringer, M.; Lorkowski, S. α-Tocopherol Long-Chain Metabolite α-13′-COOH Affects the Inflammatory Response of Lipopolysaccharide-Activated Murine RAW264.7 Macrophages. Mol. Nutr. Food Res. 2015, 59, 1524–1534. [Google Scholar] [CrossRef]
- Schubert, M.; Kluge, S.; Brunner, E.; Pace, S.; Birringer, M.; Werz, O.; Lorkowski, S. The α-Tocopherol-Derived Long-Chain Metabolite α-13′-COOH Mediates Endotoxin Tolerance and Modulates the Inflammatory Response via MAPK and NFκB Pathways. Free Radic. Biol. Med. 2022, 178, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Neukirch, K.; Alsabil, K.; Dinh, C.-P.; Bilancia, R.; Raasch, M.; Ville, A.; Cerqua, I.; Viault, G.; Bréard, D.; Pace, S.; et al. Exploration of Long-Chain Vitamin E Metabolites for the Discovery of a Highly Potent, Orally Effective, and Metabolically STable 5-LOX Inhibitor That Limits Inflammation. J. Med. Chem. 2021, 64, 11496–11526. [Google Scholar] [CrossRef] [PubMed]
- Cerqua, I.; Neukirch, K.; Terlizzi, M.; Granato, E.; Caiazzo, E.; Cicala, C.; Ialenti, A.; Capasso, R.; Werz, O.; Sorrentino, R.; et al. A Vitamin E Long-Chain Metabolite and the Inspired Drug Candidate α-Amplexichromanol Relieve Asthma Features in an Experimental Model of Allergen Sensitization. Pharmacol. Res. 2022, 181, 106250. [Google Scholar] [CrossRef]
- Mazzini, F.; Betti, M.; Netscher, T.; Galli, F.; Salvadori, P. Configuration of the Vitamin E Analogue Garcinoic Acid Extracted from Garcinia Kola Seeds. Chirality 2009, 21, 519–524. [Google Scholar] [CrossRef]
- Birringer, M.; Lington, D.; Vertuani, S.; Manfredini, S.; Scharlau, D.; Glei, M.; Ristow, M. Proapoptotic Effects of Long-Chain Vitamin E Metabolites in HepG2 Cells Are Mediated by Oxidative Stress. Free Radic. Biol. Med. 2010, 49, 1315–1322. [Google Scholar] [CrossRef]
- Willems, S.; Gellrich, L.; Chaikuad, A.; Kluge, S.; Werz, O.; Heering, J.; Knapp, S.; Lorkowski, S.; Schubert-Zsilavecz, M.; Merk, D. Endogenous Vitamin E Metabolites Mediate Allosteric PPARγ Activation with Unprecedented Co-Regulatory Interactions. Cell Chem. Biol. 2021, 28, 1489–1500.e8. [Google Scholar] [CrossRef]
- Bartolini, D.; Franco, F.; Torquato, P.; Marinelli, R.; Cerra, B.; Ronchetti, R.; Schon, A.; Fallarino, F.; Luca, A.; Bellezza, G.; et al. Garcinoic Acid Is a Natural and Selective Agonist of Pregnane X Receptor. J. Med. Chem. 2020, 63, 3701–3712. [Google Scholar] [CrossRef]
- Abraham, A.; Kattoor, A.J.; Saldeen, T.; Mehta, J.L. Vitamin E and Its Anticancer Effects. Crit. Rev. Food Sci. Nutr. 2019, 59, 2831–2838. [Google Scholar] [CrossRef]
- Jiang, Q. Natural Forms of Vitamin E and Metabolites-Regulation of Cancer Cell Death and Underlying Mechanisms. IUBMB Life 2019, 71, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Luo, P.; Zeng, Z.; Wang, H.; Malafa, M.; Suh, N. Vitamin E and Cancer Prevention: Studies with Different Forms of Tocopherols and Tocotrienols. Mol. Carcinog. 2020, 59, 365–389. [Google Scholar] [CrossRef]
- Betti, M.; Minelli, A.; Canonico, B.; Castaldo, P.; Magi, S.; Aisa, M.C.; Piroddi, M.; Di Tomaso, V.; Galli, F. Antiproliferative Effects of Tocopherols (Vitamin E) on Murine Glioma C6 Cells: Homologue-Specific Control of PKC/ERK and Cyclin Signaling. Free Radic. Biol. Med. 2006, 41, 464–472. [Google Scholar] [CrossRef]
- Jang, Y.; Park, N.-Y.; Rostgaard-Hansen, A.L.; Huang, J.; Jiang, Q. Vitamin E Metabolite 13′-Carboxychromanols Inhibit pro-Inflammatory Enzymes, Induce Apoptosis and Autophagy in Human Cancer Cells by Modulating Sphingolipids and Suppress Colon Tumor Development in Mice. Free Radic. Biol. Med. 2016, 95, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Germain, N.; Dhayer, M.; Boileau, M.; Fovez, Q.; Kluza, J.; Marchetti, P. Lipid Metabolism and Resistance to Anticancer Treatment. Biology 2020, 9, 474. [Google Scholar] [CrossRef]
- Han, J.; Kaufman, R.J. The Role of ER Stress in Lipid Metabolism and Lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the Complete Program of Cholesterol and Fatty Acid Synthesis in the Liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Ferré, P.; Foufelle, F. SREBP-1c Transcription Factor and Lipid Homeostasis: Clinical Perspective. Horm. Res. 2007, 68, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Igal, R.A. Stearoyl-CoA Desaturase-1: A Novel Key Player in the Mechanisms of Cell Proliferation, Programmed Cell Death and Transformation to Cancer. Carcinogenesis 2010, 31, 1509–1515. [Google Scholar] [CrossRef]
- Koeberle, A.; Löser, K.; Thürmer, M. Stearoyl-CoA Desaturase-1 and Adaptive Stress Signaling. Biochim. Biophys. Acta 2016, 1861, 1719–1726. [Google Scholar] [CrossRef]
- Hodson, L.; Fielding, B.A. Stearoyl-CoA Desaturase: Rogue or Innocent Bystander? Prog. Lipid Res. 2013, 52, 15–42. [Google Scholar] [CrossRef]
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res. 2019, 79, 5355–5366. [Google Scholar] [CrossRef]
- Yi, J.; Zhu, J.; Wu, J.; Thompson, C.B.; Jiang, X. Oncogenic Activation of PI3K-AKT-MTOR Signaling Suppresses Ferroptosis via SREBP-Mediated Lipogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 31189–31197. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, S.C.; Kipp, A.P.; Stuppner, H.; Koeberle, A. Ferroptosis-Modulating Small Molecules for Targeting Drug-Resistant Cancer: Challenges and Opportunities in Manipulating Redox Signaling. Med. Res. Rev. 2023, 43, 614–682. [Google Scholar] [CrossRef] [PubMed]
- Luis, G.; Godfroid, A.; Nishiumi, S.; Cimino, J.; Blacher, S.; Maquoi, E.; Wery, C.; Collignon, A.; Longuespée, R.; Montero-Ruiz, L.; et al. Tumor Resistance to Ferroptosis Driven by Stearoyl-CoA Desaturase-1 (SCD1) in Cancer Cells and Fatty Acid Biding Protein-4 (FABP4) in Tumor Microenvironment Promote Tumor Recurrence. Redox Biol. 2021, 43, 102006. [Google Scholar] [CrossRef]
- Igal, R.A. Stearoyl CoA Desaturase-1: New Insights into a Central Regulator of Cancer Metabolism. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 1865–1880. [Google Scholar] [CrossRef]
- Nile, A.H.; Hannoush, R.N. Fatty Acylation of Wnt Proteins. Nat. Chem. Biol. 2016, 12, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Magtanong, L.; Ko, P.-J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef]
- Thürmer, M.; Gollowitzer, A.; Pein, H.; Neukirch, K.; Gelmez, E.; Waltl, L.; Wielsch, N.; Winkler, R.; Löser, K.; Grander, J.; et al. PI(18:1/18:1) Is a SCD1-Derived Lipokine That Limits Stress Signaling. Nat. Commun. 2022, 13, 2982. [Google Scholar] [CrossRef] [PubMed]
- Sahani, M.H.; Itakura, E.; Mizushima, N. Expression of the Autophagy Substrate SQSTM1/P62 Is Restored during Prolonged Starvation Depending on Transcriptional Upregulation and Autophagy-Derived Amino Acids. Autophagy 2014, 10, 431–441. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef]
- Richmond, L.; Keeshan, K. Pseudokinases: A Tribble-Edged Sword. FEBS J. 2020, 287, 4170–4182. [Google Scholar] [CrossRef]
- Ku, H.-C.; Cheng, C.-F. Master Regulator Activating Transcription Factor 3 (ATF3) in Metabolic Homeostasis and Cancer. Front. Endocrinol. 2020, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid Metabolism in Cancer Cells under Metabolic Stress. Br. J. Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Walter, P. The Integrated Stress Response: From Mechanism to Disease. Science 2020, 368, eaat5314. [Google Scholar] [CrossRef]
- Ascenzi, F.; De Vitis, C.; Maugeri-Saccà, M.; Napoli, C.; Ciliberto, G.; Mancini, R. SCD1, Autophagy and Cancer: Implications for Therapy. J. Exp. Clin. Cancer Res. 2021, 40, 265. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Walsh, M.C.; Hoehn, K.L.; James, D.E.; Wherry, E.J.; Choi, Y. Regulator of Fatty Acid Metabolism, Acetyl Coenzyme a Carboxylase 1, Controls T Cell Immunity. J. Immunol. 2014, 192, 3190–3199. [Google Scholar] [CrossRef]
- Ha, J.; Lee, J.K.; Kim, K.S.; Witters, L.A.; Kim, K.H. Cloning of Human Acetyl-CoA Carboxylase-Beta and Its Unique Features. Proc. Natl. Acad. Sci. USA 1996, 93, 11466–11470. [Google Scholar] [CrossRef]
- Jensen-Urstad, A.P.L.; Semenkovich, C.F. Fatty Acid Synthase and Liver Triglyceride Metabolism: Housekeeper or Messenger? Biochim. Biophys. Acta 2012, 1821, 747–753. [Google Scholar] [CrossRef]
- Mziaut, H.; Korza, G.; Ozols, J. The N Terminus of Microsomal Delta 9 Stearoyl-CoA Desaturase Contains the Sequence Determinant for Its Rapid Degradation. Proc. Natl. Acad. Sci. USA 2000, 97, 8883–8888. [Google Scholar] [CrossRef]
- Wang, Y.; Viscarra, J.; Kim, S.-J.; Sul, H.S. Transcriptional Regulation of Hepatic Lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef]
- Aljohani, A.; Syed, D.N.; James, M. Ntambi Insights into Stearoyl-CoA Desaturase-1 Regulation of Systemic Metabolism. Trends Endocrinol. Metab. 2017, 28, 831–842. [Google Scholar] [CrossRef]
- Amemiya-Kudo, M.; Shimano, H.; Yoshikawa, T.; Yahagi, N.; Hasty, A.H.; Okazaki, H.; Tamura, Y.; Shionoiri, F.; Iizuka, Y.; Ohashi, K.; et al. Promoter Analysis of the Mouse Sterol Regulatory Element-Binding Protein-1c Gene. J. Biol. Chem. 2000, 275, 31078–31085. [Google Scholar] [CrossRef]
- Wang, X.; Sato, R.; Brown, M.S.; Hua, X.; Goldstein, J.L. SREBP-1, a Membrane-Bound Transcription Factor Released by Sterol-Regulated Proteolysis. Cell 1994, 77, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q. Natural Forms of Vitamin E as Effective Agents for Cancer Prevention and Therapy. Adv. Nutr. 2017, 8, 850–867. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-Analysis: High-Dosage Vitamin E Supplementation May Increase All-Cause Mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345.e22. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.M.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the Risk of Prostate Cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef]
- Hu, Q.; Zhang, Y.; Lou, H.; Ou, Z.; Liu, J.; Duan, W.; Wang, H.; Ge, Y.; Min, J.; Wang, F.; et al. GPX4 and Vitamin E Cooperatively Protect Hematopoietic Stem and Progenitor Cells from Lipid Peroxidation and Ferroptosis. Cell Death Dis. 2021, 12, 706. [Google Scholar] [CrossRef]
- Villalón-García, I.; Álvarez-Córdoba, M.; Povea-Cabello, S.; Talaverón-Rey, M.; Villanueva-Paz, M.; Luzón-Hidalgo, R.; Suárez-Rivero, J.M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Salas, J.J.; et al. Vitamin E Prevents Lipid Peroxidation and Iron Accumulation in PLA2G6-Associated Neurodegeneration. Neurobiol. Dis. 2022, 165, 105649. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis Turns 10: Emerging Mechanisms, Physiological Functions, and Therapeutic Applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the Crossroads of Cancer-Acquired Drug Resistance and Immune Evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef]
- Galli, F.; Stabile, A.M.; Betti, M.; Conte, C.; Pistilli, A.; Rende, M.; Floridi, A.; Azzi, A. The Effect of Alpha- and Gamma-Tocopherol and Their Carboxyethyl Hydroxychroman Metabolites on Prostate Cancer Cell Proliferation. Arch. Biochem. 2004, 423, 97–102. [Google Scholar] [CrossRef]
- Tracz-Gaszewska, Z.; Dobrzyn, P. Stearoyl-CoA Desaturase 1 as a Therapeutic Target for the Treatment of Cancer. Cancers 2019, 11, 948. [Google Scholar] [CrossRef] [PubMed]
- Sen, U.; Coleman, C.; Sen, T. Stearoyl Coenzyme A Desaturase-1: Multitasker in Cancer, Metabolism, and Ferroptosis. Trends Cancer 2023, 9, 480–489. [Google Scholar] [CrossRef]
- Mason, P.; Liang, B.; Li, L.; Fremgen, T.; Murphy, E.; Quinn, A.; Madden, S.L.; Biemann, H.-P.; Wang, B.; Cohen, A. SCD1 Inhibition Causes Cancer Cell Death by Depleting Mono-Unsaturated Fatty Acids. PLoS ONE 2012, 7, e33823. [Google Scholar] [CrossRef] [PubMed]
- Green, C.D.; Olson, L.K. Modulation of Palmitate-Induced Endoplasmic Reticulum Stress and Apoptosis in Pancreatic β-Cells by Stearoyl-CoA Desaturase and Elovl6. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E640–E649. [Google Scholar] [CrossRef] [PubMed]
- Peck, B.; Schug, Z.T.; Zhang, Q.; Dankworth, B.; Jones, D.T.; Smethurst, E.; Patel, R.; Mason, S.; Jiang, M.; Saunders, R. Inhibition of Fatty Acid Desaturation Is Detrimental to Cancer Cell Survival in Metabolically Compromised Environments. Cancer Metab. 2016, 4, 6. [Google Scholar] [CrossRef]
- Koeberle, A.; Pergola, C.; Shindou, H.; Koeberle, S.C.; Shimizu, T.; Laufer, S.A.; Werz, O. Role of P38 Mitogen-Activated Protein Kinase in Linking Stearoyl-CoA Desaturase-1 Activity with Endoplasmic Reticulum Homeostasis. FASEB J. 2015, 29, 2439–2449. [Google Scholar] [CrossRef]
- Mohammadzadeh, F.; Mosayebi, G.; Montazeri, V.; Darabi, M.; Fayezi, S.; Shaaker, M.; Rahmati, M.; Baradaran, B.; Mehdizadeh, A.; Darabi, M. Fatty Acid Composition of Tissue Cultured Breast Carcinoma and the Effect of Stearoyl-CoA Desaturase 1 Inhibition. J. Breast Cancer 2014, 17, 136–142. [Google Scholar] [CrossRef]
- Pinkham, K.; Park, D.J.; Hashemiaghdam, A.; Kirov, A.B.; Adam, I.; Rosiak, K.; da Hora, C.C.; Teng, J.; Cheah, P.S.; Carvalho, L.; et al. Stearoyl CoA Desaturase Is Essential for Regulation of Endoplasmic Reticulum Homeostasis and Tumor Growth in Glioblastoma Cancer Stem Cells. Stem Cell Rep. 2019, 12, 712–727. [Google Scholar] [CrossRef]
- de Lima Luna, A.C.; Forti, F.L. Modulation of SCD1 Activity in Hepatocyte Cell Lines: Evaluation of Genomic Stability and Proliferation. Mol. Cell. Biochem. 2021, 476, 3393–3405. [Google Scholar] [CrossRef]
- Antonny, B.; Vanni, S.; Shindou, H.; Ferreira, T. From Zero to Six Double Bonds: Phospholipid Unsaturation and Organelle Function. Trends Cell Biol. 2015, 25, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Harayama, T.; Riezman, H. Understanding the Diversity of Membrane Lipid Composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. P38(MAPK): Stress Responses from Molecular Mechanisms to Therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; So, J.-S.; Park, J.-G.; Lee, A.-H. Transcriptional Control of Hepatic Lipid Metabolism by SREBP and ChREBP. Semin. Liver Dis. 2013, 33, 301–311. [Google Scholar] [CrossRef]
- Cheng, X.; Li, J.; Guo, D. SCAP/SREBPs Are Central Players in Lipid Metabolism and Novel Metabolic Targets in Cancer Therapy. Curr. Top Med. Chem. 2018, 18, 484–493. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, J.; Su, W.; Wu, J.; Wang, C.; Kong, X.; Gustafsson, J.-Å.; Ding, J.; Ma, X.; Guan, Y. Liver X Receptor Activation Increases Hepatic Fatty Acid Desaturation by the Induction of SCD1 Expression through an LXRα-SREBP1c-Dependent Mechanism. J. Diabetes 2014, 6, 212–220. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular Mechanism of PPARα Action and Its Impact on Lipid Metabolism, Inflammation and Fibrosis in Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.-L.; Schulze, A. SREBP Activity Is Regulated by MTORC1 and Contributes to Akt-Dependent Cell Growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef]
- Tian, L.-Y.; Smit, D.J.; Jücker, M. The Role of PI3K/AKT/MTOR Signaling in Hepatocellular Carcinoma Metabolism. Int. J. Mol. Sci. 2023, 24, 2652. [Google Scholar] [CrossRef]
- Yamamoto, T.; Shimano, H.; Inoue, N.; Nakagawa, Y.; Matsuzaka, T.; Takahashi, A.; Yahagi, N.; Sone, H.; Suzuki, H.; Toyoshima, H.; et al. Protein Kinase A Suppresses Sterol Regulatory Element-Binding Protein-1C Expression via Phosphorylation of Liver X Receptor in the Liver*. J. Biol. Chem. 2007, 282, 11687–11695. [Google Scholar] [CrossRef]
- Higuchi, N.; Kato, M.; Shundo, Y.; Tajiri, H.; Tanaka, M.; Yamashita, N.; Kohjima, M.; Kotoh, K.; Nakamuta, M.; Takayanagi, R.; et al. Liver X Receptor in Cooperation with SREBP-1c Is a Major Lipid Synthesis Regulator in Nonalcoholic Fatty Liver Disease. Hepatol. Res. 2008, 38, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Knight, B.L.; Hebbachi, A.; Hauton, D.; Brown, A.-M.; Wiggins, D.; Patel, D.D.; Gibbons, G.F. A Role for PPARα in the Control of SREBP Activity and Lipid Synthesis in the Liver. Biochem. J. 2005, 389, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Yang, R.; Tarr, P.T.; Wu, P.-H.; Handschin, C.; Li, S.; Yang, W.; Pei, L.; Uldry, M.; Tontonoz, P.; et al. Hyperlipidemic Effects of Dietary Saturated Fats Mediated through PGC-1β Coactivation of SREBP. Cell 2005, 120, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, K.; Liu, X.; Huang, L.; Zhao, D.; Li, L.; Gao, M.; Pei, D.; Wang, C.; Liu, X. Srebp-1 Interacts with c-Myc to Enhance Somatic Cell Reprogramming. Stem Cells 2016, 34, 83–92. [Google Scholar] [CrossRef]
- Ponugoti, B.; Kim, D.-H.; Xiao, Z.; Smith, Z.; Miao, J.; Zang, M.; Wu, S.-Y.; Chiang, C.-M.; Veenstra, T.D.; Kemper, J.K. SIRT1 Deacetylates and Inhibits SREBP-1C Activity in Regulation of Hepatic Lipid Metabolism. J. Biol. Chem. 2010, 285, 33959–33970. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Yahagi, N.; Izumida, Y.; Nishi, M.; Kubota, M.; Teraoka, Y.; Yamamoto, T.; Matsuzaka, T.; Nakagawa, Y.; Sekiya, M.; et al. Polyunsaturated Fatty Acids Selectively Suppress Sterol Regulatory Element-Binding Protein-1 through Proteolytic Processing and Autoloop Regulatory Circuit. J. Biol. Chem. 2010, 285, 11681–11691. [Google Scholar] [CrossRef]
- Hawker, J.R. Chemiluminescence-Based BrdU ELISA to Measure DNA Synthesis. J. Immunol. Methods 2003, 274, 77–82. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Juan, G. DNA Content Measurement for DNA Ploidy and Cell Cycle Analysis. Curr. Protoc. Cytom. 2001. [Google Scholar] [CrossRef]
- Stolle, K.; Schnoor, M.; Fuellen, G.; Spitzer, M.; Cullen, P.; Lorkowski, S. Cloning, Genomic Organization, and Tissue-Specific Expression of the RASL11B Gene. Biochim. Biophys. Acta Gene Struct. Expr. 2007, 1769, 514–524. [Google Scholar] [CrossRef]
- Stolle, K.; Schnoor, M.; Fuellen, G.; Spitzer, M.; Engel, T.; Spener, F.; Cullen, P.; Lorkowski, S. Cloning, Cellular Localization, Genomic Organization, and Tissue-Specific Expression of the TGFβ1-Inducible SMAP-5 Gene. Gene 2005, 351, 119–130. [Google Scholar] [CrossRef]
- Koeberle, A.; Shindou, H.; Harayama, T.; Shimizu, T. Role of Lysophosphatidic Acid Acyltransferase 3 for the Supply of Highly Polyunsaturated Fatty Acids in TM4 Sertoli Cells. FASEB J. 2010, 24, 4929–4938. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, A.; Shindou, H.; Harayama, T.; Yuki, K.; Shimizu, T. Polyunsaturated Fatty Acids Are Incorporated into Maturating Male Mouse Germ Cells by Lysophosphatidic Acid Acyltransferase 3. FASEB J. 2012, 26, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, A.; Shindou, H.; Koeberle, S.C.; Laufer, S.A.; Shimizu, T.; Werz, O. Arachidonoyl-Phosphatidylcholine Oscillates during the Cell Cycle and Counteracts Proliferation by Suppressing Akt Membrane Binding. Proc. Natl. Acad. Sci. USA 2013, 110, 2546–2551. [Google Scholar] [CrossRef] [PubMed]
- Espada, L.; Dakhovnik, A.; Chaudhari, P.; Martirosyan, A.; Miek, L.; Poliezhaieva, T.; Schaub, Y.; Nair, A.; Döring, N.; Rahnis, N.; et al. Loss of Metabolic Plasticity Underlies Metformin Toxicity in Aged Caenorhabditis Elegans. Nat. Metab. 2020, 2, 1316–1331. [Google Scholar] [CrossRef]
- van Pijkeren, A.; Egger, A.-S.; Hotze, M.; Zimmermann, E.; Kipura, T.; Grander, J.; Gollowitzer, A.; Koeberle, A.; Bischoff, R.; Thedieck, K.; et al. Proteome Coverage after Simultaneous Proteo-Metabolome Liquid-Liquid Extraction. J. Proteome Res. 2023, 22, 951–966. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, S.; Gollowitzer, A.; Börmel, L.; Maier, C.; Gottschalk, L.; Werz, O.; Wallert, M.; Koeberle, A.; Lorkowski, S. α-Tocopherol-13′-Carboxychromanol Induces Cell Cycle Arrest and Cell Death by Inhibiting the SREBP1-SCD1 Axis and Causing Imbalance in Lipid Desaturation. Int. J. Mol. Sci. 2023, 24, 9229. https://doi.org/10.3390/ijms24119229
Liao S, Gollowitzer A, Börmel L, Maier C, Gottschalk L, Werz O, Wallert M, Koeberle A, Lorkowski S. α-Tocopherol-13′-Carboxychromanol Induces Cell Cycle Arrest and Cell Death by Inhibiting the SREBP1-SCD1 Axis and Causing Imbalance in Lipid Desaturation. International Journal of Molecular Sciences. 2023; 24(11):9229. https://doi.org/10.3390/ijms24119229
Chicago/Turabian StyleLiao, Sijia, André Gollowitzer, Lisa Börmel, Charlotte Maier, Luisa Gottschalk, Oliver Werz, Maria Wallert, Andreas Koeberle, and Stefan Lorkowski. 2023. "α-Tocopherol-13′-Carboxychromanol Induces Cell Cycle Arrest and Cell Death by Inhibiting the SREBP1-SCD1 Axis and Causing Imbalance in Lipid Desaturation" International Journal of Molecular Sciences 24, no. 11: 9229. https://doi.org/10.3390/ijms24119229