
Effect of Ionization Degree of Poly(amidoamine) Dendrimer and 5-Fluorouracil on the Efficiency of Complex Formation—A Theoretical and Experimental Approach
 , ,
, ,  ,
,  , ,
, ,  and
and
Abstract
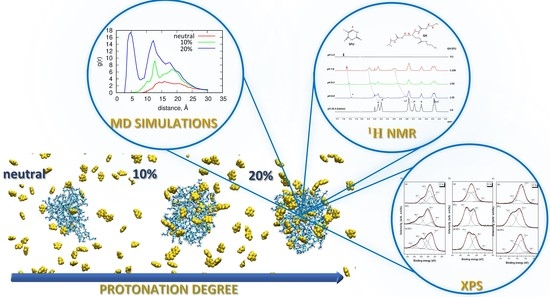
:
1. Introduction
2. Results and Discussion
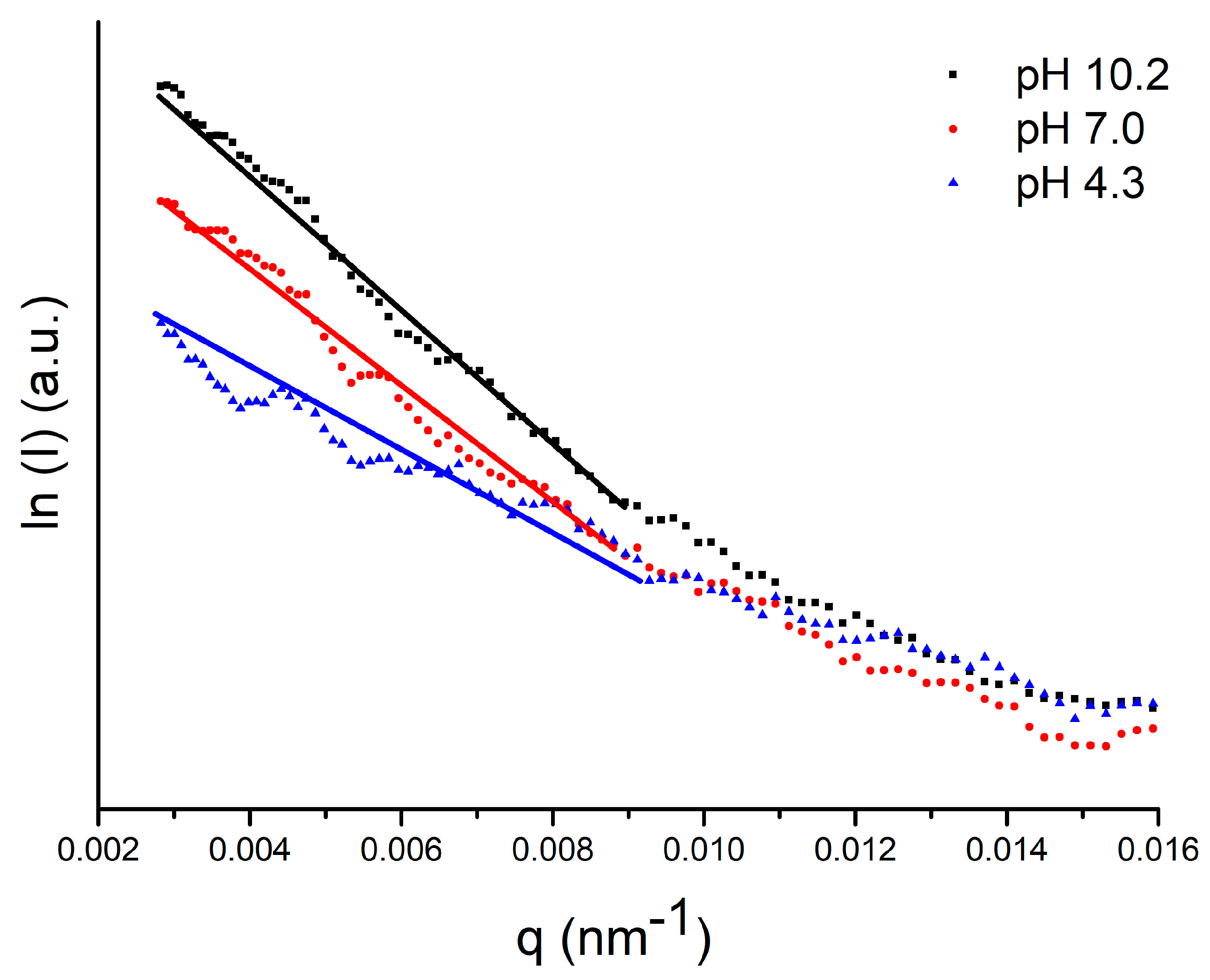
2.1. SAXS and DLS
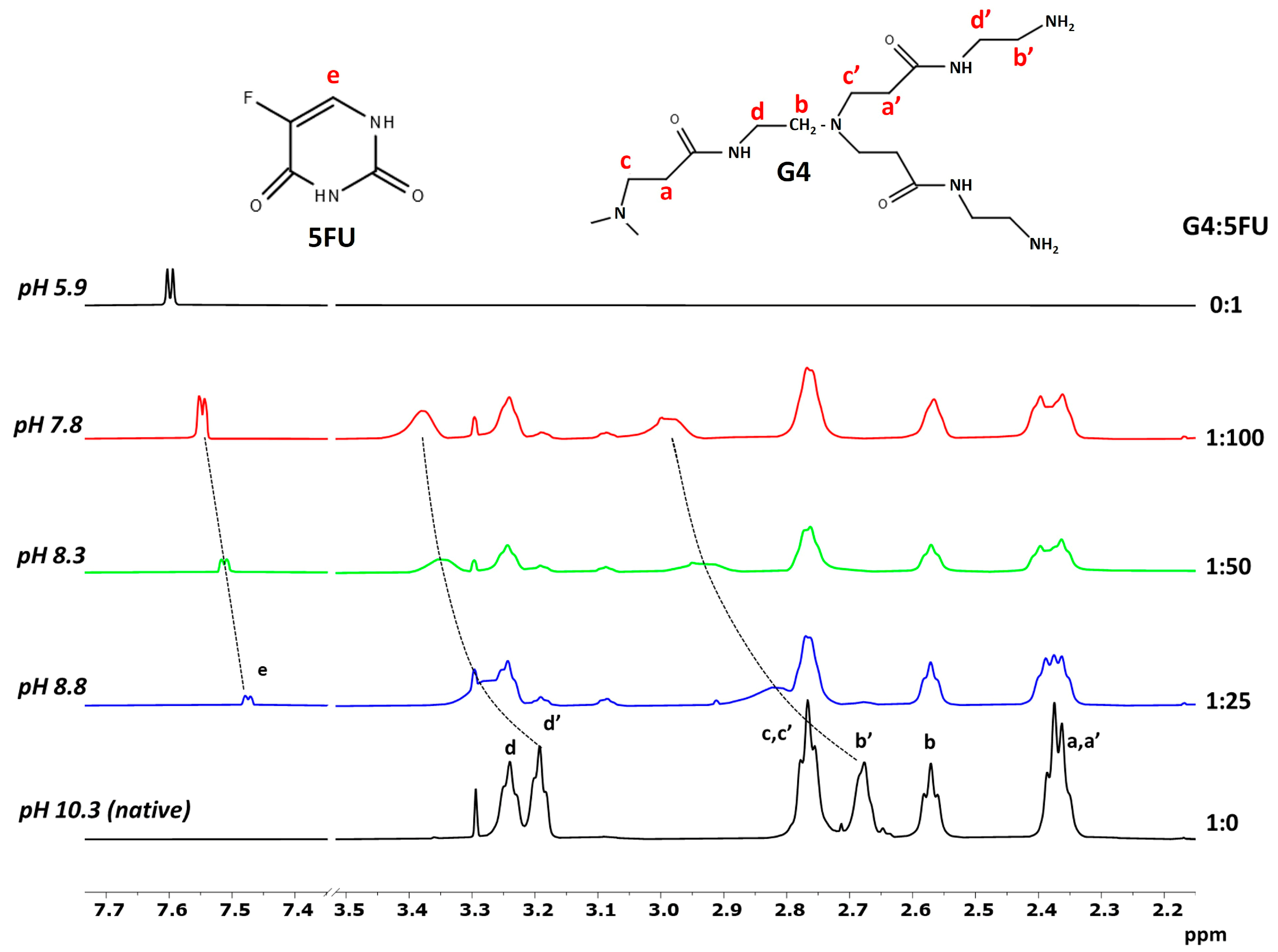
2.2. 1H NMR Spectroscopy
2.3. X-ray Photoelectron Spectroscopy (XPS)
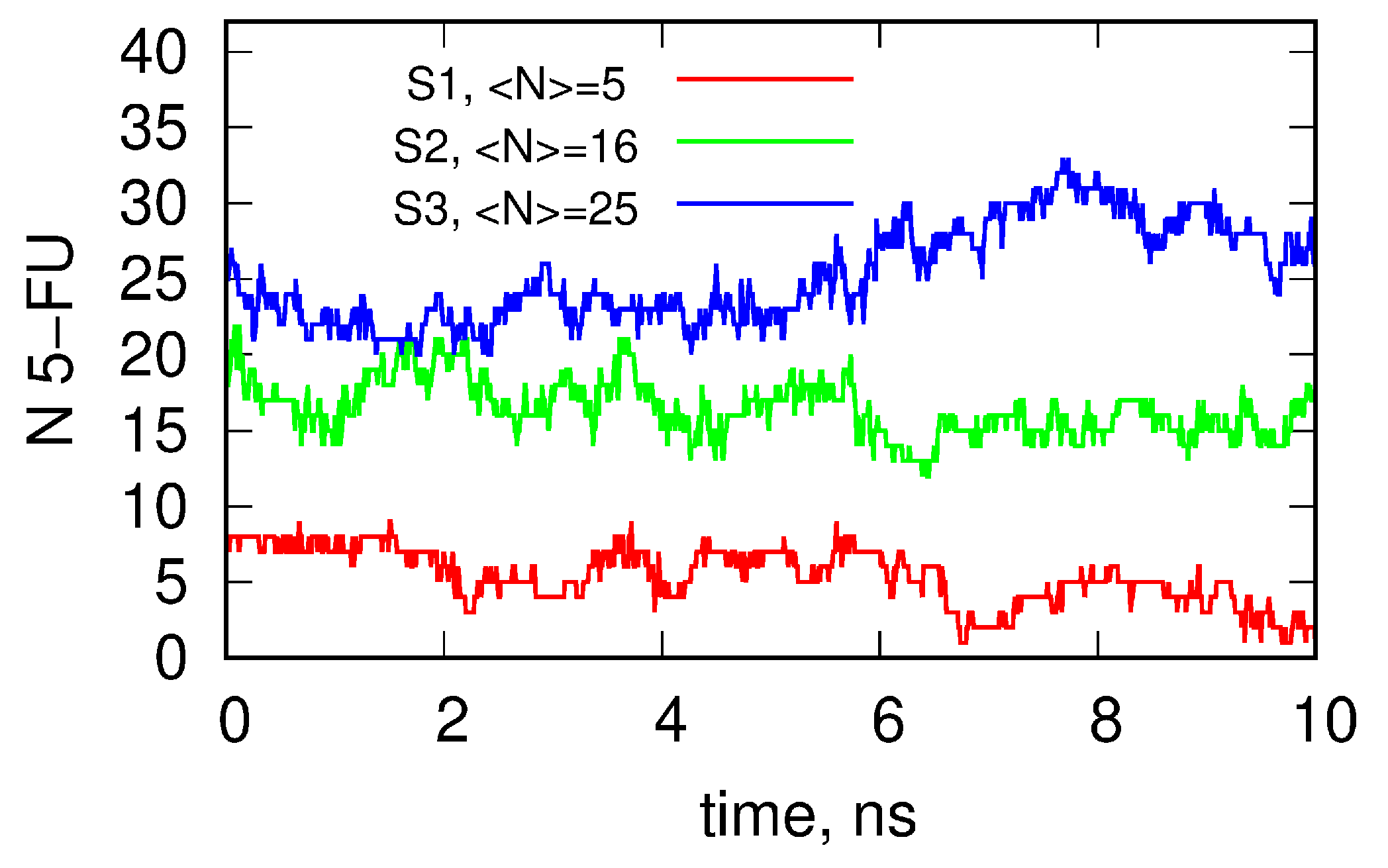
2.4. Molecular Dynamics (MD)
3. Materials and Methods
3.1. Materials
3.2. Dynamic Light Scattering (DLS)
3.3. Small Angle X-ray Scattering (SAXS)
3.4. 1H NMR Spectroscopy
3.5. X-ray Photoelectron Spectroscopy (XPS)
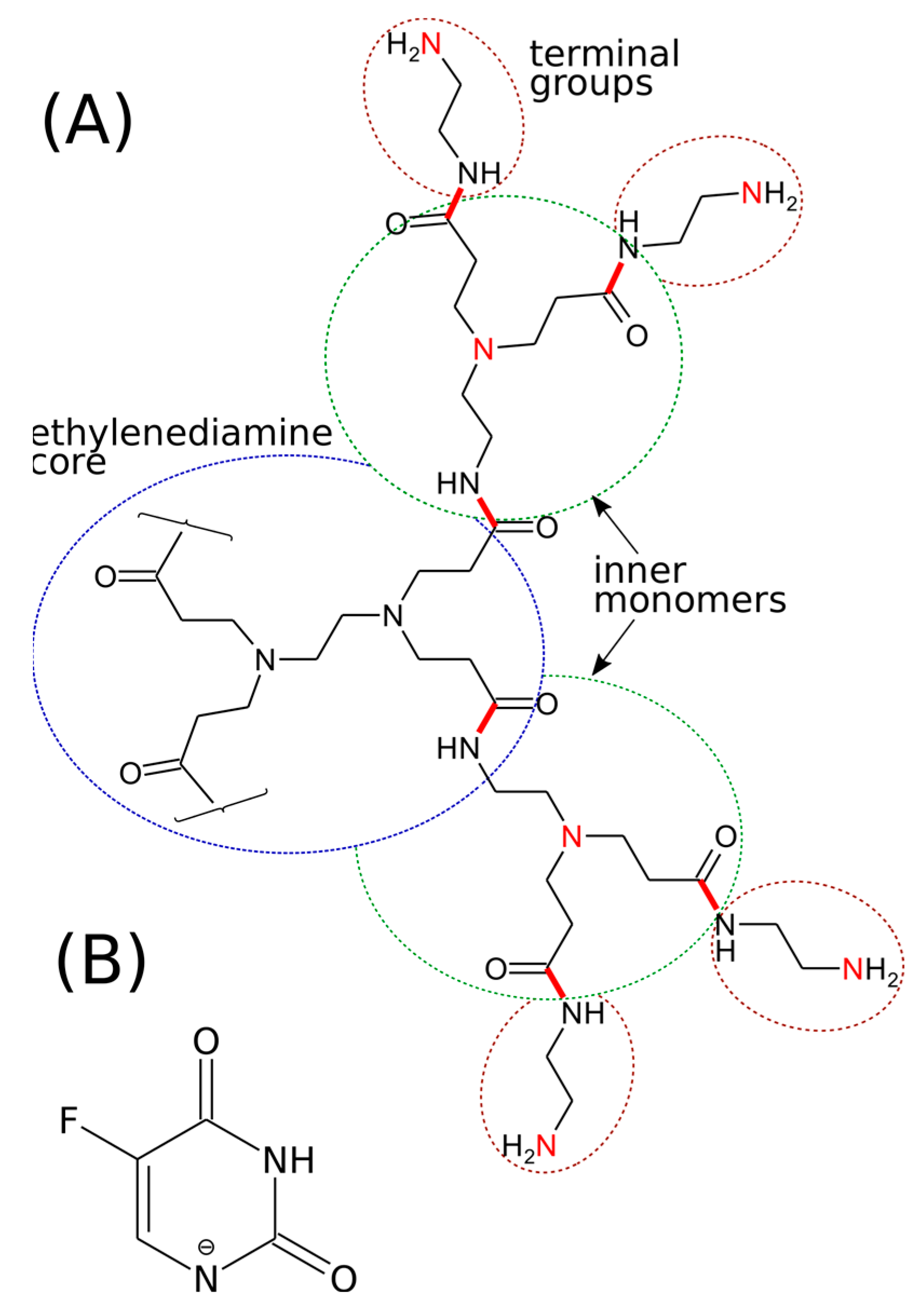
3.6. MD Simulations—Models and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PAMAM dendrimers | poly(amidoamine) dendrimers |
| G4PAMAM | poly(amidoamine) dendrimer of fourth generation |
| 5FU | 5-Fluorouracil |
| G4PAMAM–5FU | 5-Fluorouracil complex with fourth generation of dendrimer |
| Rg | gyration radius |
| RH | hydrodynamic radius |
| D | self-diffusion coefficient |
| BE | binding energy |
References
- Raj, S.; Khurana, S.; Choudhari, R.; Kesari, K.K.; Kamal, M.A.; Garg, N.; Ruokolainen, J.; Das, B.C.; Kumar, D. Specific targeting cancer cells with nanoparticles and drug delivery in cancer therapy. Semin. Cancer Biol. 2021, 69, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Souri, M.; Soltani, M.; Kashkooli, F.M.; Shahvandi, M.K.; Chiani, M.; Shariati, F.S.; Mehrabi, M.R.; Munn, L.L. Towards principled design of cancer nanomedicine to accelerate clinical translation. Mater. Today Bio 2022, 13, 100208. [Google Scholar] [CrossRef] [PubMed]
- Jachimska, B.; Łapczyńska, M.; Zapotoczny, S. Reversible swelling process of sixth-generation poly(amido amine) dendrimers molecule as determined by quartz crystal microbalance technique. J. Phys. Chem. C 2013, 117, 1136–1145. [Google Scholar] [CrossRef]
- Rae, J.M.; Jachimska, B. Analysis of dendrimer-protein interactions and their implications on potential applications of dendrimers in nanomedicine. Nanoscale 2021, 13, 2703–2713. [Google Scholar] [CrossRef]
- Mignani, S.; Shi, X.; Rodrigues, J.; Tomas, H.; Karpus, A.; Majoral, J.P. First-in-class and best-in-class dendrimer nanoplatforms from concept to clinic: Lessons learned moving forward. Eur. J. Med. Chem. 2021, 219, 113456. [Google Scholar] [CrossRef] [PubMed]
- Sathe, R.Y.; Bharatam, P.V. Drug-dendrimer complexes and conjugates: Detailed furtherance through theory and experiments. Adv. Colloid Interface Sci. 2022, 303, 102639. [Google Scholar] [CrossRef]
- Tokarczyk, K.; Jachimska, B. Characterization of G4 PAMAM dendrimer complexes with 5-fluorouracil and their interactions with bovine serum albumin. Colloids Surf. Physicochem. Eng. Asp. 2019, 561, 357–363. [Google Scholar] [CrossRef]
- Jachimska, B. Physicochemical Characterization of PAMAM Dendrimer as a Multifunctional Nanocarriers; Elsevier Inc.: Amsterdam, The Netherlands, 2019. [Google Scholar] [CrossRef]
- Caminade, A.M.; Turrin, C.O. Dendrimers for drug delivery. J. Mater. Chem. B 2014, 2, 4055–4066. [Google Scholar] [CrossRef]
- Singh, V.; Kesharwani, P. Dendrimer as a promising nanocarrier for the delivery of doxorubicin as an anticancer therapeutics. J. Biomater. Sci. Polym. Ed. 2021, 32, 1882–1909. [Google Scholar] [CrossRef]
- Surekha, B.; Kommana, N.S.; Dubey, S.K.; Kumar, A.V.P.; Shukla, R.; Kesharwani, P. PAMAM dendrimer as a talented multifunctional biomimetic nanocarrier for cancer diagnosis and therapy. Colloids Surf. B Biointerfaces 2021, 204, 111837. [Google Scholar] [CrossRef]
- Kharwade, R.; More, S.; Warokar, A.; Agrawal, P.; Mahajan, N. Starburst pamam dendrimers: Synthetic approaches, surface modifications, and biomedical applications. Arab J. Chem. 2020, 13, 6009–6039. [Google Scholar] [CrossRef]
- Kheraldine, H.; Rachid, O.; Habib, A.M.; Al Moustafa, A.E.; Benter, I.F.; Akhtar, S. Emerging innate biological properties of nano-drug delivery systems: A focus on PAMAM dendrimers and their clinical potential. Adv. Drug Deliv. Rev. 2021, 178, 113908. [Google Scholar] [CrossRef] [PubMed]
- Fox, L.J.; Richardson, R.M.; Briscoe, W.H. PAMAM dendrimer—Cell membrane interactions. Adv. Colloid Interface Sci. 2018, 257, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Szota, M.; Reczyńska-Kolman, K.; Pamuła, E.; Michel, O.; Kulbacka, J.; Jachimska, B. Poly(Amidoamine) dendrimers as nanocarriers for 5-fluorouracil: Effectiveness of complex formation and cytotoxicity studies. Int. J. Mol. Sci. 2021, 22, 11167. [Google Scholar] [CrossRef]
- Ambekar, R.S.; Choudhary, M.; Kandasubramanian, B. Recent advances in dendrimer-based nanoplatform for cancer treatment: A review. Eur. Polym. J. 2020, 126, 109546. [Google Scholar] [CrossRef]
- Gao, X.; Ma, M.; Pedersen, C.M.; Liu, R.; Zhang, Z.; Chang, H.; Qiao, Y.; Wang, Y. Interactions between PAMAM−NH2 and 6-Mercaptopurine: Qualitative and Quantitative NMR studies. Chem. Asian J. 2021, 16, 3658–3663. [Google Scholar] [CrossRef]
- Ybarra, D.E.; Calienni, M.N.; Frias, E.T.A.; Lillo, C.; Alonso, S.D.V.; Montanari, J.; Alvira, F.C. Vismodegib in PAMAM-dendrimers for potential theragnosis in skin cancer. OpenNano 2022, 7, 100053. [Google Scholar] [CrossRef]
- Mignani, S.; El Kazzouli, S.; Bousmina, M.; Majoral, J.P. Expand classical drug administration ways by emerging routes using dendrimer drug delivery systems: A concise overview. Adv. Drug Deliv. Rev. 2013, 65, 1316–1330. [Google Scholar] [CrossRef]
- Chanphai, P.; Bekale, L.; Sanyakamdhorn, S.; Agudelo, D.; Bérubé, G.; Thomas, T.; Tajmir-Riahi, H. PAMAM dendrimers in drug delivery: Loading efficacy and polymer morphology. Can. J. Chem. 2017, 95, 891–896. [Google Scholar] [CrossRef]
- Chandran, S.P.; Natarajan, S.B.; Chandraseharan, S.; Mohd Shahimi, M.S.B. Nano drug delivery strategy of 5-fluorouracil for the treatment of colorectal cancer. J. Cancer Res. Pract. 2017, 4, 45–48. [Google Scholar] [CrossRef]
- Giacchetti, S.; Perpoint, B.; Zidani, R.; Le Bail, N.; Faggiuolo, R.; Focan, C.; Chollet, P.; Llory, J.; Letourneau, Y.; Coudert, B.; et al. Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil-leucovorin as first-line treatment of metastatic colorectal cancer. J. Clin. Oncol. 2000, 18, 136. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, B.E.; Amorim, O.H.J.; Lima, L.L.; Rezende, R.A.; Mestnik, N.C.; Bagatin, E.; Leonardi, G.R. 5-Fluorouracil, innovative drug delivery systems to enhance bioavailability for topical use. J. Drug Deliv. Sci. Technol. 2021, 61, 102155. [Google Scholar] [CrossRef]
- De Souza, F.G.; Helena, C.; Giarolla, J. PAMAM Dendrimers: A review of methodologies employed in biopharmaceutical classification. J. Pharm. Sci. 2022, 11, 2662–2673. [Google Scholar] [CrossRef]
- Najlah, M.; Freeman, S.; Khoder, M.; Attwood, D.; D’Emanuele, A. In vitro evaluation of third generation PAMAM dendrimer conjugates. Molecules 2017, 22, 1661. [Google Scholar] [CrossRef] [PubMed]
- Falconieri, M.C.; Adamo, M.; Monasterolo, C.; Bergonzi, M.C.; Coronnello, M.; Bilia, A.R. New Dendrimer-Based Nanoparticles Enhance Curcumin Solubility. Planta Med. 2017, 83, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Buczkowski, A.; Sekowski, S.; Grala, A.; Palecz, D.; Milowska, K.; Urbaniak, P.; Gabryelak, T.; Piekarski, H.; Palecz, B. Interaction between PAMAM-NH2 G4 dendrimer and 5-fluorouracil in aqueous solution. Int. J. Pharm. 2011, 408, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Jain, V.; Bharatam, P.V. Pharmacoinformatic approaches to understand complexation of dendrimeric nanoparticles with drugs. Nanoscale 2014, 6, 2476–2501. [Google Scholar] [CrossRef]
- Cakara, D.; Kleimann, J.; Borkovec, M. Microscopic protonation equilibria of poly(amidoamine) dendrimers from macroscopic titrations. Macromolecules 2003, 36, 4201–4207. [Google Scholar] [CrossRef]
- Prosa, T.J.; Bauer, B.J.; Amis, E.J. From stars to spheres: A SAXS analysis of dilute dendrimer solutions. Macromolecules 2001, 34, 4897–4906. [Google Scholar] [CrossRef]
- Rathgeber, S.; Monkenbusch, M.; Kreitschmann, M.; Urban, V.; Brulet, A. Dynamics of star-burst dendrimers in solution in relation to their structural properties. J. Chem. Phys. 2002, 117, 4047–4062. [Google Scholar] [CrossRef] [Green Version]
- Prosa, T.J.; Bauer, B.J.; Amis, E.J.; Tomalia, D.A.; Scherrenberg, R. A SAXS study of the internal structure of dendritic polymer systems. J. Polym. Sci. Part B Polym. Phys. 1997, 35, 2913–2924. [Google Scholar] [CrossRef]
- Maiti, P.K.; Çaǧin, T.; Lin, S.T.; Goddard, W.A. Effect of solvent and pH on the structure of PAMAM dendrimers. Macromolecules 2005, 38, 979–991. [Google Scholar] [CrossRef]
- Smilgies, D.M.; Folta-Stogniew, E. Molecular weight-gyration radius relation of globular proteins: A comparison of light scattering, small-angle X-ray scattering and structure-based data. J. Appl. Crystallogr. 2015, 48, 1604–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikzamir, M.; Hanifehpour, Y.; Akbarzadeh, A.; Panahi, Y. Applications of Dendrimers in Nanomedicine and Drug Delivery: A Review. J. Inorg. Organomet. Polym. Mater. 2021, 31, 2246–2261. [Google Scholar] [CrossRef]
- Zhao, L.; Cheng, Y.; Hu, J.; Wu, Q.; Xu, T. Host-guest chemistry of dendrimer-drug complexes. 3. Competitive binding of multiple drugs by a single dendrimer for combination therapy. J. Phys. Chem. B 2009, 113, 14172–14179. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Gao, X.; Guo, Z.; Qiao, Y. New Insights into the Binding Site and Affinity of the Interaction between Biotin and PAMAMs-NH2via NMR Studies. J. Phys. Chem. B 2021, 125, 4076–4085. [Google Scholar] [CrossRef] [PubMed]
- Wielińska, J.; Nowacki, A.; Liberek, B. 5-fluorouracil-complete insight into its neutral and ionised forms. Molecules 2019, 24, 3683. [Google Scholar] [CrossRef] [Green Version]
- Abdrakhimova, G.S.; Ovchinnikov, M.Y.; Lobov, A.N.; Spirikhin, L.V.; Ivanov, S.P.; Khursan, S.L. 5-fluorouracil solutions: NMR study of acid-base equilibrium in water and DMSO. J. Phys. Org. Chem. 2014, 27, 876–883. [Google Scholar] [CrossRef]
- Markova, N.; Enchev, V.; Ivanova, G. Tautomeric equilibria of 5-fluorouracil anionic species in water. J. Phys. Chem. A 2010, 114, 13154–13162. [Google Scholar] [CrossRef]
- Buczkowski, A.; Waliszewski, D.; Urbaniak, P.; Palecz, B. Study of the interactions of PAMAM G3-NH2 and G3-OH dendrimers with 5-fluorouracil in aqueous solutions. Int. J. Pharm. 2016, 505, 1–13. [Google Scholar] [CrossRef]
- Viltres, H.; Odio, O.F.; Biesinger, M.C.; Montiel, G.; Borja, R.; Reguera, E. Preparation of Amine- and Disulfide-Containing PAMAM-Based Dendrons for the Functionalization of Hydroxylated Surfaces: XPS as Structural Sensor. ChemistrySelect 2020, 5, 4875–4884. [Google Scholar] [CrossRef]
- Todea, M.; Muresan-Pop, M.; Simon, S.; Moisescu-Goia, C.; Simon, V.; Eniu, D. XPS investigation of new solid forms of 5-fluorouracil with piperazine. J. Mol. Struct. 2018, 1165, 120–125. [Google Scholar] [CrossRef]
- Mazzola, F.; Trinh, T.; Cooil, S.; Østli, E.R.; Høydalsvik, K.; Skjønsfjell, E.T.B.; Kjelstrup, S.; Preobrajenski, A.; Cafolla, A.A.; Evans, A.; et al. Silver catalyzed Fluorouracil degradation; a promising new role for graphene. arXiv 2014, arXiv:1411.2096. [Google Scholar]
- Acres, R.G.; Ellis, A.V.; Alvino, J.; Lenahan, C.E.; Khodakov, D.A.; Metha, G.F.; Andersson, G.G. Molecular structure of 3-aminopropyltriethoxysilane layers formed on silanol-terminated silicon surfaces. J. Phys. Chem. C 2012, 116, 6289–6297. [Google Scholar] [CrossRef]
- Stevens, J.S.; Byard, S.J.; Schroeder, S.L.M. Characterization of Proton Transfer in Co-Crystals by X-ray Photoelectron Spectroscopy (XPS). Cryst. Growth Des. 2010, 10, 1443–1450. [Google Scholar] [CrossRef]
- Risinggård, H.K.; Cooil, S.; Mazzola, F.; Hu, D.; Kjærvik, M.; Østli, E.R.; Patil, N.; Preobrajenski, A.; Evans, D.A.; Breiby, D.W.; et al. Degradation of the chemotherapy drug 5-fluorouracil on medical-grade silver surfaces. Appl. Surf. Sci. 2018, 435, 1213–1219. [Google Scholar] [CrossRef] [Green Version]
- Jędrzak, A.; Grześkowiak, B.F.; Coy, E.; Wojnarowicz, J.; Szutkowski, K.; Jurga, S.; Jesionowski, T.; Mrówczyński, R. Dendrimer based theranostic nanostructures for combined chemo- and photothermal therapy of liver cancer cells in vitro. Colloids Surf. B Biointerfaces 2019, 173, 698–708. [Google Scholar] [CrossRef]
- Cassidy, A.; Tsud, N.; Bercha, S.; Feyer, V.; Prince, K.C.; Plekan, O. Adsorption of 5-Fluorouracil on Au(111) and Cu(111) surfaces. AIP Adv. 2019, 9, 085318. [Google Scholar] [CrossRef]
- Naumkin, A.V.; Kraut-Vass, A.; Gaarenstroom, S.W.; Powell, C.J. NIST X-ray Photoelectron Spectroscopy Database. 2012. Available online: https://srdata.nist.gov/xps/default.aspx (accessed on 15 March 2022).
- Jain, V.; Maingi, V.; Maiti, P.K.; Bharatam, P.V. Molecular dynamics simulations of PPI dendrimer-drug complexes. Soft Matter. 2013, 9, 6482–6496. [Google Scholar] [CrossRef]
- Maingi, V.; Jain, V.; Bharatam, P.V.; Maiti, P.K. Dendrimer building toolkit: Model building and characterization of various dendrimer architectures. J. Comput. Chem. 2012, 33, 1997–2011. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Dupradeau, F.-Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanquelef, E.; Simon, S.; Marquant, G.; Garcia, E.; Klimerak, G.; Delepine, J.C.; Cieplak, P.; Dupradeau, F.-Y. R.E.D. Server: A web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 2011, 39, W511–W517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa Da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://ambermd.org/AmberTools.php (accessed on 17 June 2020).
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Available online: https://www.gromacs.org/development.html (accessed on 28 November 2022).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH | [a] Rg (nm) | [b] Rg (nm) | [c] RH (nm) |
|---|---|---|---|
| SAXS | MD | DLS | |
| 10.2 | 1.87 ± 0.02 | 1.47 ± 0.01 | 2.45 ± 0.05 [12] |
| 7.0 | 2.11 ± 0.02 | - | 2.67 ± 0.05 |
| 4.3 | 2.17 ± 0.02 | - | 2.79 ± 0.05 |
| G4:5FU | pH | D (m2/s) × 10−11 | RH (nm) |
|---|---|---|---|
| 1:0 | 10.3 | 8.10 ± 0.07 | 2.97 ± 0.03 |
| 1:25 | 8.8 | 7.96 ± 0.07 | 3.03 ± 0.03 |
| 1:50 | 8.3 | 7.73 ± 0.13 | 3.12 ± 0.06 |
| 1:100 | 7.8 | 6.44 ± 0.18 | 3.7 ± 0.1 |
| Core Excitation | G4 BE (eV) Area (%) | 5FU BE (eV) Area (%) | G4-5FU BE (eV) Area (%) | Assignation |
|---|---|---|---|---|
| O 1s | 529.1 2.9 | --- | 529.7 6.8 | O–metal(support) |
| 531.0 78.2 | 531.4 52.1 | 531.0 49.8 | carboxyl in PAMAM amide group | |
| 532.0 18.9 | 533.2 47.9 | 532.1 42.5 | C=O/OH | |
| --- | --- | 534.1 0.9 | chemisorbed water | |
| N 1s | 397.4 4.1 | --- | 397.1 3.5 | =N– |
| 399.0 59.7 | 398.7 39.1 | 398.4 38.1 | C–NH–C | |
| 399.6 36.2 | 400.5 60.9 | 400.0 58.4 | N–C=O | |
| C 1s | 285.0 31.9 | 285.0 11.9 | 285.0 43.6 | C–C/C–H |
| 285.6 51.1 | 286.2 55.8 | 286.3 19.1 | C–O/C–N | |
| 287.7 17.0 | 288.4 28.3 | 288.0 30.6 | –NH–C=O | |
| --- | 290.5 4.0 | 290.0 6.7 | C–F | |
| F 1s | --- | 683.6 25.1 | 683.6 18.0 | F–metal (support) |
| 685.0 6.2 | 685.5 4.8 | F in degraded 5FU | ||
| 687.5 64.7 | 687.0 76.3 | F in 5FU | ||
| 688.8 4.0 | 689.0 0.9 | C–F |
| Protonation Degree (%) | Rg (nm) | Aspect Ratio | Asphericity Factor | ||
|---|---|---|---|---|---|
| Iz/Ix | Iz/Iy | ||||
| S1 | 0 | 1.47 ± 0.01 | 1.50 ± 0.11 | 1.20 ± 0.03 | 0.0136 |
| S2 | 10 | 1.46 ± 0.01 | 1.60 ± 0.05 | 1.12 ± 0.02 | 0.0178 |
| S3 | 20 | 1.54 ± 0.02 | 1.73 ± 0.07 | 1.19 ± 0.04 | 0.0238 |
| Property/System | S1 | S2 | S3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Energy (kJ/mol) per single 5FU molecule | Total | LJ | Coul | Total | LJ | Coul | Total | LJ | Coul |
| −4.60 | −2.74 | −1.83 | −21.47 | −7.93 | −13.54 | −34.50 | −12.10 | −22.40 | |
| N(t > 10 ns) | 0 | 6 | 12 | ||||||
| N(t < 10 ns) | 8 | 12 | 13 | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szota, M.; Wolski, P.; Carucci, C.; Marincola, F.C.; Gurgul, J.; Panczyk, T.; Salis, A.; Jachimska, B. Effect of Ionization Degree of Poly(amidoamine) Dendrimer and 5-Fluorouracil on the Efficiency of Complex Formation—A Theoretical and Experimental Approach. Int. J. Mol. Sci. 2023, 24, 819. https://doi.org/10.3390/ijms24010819
Szota M, Wolski P, Carucci C, Marincola FC, Gurgul J, Panczyk T, Salis A, Jachimska B. Effect of Ionization Degree of Poly(amidoamine) Dendrimer and 5-Fluorouracil on the Efficiency of Complex Formation—A Theoretical and Experimental Approach. International Journal of Molecular Sciences. 2023; 24(1):819. https://doi.org/10.3390/ijms24010819
Chicago/Turabian StyleSzota, Magdalena, Pawel Wolski, Cristina Carucci, Flaminia Cesare Marincola, Jacek Gurgul, Tomasz Panczyk, Andrea Salis, and Barbara Jachimska. 2023. "Effect of Ionization Degree of Poly(amidoamine) Dendrimer and 5-Fluorouracil on the Efficiency of Complex Formation—A Theoretical and Experimental Approach" International Journal of Molecular Sciences 24, no. 1: 819. https://doi.org/10.3390/ijms24010819