
Injured Endothelial Cell: A Risk Factor for Pulmonary Fibrosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
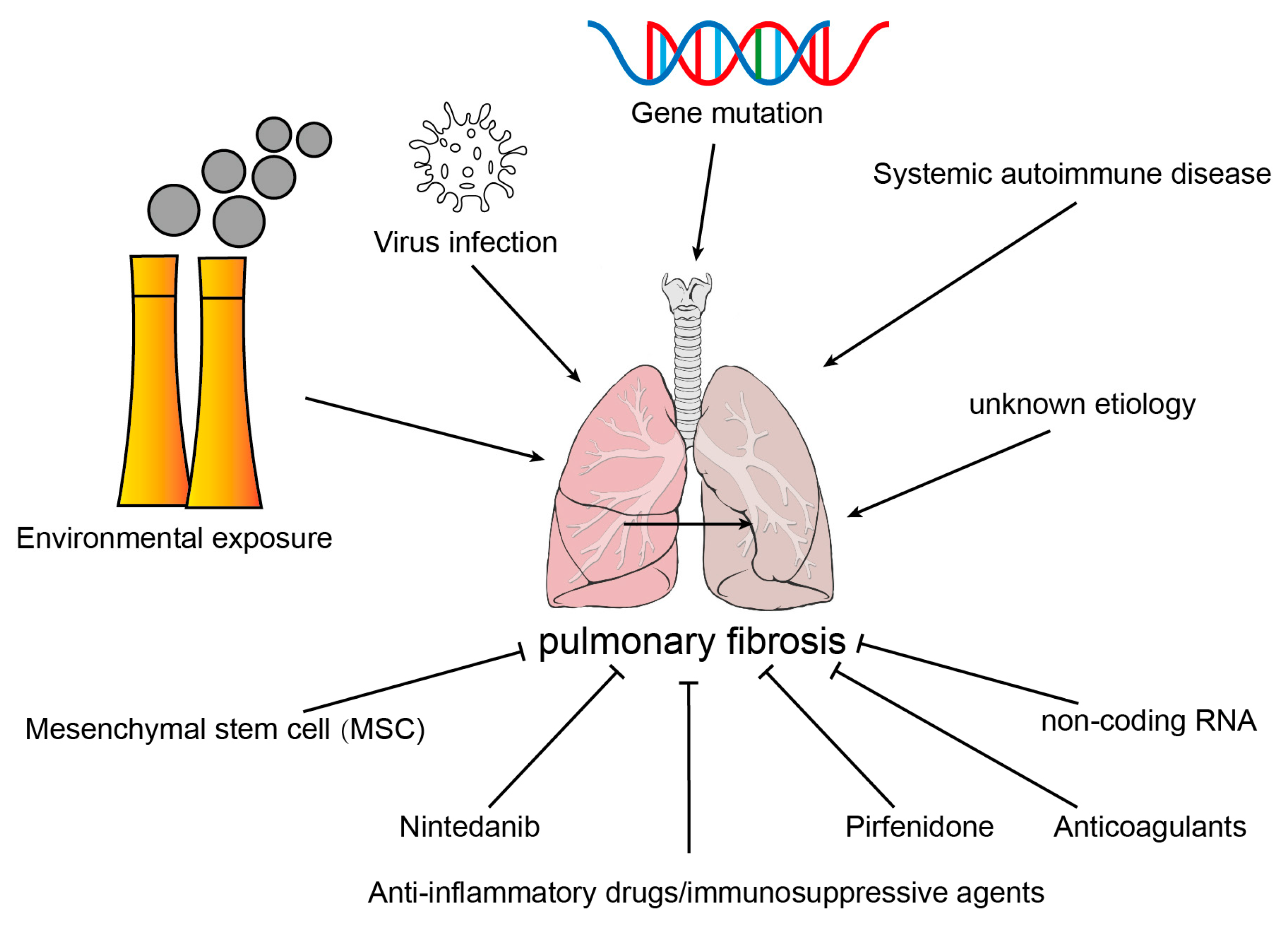
2. Pulmonary Fibrosis
2.1. Etiology of Common Pulmonary Fibrosis
2.1.1. Environmental Exposure
2.1.2. Virus Infection
2.1.3. Systemic Autoimmune Disease
2.1.4. Gene Mutation
2.1.5. Idiopathic Pulmonary Fibrosis
2.2. Source of Myofibroblasts in Fibrotic Lung Tissues
2.2.1. Interstitial Fibroblast
2.2.2. Epithelial Cell
2.2.3. Circulating Fibrocyte
2.2.4. Microvascular Pericyte
2.2.5. Macrophage
2.2.6. Endothelial Cell
3. Endothelial–Mesenchymal Transition
3.1. Endothelia Cell
3.2. Injury and Activation of Endothelial Cells
3.3. Endothelial–Mesenchymal Transition
3.4. Inducement of E(nd)MT
3.4.1. Hypoxia
3.4.2. Oxidative Stress
3.4.3. Inflammatory
3.4.4. Shear Stress
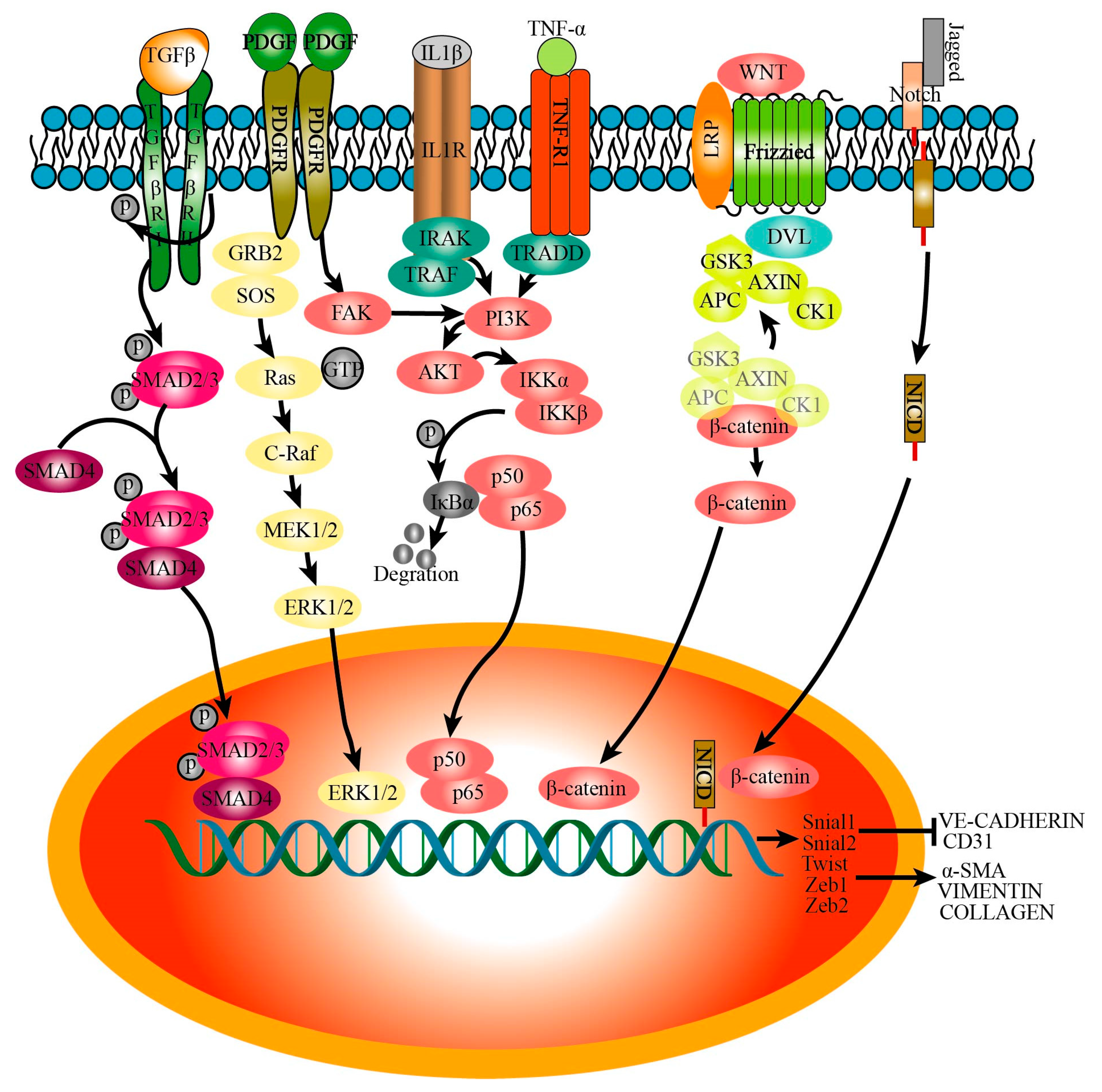
3.5. Signaling Pathways That Regulate Endothelial–Mesenchymal Transition
3.5.1. TGFβ
3.5.2. IL-1β
3.5.3. TNFα
3.5.4. Wnt
3.5.5. Notch
3.5.6. PDGF
3.5.7. Others
4. Effects of Activated Endothelial Cells on other Cells in Pulmonary Fibrosis
4.1. Recruitment of Immune Cells
4.2. Activation and Myofibroblastic Transformation of Fibroblasts
4.3. Epithelial Cell Damage and Repair
4.4. Pericytes Cell Activation
5. Drugs for IPF That Targeted Endothelial Cells
5.1. Ambrisentan, Bosentan, and Macitentan
5.2. Rapamycin
5.3. Imatinib
5.4. Relaxin
5.5. Nintedanib
5.6. Pirfenidone
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
List of Abbreviations
| PF | Pulmonary fibrosis |
| ECM | Extracellular matrix |
| E(nd)MT | Endothelial–mesenchymal transition |
| EMT | Epithelial–mesenchymal transition |
| ECs | Endothelial cells |
| AECII | Type II alveolar epithelial cells |
| MMP | Matrix metalloproteinases |
| BLM | Bleomycin |
| IPF | Idiopathic pulmonary fibrosis |
| MMT | Macrophage–mesenchymal transition |
| FMT | Fibroblast–mesenchymal transition |
References
- Cheng, L.; Wang, D.; Deng, B.; Li, J.; Zhang, J.; Guo, X.; Yan, T.; Yue, X.; An, Y.; Zhang, B.; et al. DR7dA, a Novel Antioxidant Peptide Analog, Demonstrates Antifibrotic Activity in Pulmonary Fibrosis In Vivo and In Vitro. J. Pharmacol. Exp. Ther. 2022, 382, 100–112. [Google Scholar] [CrossRef]
- Marchioni, A.; Tonelli, R.; Cerri, S.; Castaniere, I.; Andrisani, D.; Gozzi, F.; Bruzzi, G.; Manicardi, L.; Moretti, A.; Demurtas, J.; et al. Pulmonary Stretch and Lung Mechanotransduction: Implications for Progression in the Fibrotic Lung. Int. J. Mol. Sci. 2021, 22, 6443. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.M.; Li, W.C.; Liu, D.S.; Li, Y.P.; Wen, F.Q.; Feng, Y.L.; Zhang, S.F.; Huang, X.Y.; Wang, T.; Wang, K.; et al. VEGFR-2 antagonist SU5416 attenuates bleomycin-induced pulmonary fibrosis in mice. Int. Immunopharmacol. 2009, 9, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.K.; Ramesh, V.; Castro, C.A.; Kaushik, V.; Kulkarni, Y.M.; Wright, C.A.; Venkatadri, R.; Rojanasakul, Y.; Azad, N. Nitric oxide mediates bleomycin-induced angiogenesis and pulmonary fibrosis via regulation of VEGF. J. Cell. Biochem. 2015, 116, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Inui, N.; Hakamata, A.; Suzuki, Y.; Enomoto, N.; Fujisawa, T.; Nakamura, Y.; Watanabe, H.; Suda, T. Changes in pulmonary endothelial cell properties during bleomycin-induced pulmonary fibrosis. Respir. Res. 2018, 19, 127. [Google Scholar] [CrossRef]
- Hulshoff, M.S.; Del Monte-Nieto, G.; Kovacic, J.; Krenning, G. Non-coding RNA in endothelial-to-mesenchymal transition. Cardiovasc. Res. 2019, 115, 1716–1731. [Google Scholar] [CrossRef]
- Fujimoto, H.; Kobayashi, T.; Azuma, A. Idiopathic Pulmonary Fibrosis: Treatment and Prognosis. Clin. Med. Insights Circ. Respir. Pulm. Med. 2015, 9, 179–185. [Google Scholar] [CrossRef]
- Somogyi, V.; Chaudhuri, N.; Torrisi, S.E.; Kahn, N.; Muller, V.; Kreuter, M. The therapy of idiopathic pulmonary fibrosis: What is next? Eur. Respir. Rev. 2019, 28, 153. [Google Scholar] [CrossRef]
- Gao, Y.; Sun, J.; Dong, C.; Zhao, M.; Hu, Y.; Jin, F. Extracellular Vesicles Derived from Adipose Mesenchymal Stem Cells Alleviate PM2.5-Induced Lung Injury and Pulmonary Fibrosis. Med. Sci. Monit. 2020, 26, e922782. [Google Scholar] [CrossRef]
- Xu, M.; Wang, X.; Xu, L.; Zhang, H.; Li, C.; Liu, Q.; Chen, Y.; Chung, K.F.; Adcock, I.M.; Li, F. Chronic lung inflammation and pulmonary fibrosis after multiple intranasal instillation of PM2.5 in mice. Environ. Toxicol. 2021, 36, 1434–1446. [Google Scholar] [CrossRef]
- Cheresh, P.; Morales-Nebreda, L.; Kim, S.J.; Yeldandi, A.; Williams, D.B.; Cheng, Y.; Mutlu, G.M.; Budinger, G.R.; Ridge, K.; Schumacker, P.T.; et al. Asbestos-induced pulmonary fibrosis is augmented in 8-oxoguanine DNA glycosylase knockout mice. Am. J. Respir. Cell Mol. Biol. 2015, 52, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Sheng, G.; Chen, P.; Wei, Y.; Yue, H.; Chu, J.; Zhao, J.; Wang, Y.; Zhang, W.; Zhang, H.L. Viral Infection Increases the Risk of Idiopathic Pulmonary Fibrosis: A Meta-Analysis. Chest 2020, 157, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Tang, X.X. Virus infection induced pulmonary fibrosis. J. Transl. Med. 2021, 19, 496. [Google Scholar] [CrossRef] [PubMed]
- Fukui, Y.; Nakamura, K.; Hirabayashi, M.; Miyagawa, T.; Toyama, S.; Omatsu, J.; Awaji, K.; Ikawa, T.; Norimatsu, Y.; Yoshizaki, A.; et al. Serum vasohibin-1 levels: A potential marker of dermal and pulmonary fibrosis in systemic sclerosis. Exp. Dermatol. 2021, 30, 951–958. [Google Scholar] [CrossRef]
- Wang, S.; Liu, M.; Li, X.; Zhang, J.; Wang, F.; Zhang, C.; Roden, A.; Ryu, J.H.; Warrington, K.J.; Sun, J.; et al. Canonical and noncanonical regulatory roles for JAK2 in the pathogenesis of rheumatoid arthritis-associated interstitial lung disease and idiopathic pulmonary fibrosis. FASEB J. 2022, 36, e22336. [Google Scholar] [CrossRef]
- Shen, N.; Zhou, X.; Jin, X.; Lu, C.; Hu, X.; Zhang, Y.; Jiang, Y.; Xu, Q.; Xu, X.; Liu, M.; et al. MDA5 expression is associated with TGF-beta-induced fibrosis: Potential mechanism of interstitial lung disease in anti-MDA5 dermatomyositis. Rheumatology 2022, 62, 373–383. [Google Scholar] [CrossRef]
- Shi, L.; Fu, Q.; Chen, N.; Liu, R.; Zheng, Y. Angiopoietin-like protein 2 as a novel marker for patients with primary Sjogren’s syndrome-related interstitial lung disease. Clin. Exp. Med. 2020, 20, 393–399. [Google Scholar] [CrossRef]
- Caballero, I.; Ringot-Destrez, B.; Si-Tahar, M.; Barbry, P.; Guillon, A.; Lantier, I.; Berri, M.; Chevaleyre, C.; Fleurot, I.; Barc, C.; et al. Evidence of early increased sialylation of airway mucins and defective mucociliary clearance in CFTR-deficient piglets. J. Cyst. Fibros. 2021, 20, 173–182. [Google Scholar] [CrossRef]
- Pardo, A.; Selman, M. The Interplay of the Genetic Architecture, Aging, and Environmental Factors in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 64, 163–172. [Google Scholar] [CrossRef]
- Lim, M.J.; Ahn, J.; Yi, J.Y.; Kim, M.H.; Son, A.R.; Lee, S.L.; Lim, D.S.; Kim, S.S.; Kang, M.A.; Han, Y.; et al. Induction of galectin-1 by TGF-beta1 accelerates fibrosis through enhancing nuclear retention of Smad2. Exp. Cell Res. 2014, 326, 125–135. [Google Scholar] [CrossRef]
- Yao, L.; Conforti, F.; Hill, C.; Bell, J.; Drawater, L.; Li, J.; Liu, D.; Xiong, H.; Alzetani, A.; Chee, S.J.; et al. Paracrine signalling during ZEB1-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in lung fibrosis. Cell Death Differ. 2019, 26, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.Y.; Chu, P.H.; Lee, T.H. R1R2 peptide ameliorates pulmonary fibrosis in mice through fibrocyte migration and differentiation. PLoS ONE 2017, 12, e0185811. [Google Scholar] [CrossRef] [PubMed]
- Aono, Y.; Kishi, M.; Yokota, Y.; Azuma, M.; Kinoshita, K.; Takezaki, A.; Sato, S.; Kawano, H.; Kishi, J.; Goto, H.; et al. Role of platelet-derived growth factor/platelet-derived growth factor receptor axis in the trafficking of circulating fibrocytes in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2014, 51, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.E.; Johnson, J.R. Pericytes in chronic lung disease. Int. Arch. Allergy Immunol. 2014, 164, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Sava, P.; Ramanathan, A.; Dobronyi, A.; Peng, X.; Sun, H.; Ledesma-Mendoza, A.; Herzog, E.L.; Gonzalez, A.L. Human pericytes adopt myofibroblast properties in the microenvironment of the IPF lung. JCI Insight 2017, 2, 24. [Google Scholar] [CrossRef]
- Wang, Y.C.; Xie, H.; Zhang, Y.C.; Meng, Q.H.; Xiong, M.M.; Jia, M.W.; Peng, F.; Tang, D.L. Exosomal miR-107 antagonizes profibrotic phenotypes of pericytes by targeting a pathway involving HIF-1alpha/Notch1/PDGFRbeta/YAP1/Twist1 axis in vitro. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H520–H534. [Google Scholar] [CrossRef]
- Nikolic-Paterson, D.J.; Wang, S.; Lan, H.Y. Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int. Suppl. 2014, 4, 34–38. [Google Scholar] [CrossRef]
- Yang, F.; Chang, Y.; Zhang, C.; Xiong, Y.; Wang, X.; Ma, X.; Wang, Z.; Li, H.; Shimosawa, T.; Pei, L.; et al. UUO induces lung fibrosis with macrophage-myofibroblast transition in rats. Int. Immunopharmacol. 2021, 93, 107396. [Google Scholar] [CrossRef]
- Hashimoto, N.; Phan, S.H.; Imaizumi, K.; Matsuo, M.; Nakashima, H.; Kawabe, T.; Shimokata, K.; Hasegawa, Y. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 43, 161–172. [Google Scholar] [CrossRef]
- Gaikwad, A.V.; Lu, W.; Dey, S.; Bhattarai, P.; Chia, C.; Larby, J.; Haug, G.; Myers, S.; Jaffar, J.; Westall, G.; et al. Vascular remodelling in idiopathic pulmonary fibrosis patients and its detrimental effect on lung physiology: Potential role of endothelial-to-mesenchymal transition. ERJ Open Res. 2022, 8, 1. [Google Scholar] [CrossRef]
- Dugina, V.B.; Shagieva, G.S.; Shakhov, A.S.; Alieva, I.B. The Cytoplasmic Actins in the Regulation of Endothelial Cell Function. Int. J. Mol. Sci. 2021, 22, 7836. [Google Scholar] [CrossRef] [PubMed]
- Minami, T.; Aird, W.C. Endothelial cell gene regulation. Trends Cardiovasc. Med. 2005, 15, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Kim, Y.O.; Wagner, W.L.; Schuppan, D.; Valenzuela, C.D.; Mentzer, S.J.; Kreuz, S.; Stiller, D.; Wollin, L.; Konerding, M.A. Effects of nintedanib on the microvascular architecture in a lung fibrosis model. Angiogenesis 2017, 20, 359–372. [Google Scholar] [CrossRef]
- Wang, R.; Wang, Y.; Hu, L.; Lu, Z.; Wang, X. Inhibition of complement C5a receptor protects lung cells and tissues against lipopolysaccharide-induced injury via blocking pyroptosis. Aging 2021, 13, 8588–8598. [Google Scholar] [CrossRef]
- Cao, Z.; Lis, R.; Ginsberg, M.; Chavez, D.; Shido, K.; Rabbany, S.Y.; Fong, G.H.; Sakmar, T.P.; Rafii, S.; Ding, B.S. Targeting of the pulmonary capillary vascular niche promotes lung alveolar repair and ameliorates fibrosis. Nat. Med. 2016, 22, 154–162. [Google Scholar] [CrossRef]
- Liu, Q.; Shi, X.; Tang, L.; Xu, W.; Jiang, S.; Ding, W.; Feng, Q.; Chu, H.; Ma, Y.; Li, Y.; et al. Salvianolic acid B attenuates experimental pulmonary inflammation by protecting endothelial cells against oxidative stress injury. Eur. J. Pharmacol. 2018, 840, 9–19. [Google Scholar] [CrossRef]
- Pei, B.; Zhang, N.; Pang, T.; Sun, G. Linagliptin ameliorates pulmonary fibrosis in systemic sclerosis mouse model via inhibition of endothelial-to-mesenchymal transition. Mol. Cell. Biochem. 2022, 477, 995–1007. [Google Scholar] [CrossRef]
- Xu, Y.; Hu, X.; Zhang, Y.; Pan, Z.; Sun, Z.; Huang, Z.; Zheng, S.; Pan, H.; Zou, X.; Huang, P. Heterogeneous microenvironment analysis to explore the potential regulatory role of endothelial-mesenchymal transition in idiopathic pulmonary fibrosis. Ann. Transl. Med. 2022, 10, 486. [Google Scholar] [CrossRef]
- Martin, M.; Zhang, J.; Miao, Y.; He, M.; Kang, J.; Huang, H.Y.; Chou, C.H.; Huang, T.S.; Hong, H.C.; Su, S.H.; et al. Role of endothelial cells in pulmonary fibrosis via SREBP2 activation. JCI Insight 2021, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Yarnold, J.; Brotons, M.C. Pathogenetic mechanisms in radiation fibrosis. Radiother. Oncol. 2010, 97, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Hong, Z.Y.; Nam, J.K.; Lee, H.J.; Jang, J.; Yoo, R.J.; Lee, Y.J.; Lee, C.Y.; Kim, K.H.; Park, S.; et al. A Hypoxia-Induced Vascular Endothelial-to-Mesenchymal Transition in Development of Radiation-Induced Pulmonary Fibrosis. Clin. Cancer. Res. 2015, 21, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Tada, Y.; Gladson, S.; Nishimura, R.; Shimomura, I.; Karasawa, S.; Tatsumi, K.; West, J. Vildagliptin ameliorates pulmonary fibrosis in lipopolysaccharide-induced lung injury by inhibiting endothelial-to-mesenchymal transition. Respir. Res. 2017, 18, 177. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Shi, J.; Chen, L.; Lv, Z.; Chen, X.; Cao, H.; Xiang, Z.; Han, X. M2 macrophages promote myofibroblast differentiation of LR-MSCs and are associated with pulmonary fibrogenesis. Cell Commun. Signal 2018, 16, 89. [Google Scholar] [CrossRef]
- Fleckenstein, K.; Zgonjanin, L.; Chen, L.; Rabbani, Z.; Jackson, I.L.; Thrasher, B.; Kirkpatrick, J.; Foster, W.M.; Vujaskovic, Z. Temporal onset of hypoxia and oxidative stress after pulmonary irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 196–204. [Google Scholar] [CrossRef]
- Elhady, S.S.; Goda, M.S.; Mehanna, E.T.; Elfaky, M.A.; Koshak, A.E.; Noor, A.O.; Bogari, H.A.; Malatani, R.T.; Abdelhameed, R.F.A.; Wahba, A.S. Meleagrin Isolated from the Red Sea Fungus Penicillium chrysogenum Protects against Bleomycin-Induced Pulmonary Fibrosis in Mice. Biomedicines 2022, 10, 1164. [Google Scholar] [CrossRef]
- Fu, P.; Epshtein, Y.; Ramchandran, R.; Mascarenhas, J.B.; Cress, A.E.; Jacobson, J.; Garcia, J.G.N.; Natarajan, V. Essential role for paxillin tyrosine phosphorylation in LPS-induced mitochondrial fission, ROS generation and lung endothelial barrier loss. Sci. Rep. 2021, 11, 17546. [Google Scholar] [CrossRef]
- Andersson-Sjoland, A.; Karlsson, J.C.; Rydell-Tormanen, K. ROS-induced endothelial stress contributes to pulmonary fibrosis through pericytes and Wnt signaling. Lab. Investig. 2016, 96, 206–217. [Google Scholar] [CrossRef]
- Rydell-Törmänen, K.; Westergren-Thorsson, G. Wnt-imbalance within the endothelial niche contributes to pulmonary fibrosis. Lab. Investig. 2014, 2015, 1–12. [Google Scholar]
- Ma, K.L.; Liu, J.; Ni, J.; Zhang, Y.; Lv, L.L.; Tang, R.N.; Ni, H.F.; Ruan, X.Z.; Liu, B.C. Inflammatory stress exacerbates the progression of cardiac fibrosis in high-fat-fed apolipoprotein E knockout mice via endothelial-mesenchymal transition. Int. J. Med. Sci. 2013, 10, 420–426. [Google Scholar] [CrossRef]
- Perez, L.; Munoz-Durango, N.; Riedel, C.A.; Echeverria, C.; Kalergis, A.M.; Cabello-Verrugio, C.; Simon, F. Endothelial-to-mesenchymal transition: Cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth Factor Rev. 2017, 33, 41–54. [Google Scholar] [CrossRef]
- Suzuki, H.I.; Horie, M.; Mihira, H.; Saito, A. Molecular Analysis of Endothelial-mesenchymal Transition Induced by Transforming Growth Factor-beta Signaling. J. Vis. Exp. 2018, 138, e57577. [Google Scholar] [CrossRef]
- Donato, A.J.; Black, A.D.; Jablonski, K.L.; Gano, L.B.; Seals, D.R. Aging is associated with greater nuclear NF kappa B, reduced I kappa B alpha, and increased expression of proinflammatory cytokines in vascular endothelial cells of healthy humans. Aging Cell 2008, 7, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Ramadhiani, R.; Ikeda, K.; Hirata, K.I.; Emoto, N. Endothelial Cell Senescence Exacerbates Pulmonary Fibrosis Potentially Through Accelerated Endothelial to Mesenchymal Transition. Kobe J. Med. Sci. 2021, 67, E84–E91. [Google Scholar]
- Caporarello, N.; Meridew, J.A.; Aravamudhan, A.; Jones, D.L.; Austin, S.A.; Pham, T.X.; Haak, A.J.; Moo Choi, K.; Tan, Q.; Haresi, A.; et al. Vascular dysfunction in aged mice contributes to persistent lung fibrosis. Aging Cell 2020, 19, e13196. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Gorbett, D.; Mueller, J.; Perez, R.; Daniels, C.J. Pulmonary hypertension and idiopathic pulmonary fibrosis: A dastardly duo. Am. J. Med. Sci. 2013, 346, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of shear stress on endothelial cells: Go with the flow. Acta Physiol. 2017, 219, 382–408. [Google Scholar] [CrossRef]
- Wang, F.F.; Zhang, J.L.; Ji, Y.; Yan, X.J.; Sun, L.; Zhu, Y.; Jin, H. KLF2 mediates the suppressive effect of BDNF on diabetic intimal calcification by inhibiting HK1 induced endothelial-to-mesenchymal transition. Cell. Signal. 2022, 94, 110324. [Google Scholar] [CrossRef]
- Inui, N.; Sakai, S.; Kitagawa, M. Molecular Pathogenesis of Pulmonary Fibrosis, with Focus on Pathways Related to TGF-beta and the Ubiquitin-Proteasome Pathway. Int. J. Mol. Sci. 2021, 2, 6107. [Google Scholar] [CrossRef]
- Bochenek, M.L.; Leidinger, C.; Rosinus, N.S.; Gogiraju, R.; Guth, S.; Hobohm, L.; Jurk, K.; Mayer, E.; Munzel, T.; Lankeit, M.; et al. Activated Endothelial TGFbeta1 Signaling Promotes Venous Thrombus Nonresolution in Mice Via Endothelin-1: Potential Role for Chronic Thromboembolic Pulmonary Hypertension. Circ. Res. 2020, 126, 162–181. [Google Scholar] [CrossRef]
- Laakkonen, J.P.; Lappalainen, J.P.; Theelen, T.L.; Toivanen, P.I.; Nieminen, T.; Jauhiainen, S.; Kaikkonen, M.U.; Sluimer, J.C.; Yla-Herttuala, S. Differential regulation of angiogenic cellular processes and claudin-5 by histamine and VEGF via PI3K-signaling, transcription factor SNAI2 and interleukin-8. Angiogenesis 2017, 20, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, M.; Kouvaras, E.; Papamichali, R.; Samara, M.; Chiotoglou, I.; Koukoulis, G. Smad4 and epithelial-mesenchymal transition proteins in colorectal carcinoma: An immunohistochemical study. J. Mol. Histol. 2018, 49, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Okamoto, M.; Sakazaki, Y.; Kato, S.; Young, H.A.; Aizawa, H. Role of proinflammatory cytokines IL-18 and IL-1beta in bleomycin-induced lung injury in humans and mice. Am. J. Respir. Cell Mol. Biol. 2009, 41, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Hong, O.K.; Lee, S.S.; Yoo, S.J.; Lee, M.K.; Kim, M.K.; Baek, K.H.; Song, K.H.; Kwon, H.S. Gemigliptin Inhibits Interleukin-1beta-Induced Endothelial-Mesenchymal Transition via Canonical-Bone Morphogenetic Protein Pathway. Endocrinol. Metab. (Seoul) 2020, 35, 384–395. [Google Scholar] [CrossRef]
- Lee, J.G.; Kay, E.P. NF-kappaB is the transcription factor for FGF-2 that causes endothelial mesenchymal transformation in cornea. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1530–1538. [Google Scholar] [CrossRef]
- Li, X.; Zhu, X.; Li, B.; Xia, B.; Tang, H.; Hu, J.; Ying, R. Loss of alpha7nAChR enhances endothelial-to-mesenchymal transition after myocardial infarction via NF-kappaB activation. Exp. Cell Res. 2022, 419, 113300. [Google Scholar] [CrossRef]
- Lozo Vukovac, E.; Lozo, M.; Mise, K.; Gudelj, I.; Puljiz, Z.; Jurcev-Savicevic, A.; Bradaric, A.; Kokeza, J.; Mise, J. Bronchoalveolar pH and inflammatory biomarkers in newly diagnosed IPF and GERD patients: A case-control study. Med. Sci. Monit. 2014, 20, 255–261. [Google Scholar] [CrossRef]
- Zhao, T.; Li, H.; Liu, Z. Tumor necrosis factor receptor 2 promotes growth of colorectal cancer via the PI3K/AKT signaling pathway. Oncol. Lett. 2017, 13, 342–346. [Google Scholar] [CrossRef]
- Illam, S.P.; Narayanankutty, A.; Mathew, S.E.; Valsalakumari, R.; Jacob, R.M.; Raghavamenon, A.C. Epithelial Mesenchymal Transition in Cancer Progression: Prev entive Phytochemicals. Recent Pat. Anticancer. Drug Discov. 2017, 12, 234–246. [Google Scholar] [CrossRef]
- Foulquier, S.; Daskalopoulos, E.P.; Lluri, G.; Hermans, K.C.M.; Deb, A.; Blankesteijn, W.M. WNT Signaling in Cardiac and Vascular Disease. Pharmacol. Rev. 2018, 70, 68–141. [Google Scholar] [CrossRef]
- Sun, Z.; Yang, Z.; Wang, M.; Huang, C.; Ren, Y.; Zhang, W.; Gao, F.; Cao, L.; Li, L.; Nie, S. Paraquat induces pulmonary fibrosis through Wnt/beta-catenin signaling pathway and myofibroblast differentiation. Toxicol. Lett. 2020, 333, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, Q.; Chang, L.; Wei, C.; Bei, H.; Yin, Y.; Chen, M.; Wang, H.; Liang, J.; Wu, Y. LncRNA MALAT1 modulates ox-LDL induced EndMT through the Wnt/beta-catenin signaling pathway. Lipids Health Dis. 2019, 18, 62. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Wang, W.; Cui, G.; Nan, H.; Yan, L.; Zhang, W.; Zhang, S.; Wei, J. The expression levels of Notch-related signaling molecules in pulmonary microvascular endothelial cells in bleomycin-induced rat pulmonary fibrosis. Physiol. Res. 2017, 66, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Cattelino, A.; Gallini, R.; Rudini, N.; Iurlaro, M.; Piccolo, S.; Dejana, E. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J. Cell Biol. 2004, 166, 359–367. [Google Scholar] [CrossRef]
- Tsutsumi, T.; Nagaoka, T.; Yoshida, T.; Wang, L.; Kuriyama, S.; Suzuki, Y.; Nagata, Y.; Harada, N.; Kodama, Y.; Takahashi, F.; et al. Nintedanib ameliorates experimental pulmonary arterial hypertension via inhibition of endothelial mesenchymal transition and smooth muscle cell proliferation. PLoS ONE 2019, 14, e0214697. [Google Scholar] [CrossRef]
- Heldin, C.H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal 2013, 11, 97. [Google Scholar] [CrossRef]
- Jackson, A.O.; Zhang, J.; Jiang, Z.; Yin, K. Endothelial-to-mesenchymal transition: A novel therapeutic target for cardiovascular diseases. Trends Cardiovasc. Med. 2017, 27, 383–393. [Google Scholar] [CrossRef]
- Song, S.; Zhang, M.; Yi, Z.; Zhang, H.; Shen, T.; Yu, X.; Zhang, C.; Zheng, X.; Yu, L.; Ma, C.; et al. The role of PDGF-B/TGF-beta1/neprilysin network in regulating endothelial-to-mesenchymal transition in pulmonary artery remodeling. Cell. Signal. 2016, 28, 1489–1501. [Google Scholar] [CrossRef]
- Chen, P.Y.; Qin, L.; Barnes, C.; Charisse, K.; Yi, T.; Zhang, X.; Ali, R.; Medina, P.P.; Yu, J.; Slack, F.J.; et al. FGF regulates TGF-beta signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep. 2012, 2, 1684–1696. [Google Scholar] [CrossRef]
- Correia, A.C.; Moonen, J.R.; Brinker, M.G.; Krenning, G. FGF2 inhibits endothelial-mesenchymal transition through microRNA-20a-mediated repression of canonical TGF-beta signaling. J. Cell Sci. 2016, 129, 569–579. [Google Scholar] [CrossRef]
- Laddha, A.P.; Kulkarni, Y.A. VEGF and FGF-2: Promising targets for the treatment of respiratory disorders. Respir. Med. 2019, 156, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Illigens, B.M.; Casar Berazaluce, A.; Poutias, D.; Gasser, R.; Del Nido, P.J.; Friehs, I. Vascular Endothelial Growth Factor Prevents Endothelial-to-Mesenchymal Transition in Hypertrophy. Ann. Thorac. Surg. 2017, 104, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling-in control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Ji, Y.; Zhang, G.; Qu, Y.; Zhang, L.; Jiang, W. Ponatinib attenuates experimental pulmonary arterial hypertension by modulating Wnt signaling and vasohibin-2/vasohibin-1. Life Sci. 2016, 148, 1–8. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Frid, M.G.; Gerasimovskaya, E.; Zhang, H.; McCarthy, M.K.; Thurman, J.M.; Morrison, T.E. Mechanisms of SARS-CoV-2-induced lung vascular disease: Potential role of complement. Pulm. Circ. 2021, 11, 20458940211015799. [Google Scholar] [CrossRef]
- Leach, H.G.; Chrobak, I.; Han, R.; Trojanowska, M. Endothelial cells recruit macrophages and contribute to a fibrotic milieu in bleomycin lung injury. Am. J. Respir. Cell Mol. Biol. 2013, 49, 1093–1101. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, L.; Yang, J.; Chen, X.; Guo, X.; Xu, K.; Wang, N.; Zhao, W.; Xia, C.; Lian, H.; et al. Endothelial cell-derived MMP19 promotes pulmonary fibrosis by inducing E(nd)MT and monocyte infiltration. Cell Commun. Signal 2023, 21, 56. [Google Scholar] [CrossRef]
- Baba, O.; Huang, L.H.; Elvington, A.; Szpakowska, M.; Sultan, D.; Heo, G.S.; Zhang, X.; Luehmann, H.; Detering, L.; Chevigne, A.; et al. CXCR4-Binding Positron Emission Tomography Tracers Link Monocyte Recruitment and Endothelial Injury in Murine Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 822–836. [Google Scholar] [CrossRef]
- Yi, M.; Liu, B.; Tang, Y.; Li, F.; Qin, W.; Yuan, X. Irradiated Human Umbilical Vein Endothelial Cells Undergo Endothelial-Mesenchymal Transition via the Snail/miR-199a-5p Axis to Promote the Differentiation of Fibroblasts into Myofibroblasts. Biomed. Res. Int. 2018, 2018, 4135806. [Google Scholar] [CrossRef]
- Yin, Q.; Nan, H.Y.; Zhang, W.H.; Yan, L.F.; Cui, G.B.; Huang, X.F.; Wei, J.G. Pulmonary microvascular endothelial cells from bleomycin-induced rats promote the transformation and collagen synthesis of fibroblasts. J. Cell Physiol. 2011, 226, 2091–2102. [Google Scholar] [CrossRef]
- Namba, Y.; Kurdak, S.S.; Fu, Z.; Mathieu-Costello, O.; West, J.B. Effect of reducing alveolar surface tension on stress failure in pulmonary capillaries. J. Appl. Physiol. 1995, 79, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.; Washington, K.; Selva, C.; Grunwell, J.; Tirouvanziam, R.; Takayama, S. A High-Throughput Distal Lung Air-Blood Barrier Model Enabled By Density-Driven Underside Epithelium Seeding. Adv. Healthc. Mater. 2021, 10, e2100879. [Google Scholar] [CrossRef] [PubMed]
- Jiao, K.; Zhen, J.; Wu, M.; Teng, M.; Yang, K.; Zhou, Q.; Hu, C.; Zhou, M.; Li, Y.; Li, Z. 27-Hydroxycholesterol-induced EndMT acts via STAT3 signaling to promote breast cancer cell migration by altering the tumor microenvironment. Cancer Biol. Med. 2020, 17, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Ouyang, Z.; Tang, H.H. Inhibiting the proliferation and metastasis of hilar cholangiocarcinoma cells by blocking the expression of vascular endothelial growth factor with small interfering RNA. Oncol. Lett. 2018, 16, 1841–1848. [Google Scholar] [CrossRef]
- Chen, Z.; Guo, P.; Xie, X.; Yu, H.; Wang, Y.; Chen, G. The role of tumour microenvironment: A new vision for cholangiocarcinoma. J. Cell. Mol. Med. 2019, 23, 59–69. [Google Scholar] [CrossRef]
- Sweeney, M.; Foldes, G. It Takes Two: Endothelial-Perivascular Cell Cross-Talk in Vascular Development and Disease. Front Cardiovasc. Med. 2018, 5, 154. [Google Scholar] [CrossRef]
- Xie, H.; Gao, Y.M.; Zhang, Y.C.; Jia, M.W.; Peng, F.; Meng, Q.H.; Wang, Y.C. Low let-7d exosomes from pulmonary vascular endothelial cells drive lung pericyte fibrosis through the TGFbetaRI/FoxM1/Smad/beta-catenin pathway. J. Cell. Mol. Med. 2020, 24, 13913–13926. [Google Scholar] [CrossRef]
- Park, S.H.; Saleh, D.; Giaid, A.; Michel, R.P. Increased endothelin-1 in bleomycin-induced pulmonary fibrosis and the effect of an endothelin receptor antagonist. Am. J. Respir. Crit. Care Med. 1997, 156, 600–608. [Google Scholar] [CrossRef]
- Raghu, G.; Behr, J.; Brown, K.K.; Egan, J.J.; Kawut, S.M.; Flaherty, K.R.; Martinez, F.J.; Nathan, S.D.; Wells, A.U.; Collard, H.R.; et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: A parallel, randomized trial. Ann. Intern. Med. 2013, 158, 641–649. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Brown, K.K.; Raghu, G.; du Bois, R.M.; Lynch, D.A.; Martinez, F.; Valeyre, D.; Leconte, I.; Morganti, A.; Roux, S.; et al. BUILD-3: A randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 92–99. [Google Scholar] [CrossRef]
- Raghu, G.; Million-Rousseau, R.; Morganti, A.; Perchenet, L.; Behr, J.; Group, M.S. Macitentan for the treatment of idiopathic pulmonary fibrosis: The randomised controlled MUSIC trial. Eur. Respir. J. 2013, 42, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Lukey, P.T.; Harrison, S.A.; Yang, S.; Man, Y.; Holman, B.F.; Rashidnasab, A.; Azzopardi, G.; Grayer, M.; Simpson, J.K.; Bareille, P.; et al. A randomised, placebo-controlled study of omipalisib (PI3K/mTOR) in idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 53, 3. [Google Scholar] [CrossRef] [PubMed]
- Daniels, C.E.; Lasky, J.A.; Limper, A.H.; Mieras, K.; Gabor, E.; Schroeder, D.R.; Imatinib, I.P.F.S.I. Imatinib treatment for idiopathic pulmonary fibrosis: Randomized placebo-controlled trial results. Am. J. Respir. Crit. Care Med. 2010, 181, 604–610. [Google Scholar] [CrossRef]
- Seibold, J.R.; Korn, J.H.; Simms, R.; Clements, P.J.; Moreland, L.W.; Mayes, M.D.; Furst, D.E.; Rothfield, N.; Steen, V.; Weisman, M.; et al. Recombinant human relaxin in the treatment of scleroderma. A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2000, 132, 871–879. [Google Scholar] [CrossRef]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef]
- Yu, W.K.; Chen, W.C.; Su, V.Y.; Shen, H.C.; Wu, H.H.; Chen, H.; Yang, K.Y. Nintedanib Inhibits Endothelial Mesenchymal Transition in Bleomycin-Induced Pulmonary Fibrosis via Focal Adhesion Kinase Activity Reduction. Int. J. Mol. Sci. 2022, 23, 8193. [Google Scholar] [CrossRef]
- De Biasi, S.; Cerri, S.; Bianchini, E.; Gibellini, L.; Persiani, E.; Montanari, G.; Luppi, F.; Carbonelli, C.M.; Zucchi, L.; Bocchino, M.; et al. Levels of circulating endothelial cells are low in idiopathic pulmonary fibrosis and are further reduced by anti-fibrotic treatments. BMC Med. 2015, 13, 277. [Google Scholar] [CrossRef]
- Adams, T.N.; Eiswirth, C.; Newton, C.A.; Battaile, J.T. Pirfenidone for Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 194, 374–376. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, M.; Leng, C.; Blokzijl, T.; Jansen, B.H.; Dijkstra, G.; Faber, K.N. Pirfenidone Inhibits Cell Proliferation and Collagen I Production of Primary Human Intestinal Fibroblasts. Cells 2020, 9, 775. [Google Scholar] [CrossRef]
- Tang, Q.; Xing, C.; Li, M.; Jia, Q.; Bo, C.; Zhang, Z. Pirfenidone ameliorates pulmonary inflammation and fibrosis in a rat silicosis model by inhibiting macrophage polarization and JAK2/STAT3 signaling pathways. Ecotoxicol. Environ. Saf. 2022, 244, 114066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Tan, Y.; Yong, C.; Jiao, Y.; Tang, X.; Wang, D. Pirfenidone ameliorates early pulmonary fibrosis in LPS-induced acute respiratory distress syndrome by inhibiting endothelial-to-mesenchymal transition via the Hedgehog signaling pathway. Int. Immunopharmacol. 2022, 109, 108805. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Fang, M.; Hang, Q.Q.; Chen, Y.; Qian, X.; Chen, M. Pirfenidone modulates macrophage polarization and ameliorates radiation-induced lung fibrosis by inhibiting the TGF-beta1/Smad3 pathway. J. Cell. Mol. Med. 2021, 25, 8662–8675. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, W.; Wang, L.; Wang, Y.; Yuan, H.; Zhao, M.; Lian, H.; Ma, S.; Xu, K.; Li, Z.; Yu, G. Injured Endothelial Cell: A Risk Factor for Pulmonary Fibrosis. Int. J. Mol. Sci. 2023, 24, 8749. https://doi.org/10.3390/ijms24108749
Zhao W, Wang L, Wang Y, Yuan H, Zhao M, Lian H, Ma S, Xu K, Li Z, Yu G. Injured Endothelial Cell: A Risk Factor for Pulmonary Fibrosis. International Journal of Molecular Sciences. 2023; 24(10):8749. https://doi.org/10.3390/ijms24108749
Chicago/Turabian StyleZhao, Weiming, Lan Wang, Yaxuan Wang, Hongmei Yuan, Mengxia Zhao, Hui Lian, Shuaichen Ma, Kai Xu, Zhongzheng Li, and Guoying Yu. 2023. "Injured Endothelial Cell: A Risk Factor for Pulmonary Fibrosis" International Journal of Molecular Sciences 24, no. 10: 8749. https://doi.org/10.3390/ijms24108749