
Inhibition of Oncogenic Src Ameliorates Silica-Induced Pulmonary Fibrosis via PI3K/AKT Pathway
 , and
, and 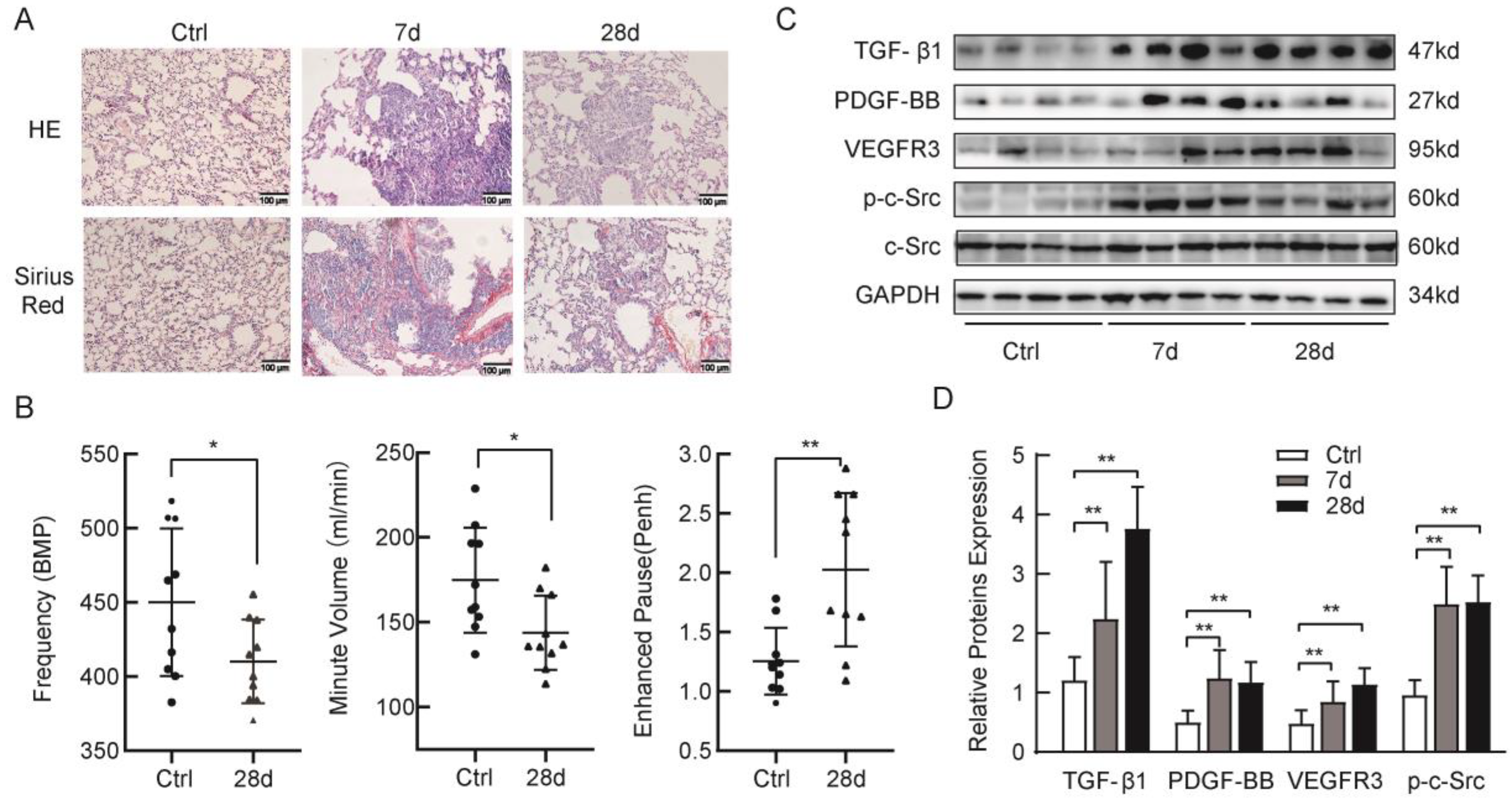{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Silica-induced Lung Fibrosis and Upregulation of p-c-Src Tyrosine Kinases, TGF-β1, and Profibrotic Factors in Mice
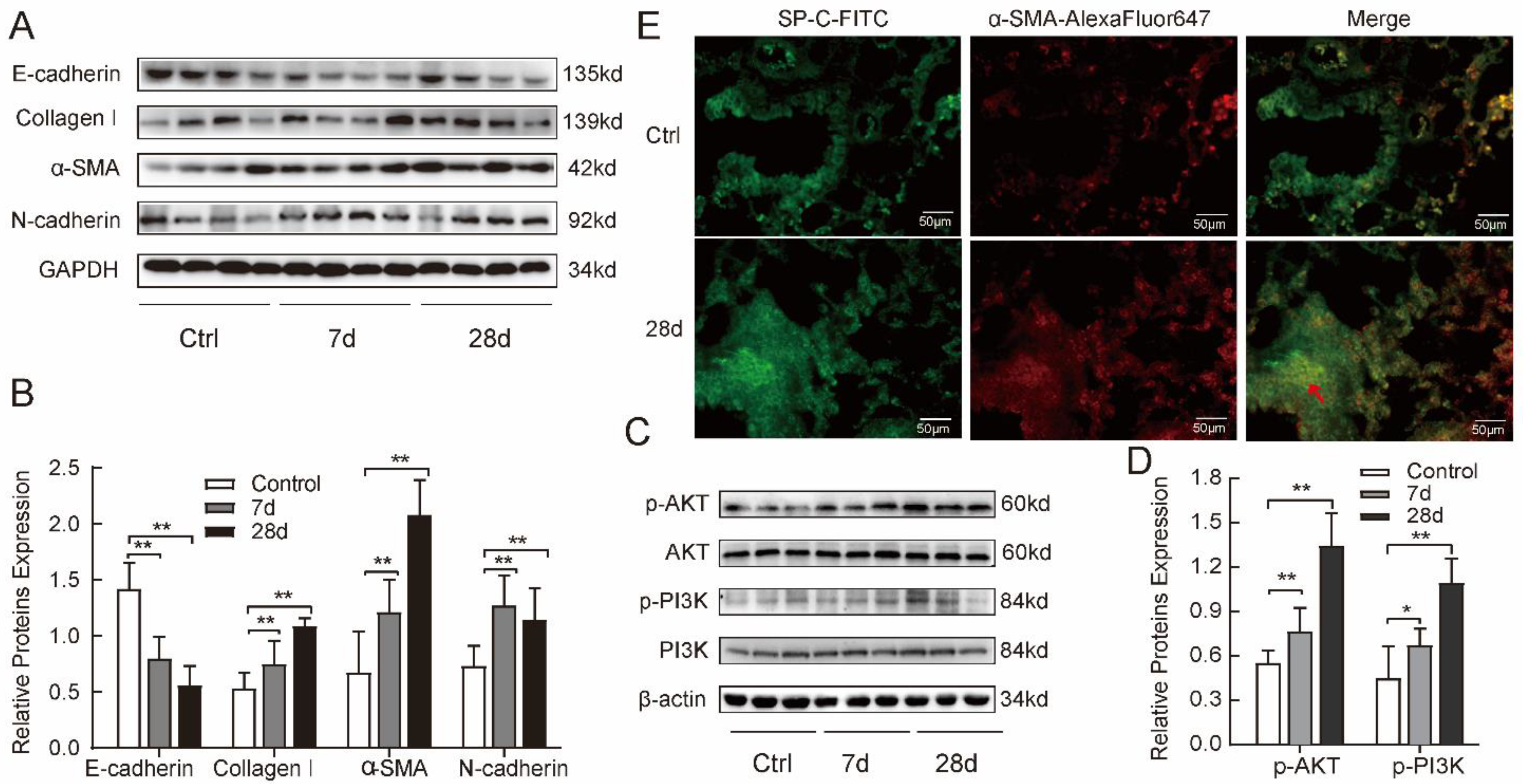
2.2. Silica-induced Fibrosis Was Associated with Upregulation of PI3K/AKT Pathway in Mice
2.3. Silica Suspension Induced Increase of Profibrotic Cytokine Tgf-β1 and Tgf-β1 Promoted Phosphorylation of c-Src Protein In Vitro
2.4. TGF-β1 Induced Mesenchymal Phenotype via the Activated PI3K/AKT Pathway in MLE-12 Cells
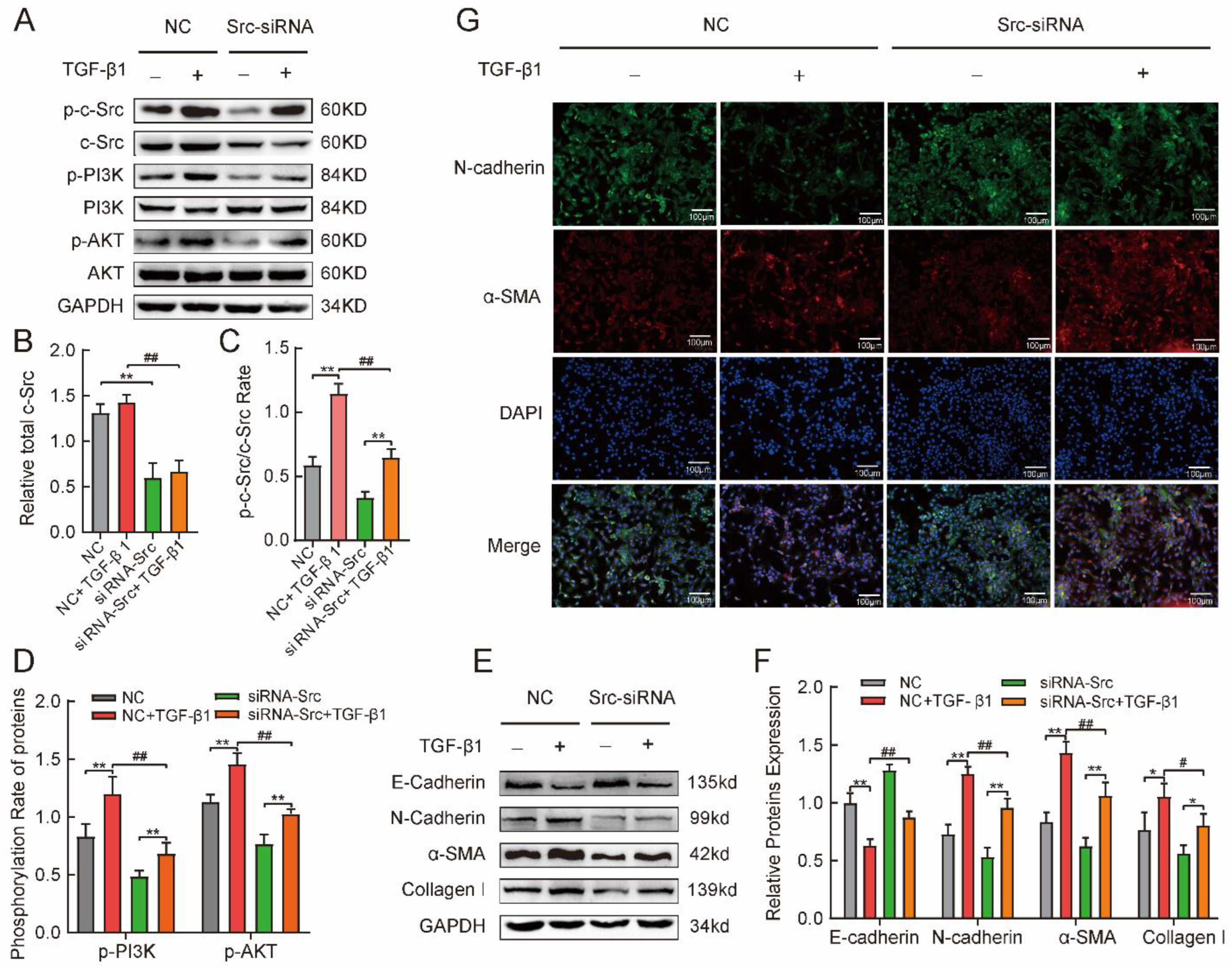
2.5. Phosphorylated c-Src Activated the PI3K/AKT Pathway Involved in TGFβ1-Induced Mesenchymal Phenotype in MLE-12 Cells
2.6. SU6656 Inhibited p-c-Src to Suppress PI3K/AKT Pathway Activation to Prevent Fibrosis in Mice Model of Silicosis
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals and Groups
4.3. Cell Culture
4.4. Pulmonary Function Tests
4.5. Histopathology and Sirius Red Staining
4.6. Cells or Tissues Immunofluorescence
4.7. Western Blotting
4.8. siRNA Transfection
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoy, R.F.; Chambers, D.C. Silica-related diseases in the modern world. Allergy 2020, 75, 2805–2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, H.; Goh, N.S.L.; Leong, T.L.; Hoy, R. Silica-associated lung disease: An old-world exposure in modern industries. Respirology 2019, 24, 1165–1175. [Google Scholar] [CrossRef] [Green Version]
- Jagirdar, J.; Begin, R.; Dufresne, A.; Goswami, S.; Lee, T.C.; Rom, W.N. Transforming Growth Factor-~ (TGF-J3) in Silicosis. Am. J. Respir. Crit. Care Med. 1996, 154, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Xu, D.; Li, S.; Wei, Z.; Li, S.; Cai, W.; Mao, N.; Jin, F.; Li, Y.; Yi, X.; et al. Pulmonary Silicosis Alters MicroRNA Expression in Rat Lung and miR-411-3p Exerts Anti-fibrotic Effects by Inhibiting MRTF-A/SRF Signaling. Mol. Ther. Nucleic Acids 2020, 20, 851–865. [Google Scholar] [CrossRef] [PubMed]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barratt, S.L.; Flower, V.A.; Pauling, J.D.; Millar, A.B. VEGF (Vascular Endothelial Growth Factor) and Fibrotic Lung Disease. Int. J. Mol. Sci. 2018, 19, 1269. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-b1 Signaling and Tissue Fibrosis. Cold Spring Harbor Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [Green Version]
- Katzen, J.; Beers, M.F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J Clin. Investig. 2020, 130, 5088–5099. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Li, Y.; Xu, H.; Wei, Z.; Yang, Y.; Jin, F.; Zhang, M.; Wang, C.; Song, W.; Huo, J.; et al. ACE2 Attenuates Epithelial-Mesenchymal Transition in MLE-12 Cells Induced by Silica. Drug. Dev. Res. 2020, 14, 1547–1559. [Google Scholar] [CrossRef]
- Deng, H.; Xu, H.; Zhang, X.; Sun, Y.; Wang, R.; Brann, D.; Yang, F. Protective effect of Ac-SDKP on alveolar epithelial cells through inhibition of EMT via TGF-beta1/ROCK1 pathway in silicosis in rat. Toxicol. Appl. Pharmacol. 2016, 294, 1–10. [Google Scholar] [CrossRef]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Saifi, M.A.; Pulivendala, G.; Godugu, C.; Talla, V. Ferulic acid ameliorates the progression of pulmonary fibrosis via inhibition of TGF-β/smad signalling. Food. Chem. Toxicol. 2021, 149, 111980. [Google Scholar] [CrossRef]
- Hassan, B.; Akcakanat, A.; Holder, A.M.; Meric-Bernstam, F. Targeting the PI3-kinase/Akt/mTOR signaling pathway. Surg. Oncol. Clin. N. Am. 2013, 22, 641–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Alam, A.; Pac-Soo, A.; Chen, Q.; Shang, Y.; Zhao, H.; Yao, S.; Ma, D. Pretreatment with valproic acid alleviates pulmonary fibrosis through epithelial-mesenchymal transition inhibition in vitro and in vivo. Lab. Investig. 2021, 101, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Matozaki, T.; Kotani, T.; Murata, Y.; Saito, Y. Roles of Src family kinase, Ras, and mTOR signaling in intestinal epithelial homeostasis and tumorigenesis. Cancer. Sci. 2021, 112, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Amatya, N.; Lin, D.Y.; Andreotti, A.H. Dynamic regulatory features of the protein tyrosine kinases. Biochem. Soc. Trans. 2019, 47, 1101–1116. [Google Scholar] [CrossRef]
- Yan, Y.; Ma, L.; Zhou, X.; Ponnusamy, M.; Tang, J.; Zhuang, M.A.; Tolbert, E.; Bayliss, G.; Bai, J.; Zhuang, S. Src inhibition blocks renal interstitial fibroblast activation and ameliorates renal fibrosis. Kidney Int. 2016, 89, 68–81. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Che, P.; Han, X.; Cai, G.Q.; Liu, G.; Antony, V.; Luckhardt, T.; Siegal, G.P.; Zhou, Y.; Liu, R.M.; et al. Therapeutic targeting of SRC kinase in myofibroblast differentiation and pulmonary fibrosis. J. Pharmacol. Exp. Ther. 2014, 351, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Grimminger, F.; Gunther, A.; Vancheri, C. The role of tyrosine kinases in the pathogenesis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1426–1433. [Google Scholar] [CrossRef]
- Rout-Pitt, N.; Farrow, N.; Parsons, D.; Donnelley, M. Epithelial mesenchymal transition (EMT): A universal process in lung diseases with implications for cystic fibrosis pathophysiology. Resp. Res. 2018, 19, 136. [Google Scholar] [CrossRef]
- Tian, W.; Li, J.; Wang, Z.; Zhang, T.; Han, Y.; Liu, Y.; Chu, W.; Liu, Y.; Yang, B. HYD-PEP06 suppresses hepatocellular carcinoma metastasis, epithelial-mesenchymal transition and cancer stem cell-like properties by inhibiting PI3K/AKT and WNT/beta-catenin signaling activation. Acta Pharm. Sin. B 2021, 11, 1592–1606. [Google Scholar] [CrossRef]
- Benmerzoug, S.; Rose, S.; Bounab, B.; Gosset, D.; Duneau, L.; Chenuet, P.; Mollet, L.; Le Bert, M.; Lambers, C.; Geleff, S.; et al. STING-dependent sensing of self-DNA drives silica-induced lung inflammation. Nat. Commun. 2018, 9, 5226. [Google Scholar] [CrossRef] [Green Version]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.; Conforti, F.; Hill, C.; Bell, J.; Drawater, L.; Li, J.; Liu, D.; Xiong, H.; Alzetani, A.; Chee, S.J.; et al. Paracrine signalling during ZEB1-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in lung fibrosis. Cell Death Differ. 2019, 26, 943–957. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Massague, J. TGF-β in developmental and fibrogenic EMTs. Semin. Cancer Biol. 2022, 86 Pt 2, 136–145. [Google Scholar] [CrossRef]
- Qi, Y.; Zhao, A.; Yang, P.; Jin, L.; Hao, C. miR-34a-5p Attenuates EMT through targeting SMAD4 in silica-induced pulmonary fibrosis. J. Cell. Mol. Med. 2020, 24, 12219–12224. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Morgani, S.M.; David, C.J.; Wang, Q.; Er, E.E.; Huang, Y.H.; Basnet, H.; Zou, Y.; Shu, W.; Soni, R.K.; et al. TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 2020, 577, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Qiu, Y.B.; Gao, Z.Q.; Wu, Y.X.; Wan, B.B.; Liu, G.; Chen, J.L.; Zhou, Q.; Yu, R.Q.; Pang, Q.F. Sodium Propionate Attenuates the Lipopolysaccharide-Induced Epithelial-Mesenchymal Transition via the PI3K/Akt/mTOR Signaling Pathway. J. Agric. Food Chem. 2020, 68, 6554–6563. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Luo, M.; Cai, W.; Zhou, S.; Feng, D.; Xu, C.; Wang, H. Runt-Related Transcription Factor 1 (RUNX1) Promotes TGF-beta-Induced Renal Tubular Epithelial-to-Mesenchymal Transition (EMT) and Renal Fibrosis through the PI3K Subunit p110delta. Ebiomedicine 2018, 31, 217–225. [Google Scholar] [CrossRef]
- Lee, J.H.; Chinnathambi, A.; Alharbi, S.A.; Shair, O.H.M.; Sethi, G.; Ahn, K.S. Farnesol abrogates epithelial to mesenchymal transition process through regulating Akt/mTOR pathway. Pharmacol. Res. 2019, 150, 104504. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhu, L.B.; Peng, A.F.; Wang, T.F.; Long, X.H.; Gao, S.; Zhou, R.P.; Liu, Z.L. LY294002 inhibits the malignant phenotype of osteosarcoma cells by modulating the phosphatidylinositol 3 kinase/Akt/fatty acid synthase signaling pathway in vitro. Mol. Med. Rep. 2015, 11, 1352–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, C.; Distler, J.H.W. Tyrosine kinase signaling in fibrotic disorders Translation of basic research to human disease, Tyrosine kinase signaling in fibrotic disorders: Translation of basic research to human disease. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 897–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhao, C.; Tian, Y.; Lu, J.; Zhang, G.; Liang, S.; Chen, D.; Liu, X.; Kuang, W.; Zhu, M. Src family kinases and pulmonary fibrosis: A review. Biomed. Pharmacother. 2020, 127, 110183. [Google Scholar] [CrossRef]
- Mishra, R.; Zhu, L.; Eckert, R.L.; Simonson, M.S. TGF-β-regulated collagen type I accumulation: Role of Src-based signals. Am. J. Physiol. Cell Physiol. 2007, 292, C1361–C1369. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Davies, K.J.; Forman, H.J. TGFβ1 rapidly activates Src through a non-canonical redox signaling mechanism. Arch. Biochem. Biophys. 2015, 568, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Ahangari, F.; Becker, C.; Foster, D.G.; Chioccioli, M.; Nelson, M.; Beke, K.; Wang, X.; Justet, A.; Adams, T.; Readhead, B.; et al. Saracatinib, a Selective Src Kinase Inhibitor, Blocks Fibrotic Responses in Preclinical Models of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2022, 206, 1463–1479. [Google Scholar] [CrossRef]
- Li, L.F.; Kao, K.C.; Liu, Y.Y.; Lin, C.W.; Chen, N.H.; Lee, C.S.; Wang, C.W.; Yang, C.T. Nintedanib reduces ventilation-augmented bleomycin-induced epithelial-mesenchymal transition and lung fibrosis through suppression of the Src pathway. J. Cell. Mol. Med. 2017, 21, 2937–2949. [Google Scholar] [CrossRef]
- Iida, M.; Sahashi, K.; Kondo, N.; Nakatsuji, H.; Tohnai, G.; Tsutsumi, Y.; Noda, S.; Murakami, A.; Onodera, K.; Okada, Y.; et al. Src inhibition attenuates polyglutamine-mediated neuromuscular degeneration in spinal and bulbar muscular atrophy. Nat. Commun. 2019, 10, 4262. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, X.; Jin, Y.; Zhang, Y.; Li, S.; Cui, J.; He, H.; Guo, L.; Yang, F.; Liu, H. Inhibition of Oncogenic Src Ameliorates Silica-Induced Pulmonary Fibrosis via PI3K/AKT Pathway. Int. J. Mol. Sci. 2023, 24, 774. https://doi.org/10.3390/ijms24010774
Hao X, Jin Y, Zhang Y, Li S, Cui J, He H, Guo L, Yang F, Liu H. Inhibition of Oncogenic Src Ameliorates Silica-Induced Pulmonary Fibrosis via PI3K/AKT Pathway. International Journal of Molecular Sciences. 2023; 24(1):774. https://doi.org/10.3390/ijms24010774
Chicago/Turabian StyleHao, Xiaohui, Yixuan Jin, Yiyang Zhang, Shifeng Li, Jie Cui, Hailan He, Lingli Guo, Fang Yang, and Heliang Liu. 2023. "Inhibition of Oncogenic Src Ameliorates Silica-Induced Pulmonary Fibrosis via PI3K/AKT Pathway" International Journal of Molecular Sciences 24, no. 1: 774. https://doi.org/10.3390/ijms24010774