1. Introduction
Metronomic or extended exposure (EE) dosing of chemotherapeutics was first introduced as an antiangiogenic therapy by Dr. Folkman, Dr. Browder, and Dr. Kerbel in 2000 [
1,
2,
3]. In contrast to maximum tolerable dosing (MTD), which is usually administered as a large single-dose or a short course of therapy at a level just below life-threatening toxicities, EE dosing is usually administered more frequently at much lower doses and at a cumulative dose that may be at or significantly below MTD. It was hypothesized that EE dosing would more effectively target endothelial cells and would prevent the reflexive regeneration of endothelial cells that can occur during the drug-free periods of conventional therapy. It was also thought that endothelial cells would not develop resistance because they were genetically stable. The antiangiogenic mechanism of EE chemotherapy would later be confirmed with multiple agents and in multiple cancer types; however, malignant cells proved more versatile than anticipated and, in many instances, developed drug resistance [
3,
4,
5,
6,
7]. It was later revealed that the mechanism of action of EE therapy was likely multimodal. Some major mechanisms that have been identified include inhibiting angiogenesis, normalizing existing vasculature, activating the immune system, and inducing tumor dormancy and senescence, but complete understanding is lacking [
3,
5,
8,
9].
We first investigated EE dosing of topotecan using an in vivo xenograft model of subcutaneously implanted human metastatic prostate adenocarcinoma (PC3) cells. EE topotecan, which was administered using a subcutaneously implanted osmotic pump, was compared to MTD topotecan, which was administered as a bolus dose using tail vein injections. In this experiment, EE topotecan significantly reduced tumor growth relative to MTD topotecan. Importantly, we used an athymic mouse model, which should have limited most immune-related mechanisms of EE topotecan. We also did not find any significant differences in the tumor vasculature density for any treatment group [
10,
11,
12]. In our in vitro experiments, when maintaining equivalent cumulative exposure, clinically meaningful changes to the IC50 could not be produced over a 72 h timepoint (MTD IC50 190 nM, EE IC50 177 nM). Therefore, seemingly, the three major mechanisms of EE dosing (angiogenesis, immunity, and direct effects) could not adequately explain our results.
Thus, EE topotecan, somewhat paradoxically, produced similar short-term efficacy and greater long-term efficacy relative to MTD topotecan. We determined the most plausible explanation for these seemingly incongruous results was a change in drug sensitivity over time by the underlying malignant cell population, or, said in another way, that MTD topotecan led to rapid regimen crippling resistance, which was attenuated by EE topotecan. This article describes the methods used and the evidence obtained to determine if alternative dosing schedules of chemotherapeutics can change the phenotypic characteristics of surviving cell populations, thus altering their long-term sensitivity.
4. Discussion
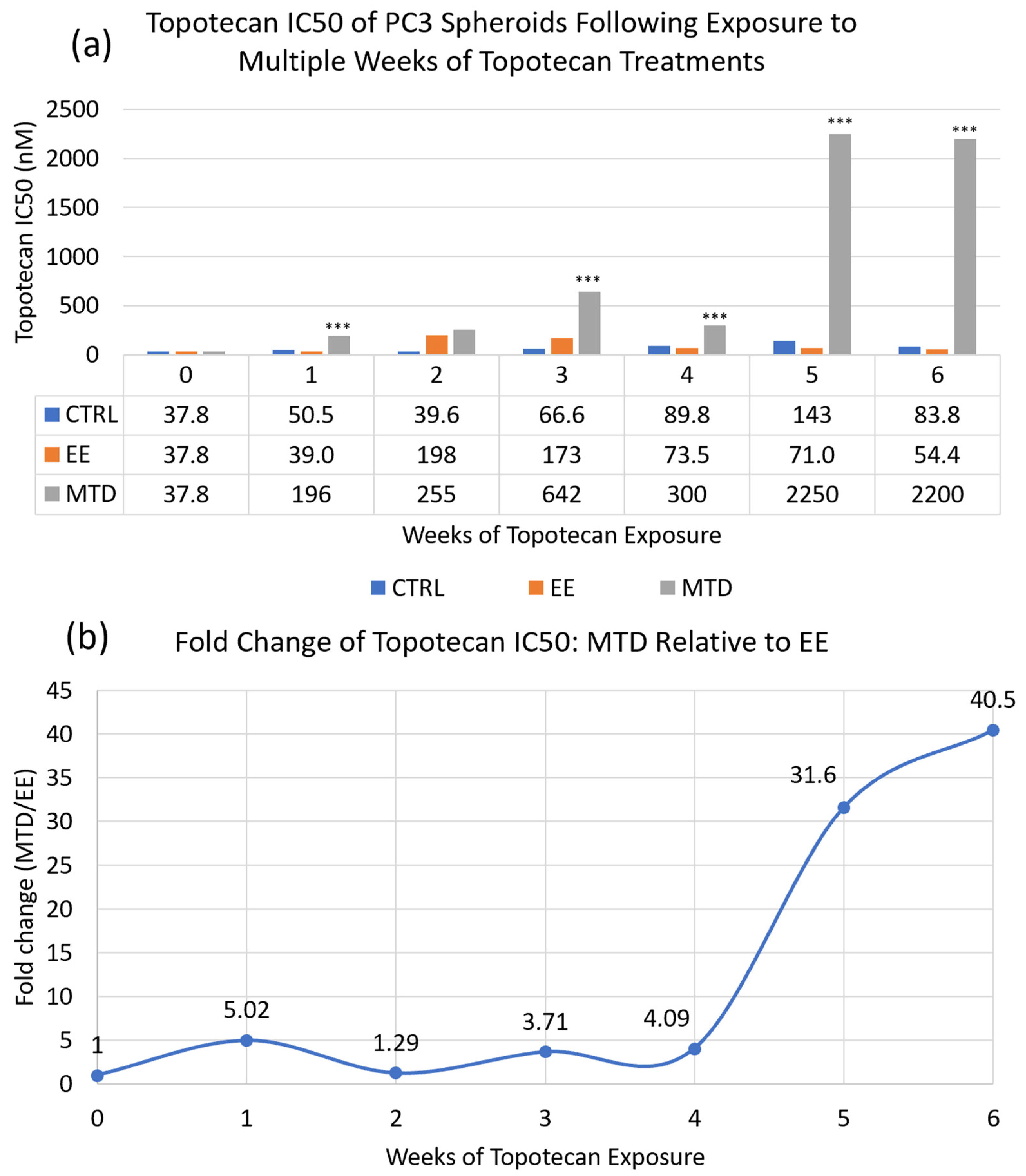
Metronomic or extended exposure dosing of oncologic agents is a relatively new paradigm with the potential to improve efficacy and reduce toxicity in some patients. To date, this treatment modality has demonstrated the ability to impact angiogenic and immunologic targets. We investigated a potentially novel mechanism impacting drug resistance. In
Figure 2, we used a long-term spheroidal model of PC3 cells to investigate the potency of topotecan over time after multiple weeks of treatment with either MTD- or EE-dosed topotecan. After 6 full weeks of drug exposure or approximately 3–4 months total, we demonstrated demonstrably decreased potency by the MTD-treated cells. On the other hand, the EE-treated cells maintained potency in line with the control cells. These data suggest that drug dosing can have a substantial impact on the underlying cell populations, which can significantly affect efficacy. This also underscores the need to better understand how therapeutics impact tumor cells and whether, long-term, we are creating more aggressive and resistant tumor cells to acutely reduce tumor volume. It also suggests that drug screening and selection should occur in longer-term model systems to appropriately identify treatments that can achieve sustained success. If we were to convert the potency data for MTD topotecan into a clinical scenario, it would suggest that after a single treatment, a patient would require approximately 4–5× the initial dose to have a similar impact on tumor cells. After 5 weeks of treatment, a patient would require 40× the initial dose, which is clinically unfeasible. If a treatment cannot eliminate tumor cells completely, which is currently true for almost all oncologic therapeutics, then maintaining a sensitive cancer cell population is vitally important.
To appropriately evaluate the impact of drug dosing on treatment resistance, we required a model system with adequate exposure duration, variable individual cell exposure through physical barriers and treatment gradients, and increased intratumor-like heterogeneity through added model complexity. Using scRNAseq (
Figure 3), we evaluated the impact of each treatment as well as the model system on population heterogeneity. Although the 3D spheroid did show increased heterogeneity relative to the 2D cells, it was less than initially expected. This was most likely caused by carryover effects from prior exposure to the spheroid model, similar to what was shown in the EE and MTD drug-free interval samples. Regardless, the most striking results from this experiment were found in the EE- and MTD-treated 3D samples. The MTD-treated 3D samples significantly increased heterogeneity, even relative to the elevated level of heterogeneity found in the underlying 2D drug-free interval MTD cells. Although the 2D drug-free interval EE cells were found to possess similar heterogeneity to the 2D drug-free interval MTD cells, the EE-treated 3D cells displayed significantly reduced heterogeneity. These cells seemed to phenotypically align in response to a more drawn-out topotecan exposure. This result highlights the potential role of EE topotecan as a modulator of cancer cell heterogeneity. Because increased heterogeneity has been shown to increase drug resistance and lead to poor clinical outcomes, reducing the genetic diversity of cancer cells prior to therapy might increase the efficacy of combination therapeutics [
21,
22,
23]. These results also highlight the need to further understand the impact of other therapeutics on cancer cell heterogeneity. Doing so may permit the ranking of therapeutics based on their impact on cellular heterogeneity, allowing clinicians to select more effective regimens.
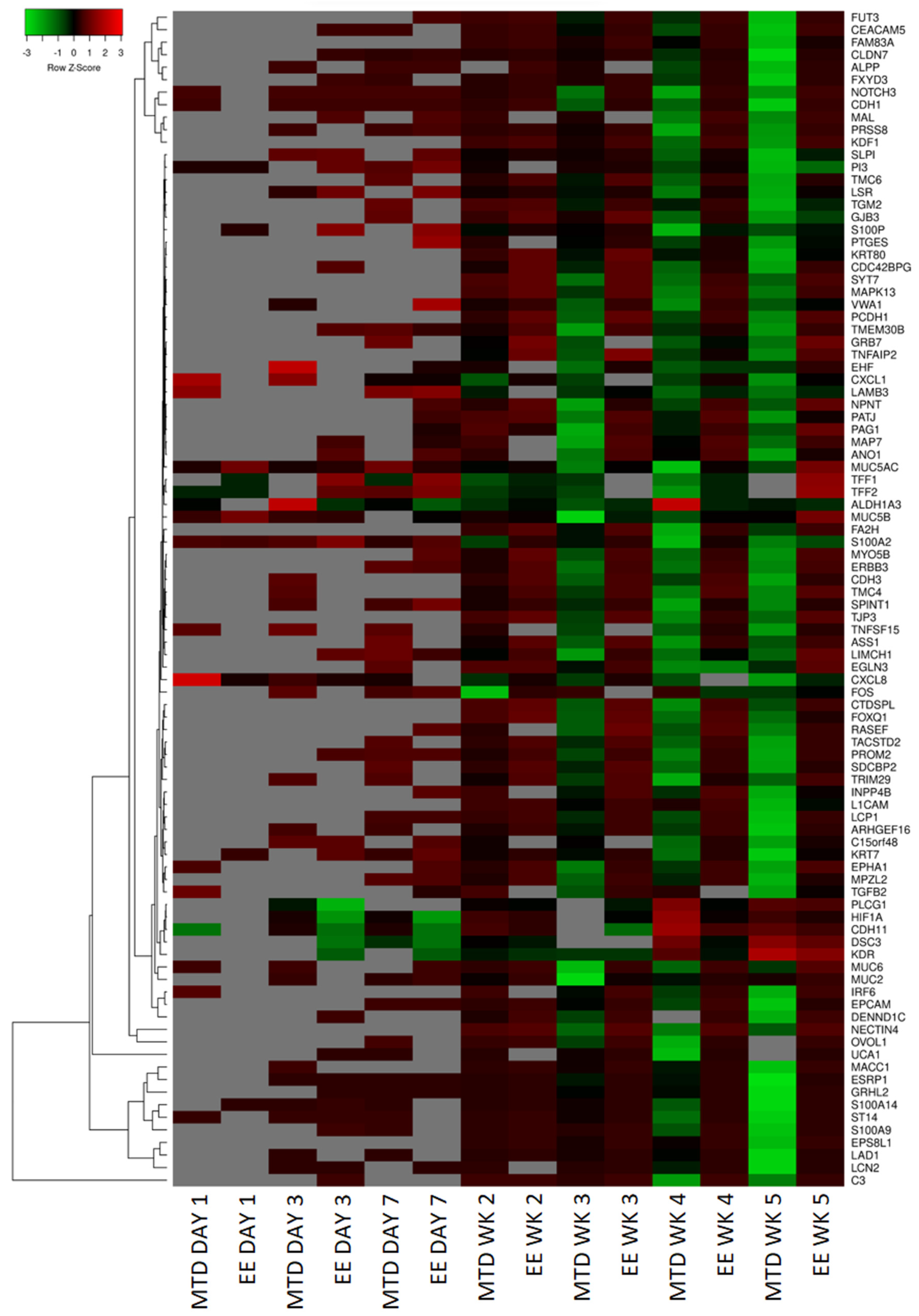
To further understand why MTD-dosed topotecan led to such a divergent potency response, we used RNAseq to help identify the top differentially expressed genes from the EE and MTD-treated cells. A summary of these results is presented as a heatmap (
Figure 4). For this set of genes, both treatment groups remained relatively stable after the first and second weeks of exposure, however, by week 3, significant changes to the MTD-treated cells could be seen, which further progressed over weeks 4 and 5. These changes also correlated well with our IC50 data, which supported further probing to determine each gene function and to determine if a mechanism of resistance could be identified.
A pattern emerged and EMT appeared most likely to cause the potency differences found between EE and MTD topotecan-treated cells. Genes that support this hypothesis have been identified in
Figure 5,
Figure 6 and
Figure 7. EMT usually involves the loss of epithelial markers and the gain of mesenchymal markers. The mRNA isolated from the MTD-treated cells showed significant downregulation of keratins, which are found in cornified and stratified epithelial cells and are known to be inhibited in EMT [
17,
18,
26]. They also downregulated each of the secreted mucins (
MUC2,
MUC5AC,
MUC5B, and
MUC6). Of these, MUC2 has been found to be most important in oncologic disease. Loss of MUC2 expression in mice is associated with increased proliferation and survival of intestinal epithelial cells and is associated with invasive adenocarcinomas [
27]. The MTD-treated cells also downregulated a substantial number of genes associated with cell adhesion, which is a common sign of EMT. CDH1 or E-Cadherin is the most common EMT-associated adhesion protein and is almost always downregulated during EMT [
13,
14,
15,
17,
18]. The MTD-treated cells reduced
CDH1 expression 68.5-fold relative to the control cells compared to a 3.6-fold decrease by the EE-treated cells. The MTD-treated cells also downregulated a number of other adhesion genes such as
EPCAM [
28,
29],
CEACAM5 [
30],
GJB3 [
31],
TJP3 [
32],
LAD1 [
33],
MPZL2 [
34], and
LSR [
35].
Furthermore, other genes associated with epithelial cells were significantly perturbed by the MTD-treated cells. For instance, EPHA1 is associated with ephrin signaling, which helps regulate the actin cytoskeleton, and is localized to epithelial junctions by E-cadherin. The loss of E-cadherin by the MTD-treated cells caused the downregulation of the
EPHA1 gene, further supporting the loss of an epithelial phenotype [
36]. The
ERBB3 gene, which encodes a well-known growth factor receptor in cancer also showed significant downregulation in MTD treated cells [
32]. The
MYO5B gene, which encodes a protein associated with apical–basolateral polarization, is also downregulated [
37]. Similarly,
PATJ, a gene that encodes a protein that regulates tight junction formation and polarization, is downregulated [
25]. The
PRSS8 gene produces a glycosylphosphatidylinositol-anchored epithelial extracellular membrane serine protease prostasin, which is expressed abundantly in normal epithelial cells and is essential for terminal epithelial differentiation, but is downregulated by MTD-treated cells. Downregulation of this protein has been associated with EMT in human bladder carcinomas [
38] and is associated with increased growth and metastasis in hepatocellular carcinoma [
39]. The
LCN2 gene produces a protein within the lipocalin superfamily and has been found to be expressed highly in early-stage colorectal cancer, but is downregulated significantly in metastatic or advanced-stage colorectal cancer, which may suggest that the MTD-treated cells are not only transitioning into a more mesenchymal phenotype but are also significantly more aggressive [
40]. The loss of any single epithelial gene would not support an EMT hypothesis; however, the consistent downregulation of many epithelial genes simultaneously indicates that the MTD-treated cells are most likely undergoing EMT, while the EE-treated cells appear to remain relatively phenotypically stable.
We also analyzed established EMT regulatory genes (
Figure 6). Of these, Snail (
SNAI1), Slug (
SNAI2),
TWIST1,
TWIST2,
ZEB1, and
ZEB2 are the most well-known; however, many of these factors do not appear to play a significant role in the EMT transition of the MTD-treated cells based on our RNAseq expression data [
13,
14,
15,
17,
18]. Although CXCL8 is does not strictly play a regulatory role in EMT and is more accurately classified as an EMT trigger, we included its expression profile in this figure to highlight its initial burst of expression during the first day of exposure and consistent decline in expression during the remainder of the experiment. In addition to
CXCL1 to a lesser extent (
Figure 4),
CXCL8 is one of the few genes (3 in total using our criteria) with significant early altered expression relative to the control. CXCL8 is known to promote proliferation, inhibit apoptosis, increase heterogeneity, and stimulate EMT [
41]. Additionally, elevated CXCL8 expression is correlated with high Gleason scores and elevated PSA [
42]. Based on our data and CXCL8’s known role in EMT, it is likely that elevated CXCL8 expression is an important early trigger of MTD topotecan-induced EMT and drug resistance.
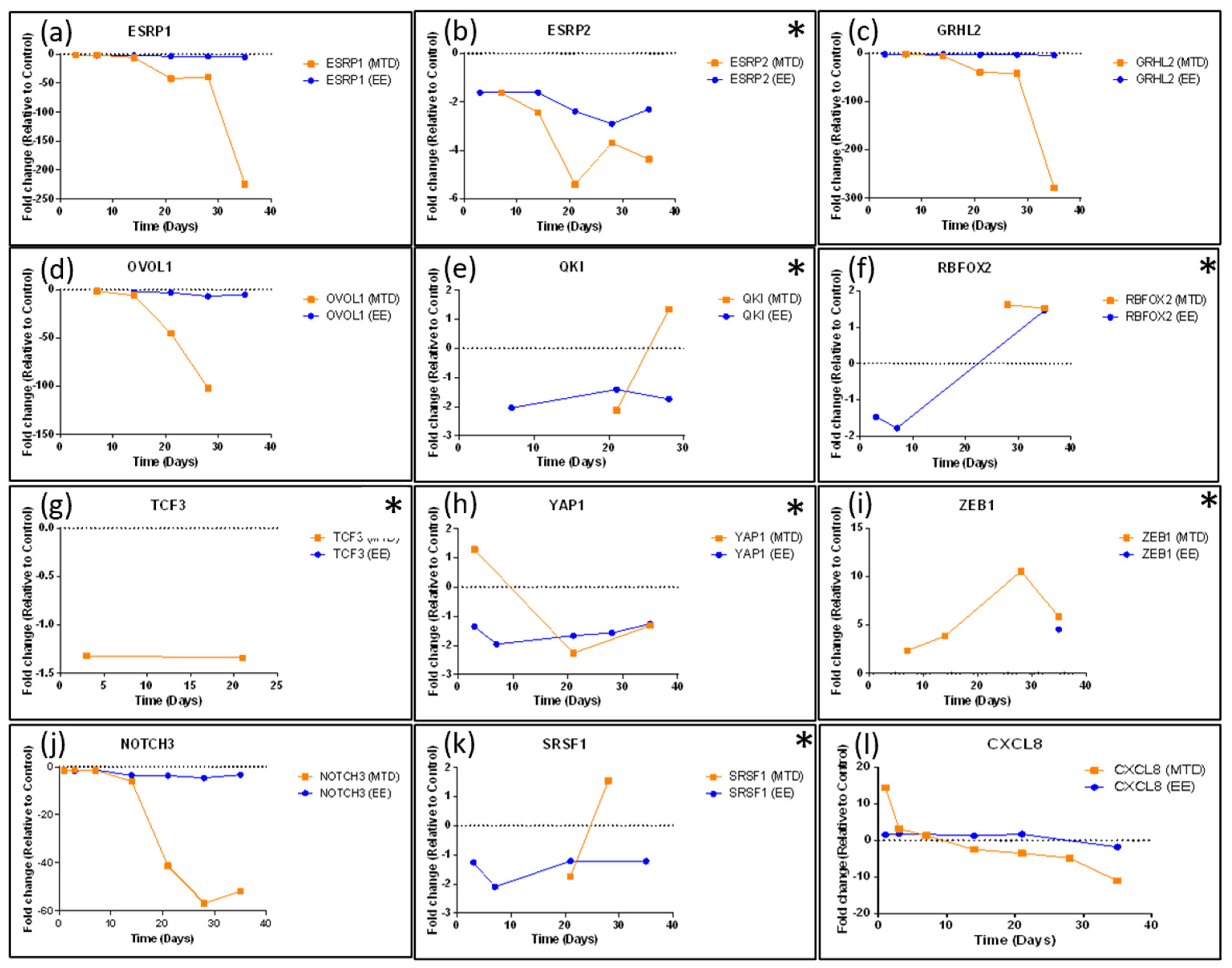
ZEB1 was not originally identified based on the selection criteria; however, after further evaluation, it appears to be significantly altered by the MTD-treated cells and is not consistently altered by the EE-treated cells. Further supporting ZEB1’s role,
TRIM29 was downregulated in MTD-treated cells and its protein is associated with increased ZEB1 expression and EMT in cervical cancer cells [
43]. Additionally, MTD-treated cells downregulated
FXYD3, which was found to be downregulated in mammary epithelial cells because of TGFβ and ZEB1 signaling, supporting ZEB1’s role in the EMT of MTD-treated cells [
44]. NOTCH3 is another important regulator associated with chemotherapy resistance in esophageal cancer cells when downregulated. In this study of esophageal cancer, silencing NOTCH3 resulted in increased production of VIM and resulted in increased chemotherapy resistance [
45]. In another study, NOTCH3 was found to inhibit EMT in breast cancer by activating downstream transcription complexes [
46]. Our results also highlight the important role of
NOTCH3 in regulating EMT as it was one of the first regulatory genes to become significantly downregulated (41.2-fold by week 3) by the MTD-treated cells. ESRP1 and ESRP2 are epithelial splicing regulatory proteins that regulate alternative splicing events associated with epithelial phenotypes and are significantly downregulated during EMT [
47]. Further supporting this finding,
OVOL1 was significantly downregulated in MTD-treated cells. OVOL1 has been found to induce mesenchymal–epithelial transition (MET) by upregulating ESRP1. OVOL1 is also a part of a regulatory feedback loop with ZEB1. Thus, its downregulation correlates with a downregulation of ESRP1 and an upregulation of ZEB1 [
48]. Lastly, a recent article highlights the EMT suppressor role of Grainyhead-like 2 in ovarian cancer cells.
GRHL2 was significantly downregulated by the MTD-treated cells (279-fold by week 5), and many of the genes identified in the article were also altered by the MTD-treated cells (
KRTs,
GRHL2,
ESRP1/2,
EPCAM,
CDH1,
CDH3,
ERBB3,
ZEB1,
CLDNs,
PROM2,
S100A14,
SPRINT1,
LAD1, and
ST14) [
33]. GRHL2 knockdown was found to result in genome-wide epigenetic remodeling through increased methylation of CpG sites and through nucleosome remodeling. It was found that GRHL2 most likely regulated the CpG methylation of epithelial genes at its binding sites. It was also found that the GRHL2 knockdown would most likely cause an intermediate form of EMT [
33]. Our results are consistent with their findings of the widespread knockdown of epithelial genes in response to a significant knockdown of GRHL2. The MTD-treated cells are likely in an intermediate stage of EMT as significant losses of epithelial markers are evident, but significant gains in mesenchymal markers are not evident (
Figure 7).
The EE-treated cells did not lose
OVOL1,
ESRP1,
GRHL2, or
NOTCH3 expression, nor did they significantly upregulate
ZEB1 expression consistently. These cells also did not significantly upregulate mesenchymal markers and did not significantly downregulate their epithelial markers. Importantly, long-term fractionated dosing of topotecan appeared to prevent EMT within these cells while still maintaining efficacy, which prevented EMT-induced drug resistance. This conclusion is further supported in
Figure 8 and
Figure 9, which show that EE-treated cells maintained similar expression patterns of efflux pumps and topoisomerase genes compared to control cells. On the other hand, a greater proportion of MTD-treated cells expressed efflux pumps and alternative topoisomerase genes. Each of these mechanisms could reduce the exposure or efficacy of topotecan and likely contribute to the reduction in IC50 potency (
Figure 2).
We demonstrated that alternative dosing strategies can have a substantial impact on underlying cell populations, which can directly affect treatment outcomes. These results also support the need for frequent genetic testing when administering oncologic therapeutics to quickly identify failed therapies and avoid harming patients. Finally, these results call into question the use of short-term efficacy models as drug-screening tools and support the need to better understand the temporal impact of oncologic medications on surviving cell populations.
5. Materials and Methods
5.1. Cell Line and Cell Culture
The human prostate cancer (PC3) cell line was obtained from ATCC and was maintained as monolayers in complete medium using F12K (Corning, Corning, NY, USA) and 10% (v/v) FBS (Hyclone, Logan, UT, USA) at 37 °C in a 5% CO2 atmosphere using a Heracell bios 160i incubator (Thermo Fisher Scientific, Waltham, MA, USA). During the experiment, the PC3 cell line was divided into multiple sub-cell lines according to the treatment group, which will be described in greater detail below. Each of these sub-cell lines were treated as a unique cell line (separate flasks, no mixing, etc.) throughout the experiment using the same methods described above.
5.2. Spheroid Formation
The spheroid protocol was largely adapted from a high-throughput liquid overlay technique developed by Metzger et al. [
49]. This technique rapidly generates many spheroids with minimal incubation time (24 h), which is necessary for drug-screening protocols. Briefly, 96-well U bottom plates (Grenier bio-one, Monroe, LA, USA) were coated with a 1.2% (
w/
v) poly-HEMA (Sigma Aldrich, St. Louis, MO, USA) solution in 95% (
v/
v) ethanol. This solution was produced by incubating poly-HEMA crystals overnight with a magnetic stir rod at 80 °C to ensure full dissolution. The poly-HEMA solution was kept warm throughout the coating process to prevent precipitation during the evaporation step. A 60 μL volume of the poly-HEMA solution was added to each well and the plates were heated using a hot plate (VWR, Radnor, PA, USA). Plates were left on the hot plate for approximately 1 h with the lid raised to evaporate the ethanol. Plates were then sealed using Parafilm (Bemis, Neenah, WI, USA) for future use. After cells were passaged and placed into a separate conical tube, they were mixed thoroughly, and a small sample was removed for counting using a TC10 automated cell counter (Biorad, Hercules, CA, USA). A minimum of two counts were taken per cell line to ensure accurate counts for cell seeding. Cells were diluted to achieve a concentration of 50,000 cells per mL and placed on ice. A total of 2.5% (
v/
v) of Matrigel (Corning, Corning, NY, USA) was added to the cell suspension using an ice-cold syringe and needle. The cells were then plated using 100 μL of the cell suspension to attain 5000 cells per well. The plates were then centrifuged at 400 g for 5 to 10 min at 4°C. This protocol rapidly generates fully formed spheroids within 24 h for the PC3 cell line.
5.3. Dosing and Spheroid Handling
Two days after initial seeding and spheroid formation, an additional 100 μL of media +/− drug was added to achieve a total volume 200 μL for the remainder of the experiment. On days 3 and 5, a media exchange was performed by removing 100 μL of media per well and replacing it with 100 μL of fresh complete media +/− drug. Limiting the media exchanges and leaving some residual, old media prevented spheroid loss throughout the experiment. On off-media exchange days, 10 μL of media was removed and replaced with 10 μL of media or treatment solution according to the treatment group. Dosing of topotecan (Chempac, Synder, TX, USA) and docetaxel (Fluka, Charlotte, NC, USA) occurred using 20× concentrated solutions, which could be directly spiked into the wells at 10 μL in 190 μL of media. The conventional (MTD) treatment was given as a bolus dose on day 0. Metronomic or EE treatment was given daily as a fractionated dose at 1/7th the MTD. The cumulative dose for the MTD and EE treatments was equal throughout the experiment. In total, there were 3 treatment groups: control, MTD topotecan, and EE topotecan. Topotecan dosing occurred at 100 nM during each week of therapy and occurred between 1 and 100,000 nM for the IC50 assays.
5.4. Study Protocol
Spheroids were generated and grown for approximately 2–3 days to allow size-dependent drug barriers to form. Spheroids were then dosed for a total of 7 days. During the first week of exposure, samples were taken for genomic and proteomic analysis on days 0, 1, 3, and 7. The remaining spheroids were saved for future weeks following digestion using Accumax (Innovative cell technologies, San Diego, CA, USA) for approximately 1 h until a single-cell suspension was achieved. A total of 3 treatment groups generated 3 unique cell lines that were maintained throughout the experiment: PC3-Control, PC3-EE-Topotecan, and PC3-MTD-Topotecan. The digested spheroids were grown in 2D for approximately 1–2 weeks until the cell population was replenished sufficiently to plate additional spheroids. Each cell population was then used to generate two groups of spheroids. One group (3D) was exposed to an additional week of treatment and one group (3D) was used to assess the resulting sensitivity of the drug (Topotecan) from the previous week(s) of drug exposure. After another full week of exposure, some spheroids were harvested for genomic and proteomic analysis, and some were digested to prepare for another week of exposure and analysis. This cycle was repeated throughout the experiment. A schematic is depicted (
Figure A3 and
Figure A4) to help illustrate the study protocol. For scRNAseq, we analyzed digested spheroids from week 5 (2D) and treated 3D samples from week 6. Week 5 samples had been grown in 2D for approximately 1 to 2 weeks in drug-free media before analysis.
5.5. Resazurin Assay (Cytotoxicity)
Resazurin was used to measure the mitochondrial activity of the cells as a surrogate for cell viability because the reductive conversion of resazurin to resorufin creates a water-soluble end product. This prevents the need for a solubilizing step, which would be untenable in a 3D format. Resazurin (Alfa Aesar, Haverhill, MA, USA) was made fresh for each assay at a 0.015% (w/v) concentration in PBS and was sterilized using a 0.22 μm filter. Before resazurin was added to the spheroids, the spheroids were moved from U bottom 96-well plates to flat bottom black, fluorescent plates (Grenier bio-one, Monroe, LA, USA). This was accomplished using a 1 mL pipette tip to move the spheroid and 100 μL media. Moving the spheroids increased the accuracy of the imaging and spectrophotometry. This also ensured that well volume variability from inconsistent evaporation dynamics that occur over the duration of the experiment would not alter the resorufin concentrations. Resazurin was added at a ratio of 10 μL per 100 μL of media and was incubated for 4 to 12 h with readings taken over time (2, 4, 6, 8, 12). Generally, 4-6 h was the most appropriate time point and achieved the lowest variation (CV values) with the greatest sensitivity and limited assay saturation. Fluorescent measurements for each plate were read using a Cytation 5 plate reader (BioTek, Winooski, VT, USA) with excitation set at 560 nm and emission set at 590 nm.
5.6. RNA Storage Protocol
Cells and spheroids were separated into individual microfuge tubes at approximately 1,000,000 cells/mL and washed 2× using PBS (Wards science, Rochester, NY, USA) and the Heraeus Fresco 21 microcentrifuge (Thermo Fisher Scientific, Waltham, MA, USA) set at 400 g and 4 °C for 10 min. Samples were maintained on ice for the duration of the protocol. PBS was aspirated and replaced with 300 μL of RNA later (Qiagen, Venlo, Netherlands). Samples were stored overnight (24 h) at 4 °C before moving to −80 °C for long-term storage. ScRNAseq samples were cryopreserved using 10% DMSO in complete media and stored in liquid nitrogen.
5.7. RNA Isolation
Total RNA was isolated from cultured cells and 3D spheroid model using standard RNA extraction kits (RNeasy Kits QIAGEN, Venlo, Netherlands). RNA concentration and integrity were estimated using a NanoDrop 2000 UV-Vis spectrophotometer (Thermo Scientific, Waltham, MA, USA), Qubit® 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA), and Agilent 2100 Bioanalyzer (Applied Biosystems, Carlsbad, CA, USA). RNA integrity number threshold of eight was used for RNAseq analysis.
5.8. RNAseq
RNAseq libraries were constructed using Illumina TruSeq RNA Sample Preparation Kit v2. Libraries were then size selected to generate inserts of approximately 200 bp. RNA sequencing was performed on llumina’s NovaSeq next-generation high-throughput sequencing system using 150 bp paired-end protocol with a depth of more than 20 million reads per sample. The average quality scores were above Q30 for all libraries in both R1 and R2.
5.9. RNAseq Data Processing
RNAseq data were normalized, and fragments per kilobase million values were used in further analysis using Partek Genomics Suite and Galaxy data analysis software, an open source, web-based platform that provides tools necessary to create and execute RNAseq analysis. In brief, RNAseq data analysis pipeline was developed using Galaxy software workflow. Quality control (QC) check on the RNAseq raw reads was performed using the FastQC tool, followed by read trimming to remove base positions with a low median (or bottom quartile) score. Tophat2 Aligner tool mapped processed RNAseq reads to the hg19 human genome build. Picard’s CollectInsertSizeMetrics tool was applied on the initial tophat2 run to obtain estimated insert sizes, which was then used to calculate mean inner distance between mate pairs (mean = estimated_insert-size − 2 × read_length). Tophat2 was re-run using corrected mean value, and Cufflinks tool was used to assemble the reads into transcripts.
5.10. Bioinformatics Analysis
Gene expression data were filtered using the following criteria: genes with mean FPKM < 1 were removed. Global gene expression profile (GEP) data were analyzed further using a combination of R and Partek Flow to perform differential expression testing to identify GEP signatures of drug response. Mean fold change >j1j and
p < 0.05 were considered thresholds for reporting significant differential gene expression. Differentially expressed gene analysis was performed between two groups of gene expression datasets (e.g., treated vs. untreated). Heatmaps were generated using unsupervised hierarchical clustering analysis based on the DEGs. Owing to the small sample size, Limma, an empirical Bayesian method, was used to detect DEGs, obtain
p-values, and further provided a false discovery rate based on the
p-value using the Benjamini–Hochberg procedure to detect the DEGs [
50]. The advantage of using Limma compared with a traditional t-test is that it provides a moderated t-test statistic by shrinking the variance statistics and therefore improves the statistical power.
All samples were initially normalized to control day 0. Then, each MTD and EE timepoint was normalized to the corresponding control timepoint, e.g., day 7 MTD and EE samples were normalized to day 7 control. After normalization, the top 1000 genes with the lowest
p-values were selected. Then, MTD and EE samples with a relative fold change difference less than 2 were removed. Finally, each gene required at least 2 timepoints with a fold change difference greater than 1.5 to remove one-off gene changes. Each gene was then manually investigated to determine its role and function using databases such as GeneCards as well as literature searches using PubMed [
51]. Genes without a well-defined function or genes without a clear role were labeled as unknown and removed from the list (
Figure 10). The complete criteria list without manual adjustments can be found in the
Supplementary Materials. Heatmaps were generated using heatmapper, a web-based tool [
52].
5.11. Ingenuity Pathway Analysis (IPA)
IPA is a web-based software application that integrates and interprets the data derived from differential mRNA expression analysis. (IPA) software (QIAGEN, Venlo, Netherlands) was used to identify the most significantly affected (1) molecular pathways predicted to be activated or inhibited, (2) upstream regulator molecule such as miRNA, transcription factors, and (3) downstream effects and biologic processes that were increased or decreased, and (4) to predict causal networks, relationships, mechanisms, and functions relevant to changes observed in our dataset and (5) to perform predictive toxicology analysis using toxicogenomic approaches (IPA-Tox) [
53].
5.12. scRNAseq
The presence of drug-resistant single-cell subpopulations (subclones) may have influenced differential responses to METRO therapy in PCa tumors. Therefore, we performed single-cell transcriptomics to identify resistant and sensitive subclones based on single-cell GEP signatures. Briefly, automated single-cell capture, and cDNA synthesis, were performed at ~5000 tumor cells/sample using 10X Genomics Chromium platform. Single-cell RNAseq-based gene expression analysis was performed on the Illumina HiSeq 2500 NGS platform (paired end. 2 × 125 bp, 100 cycles. v3 chemistry) at ~5 million reads per sample. scRNAseq data were analyzed using R, Seurat, and Partek Flow software packages. All statistical analyses were performed using the R statistical package, and GraphPad Prism with a two-sided p-value < 0.05 considered as statistically significant. Total sample numbers and replicates were determined by performing a power analysis with an effect size of 0.25 and a significance level of 0.05 with a power of 80%. IPA analysis was performed to identify regulators, relationships, mechanisms, functions, and pathways relevant to changes observed in our dataset.
5.13. Statistical Analysis
All statistical analyses were performed using R for statistical computing and graphics, v3.4.2, and GraphPad Prism v7.0. We used parametric methods to analyze differences between two groups of cells. If the assumption appeared violated, appropriate nonparametric procedures were used. All tests were two-sided, and differences with a p < 0.05 were considered statistically significant. The curve fitting and statistical analysis of the IC50 data were performed using Graphpad Prism (Dotmatics, Boston, MA, USA). The IC50 was determined at ½ of fitted maximal activity. Usually, an extra sum-of-squares F test was used to compare IC50 values between treated and control samples.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








