
Synthesis, Crystallographic Structure, Theoretical Analysis, Molecular Docking Studies, and Biological Activity Evaluation of Binuclear Ru(II)-1-Naphthylhydrazine Complex
 , , , ,
, , , , 
Abstract
:1. Introduction
2. Results and Discussion
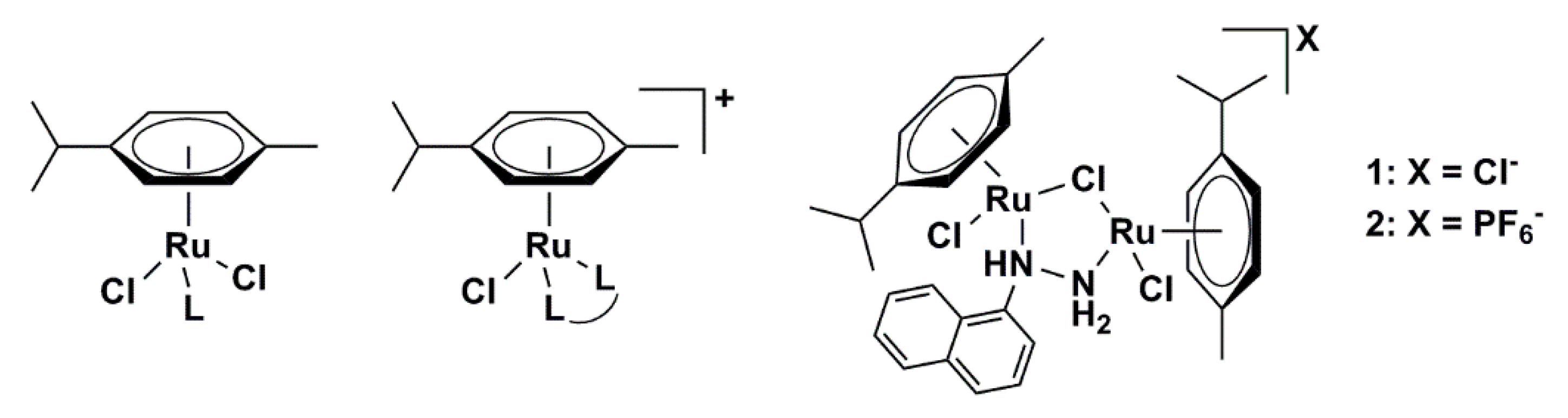
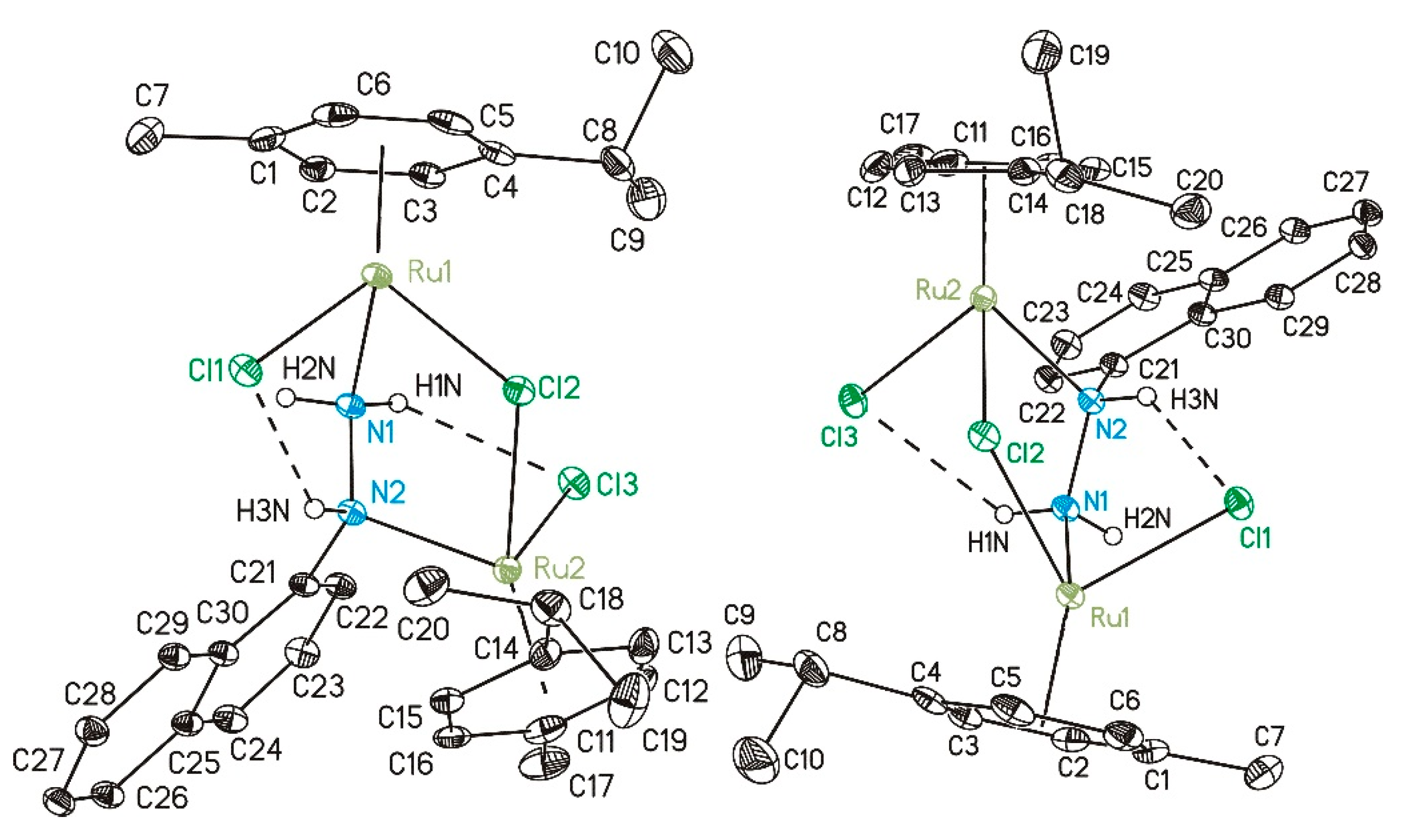
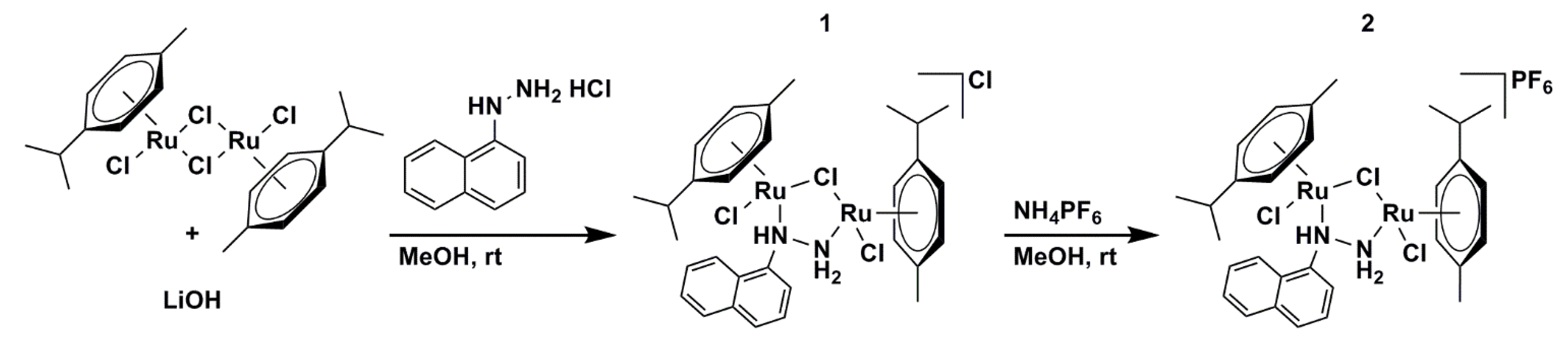
2.1. Synthetic Procedure and Crystallographic Structure
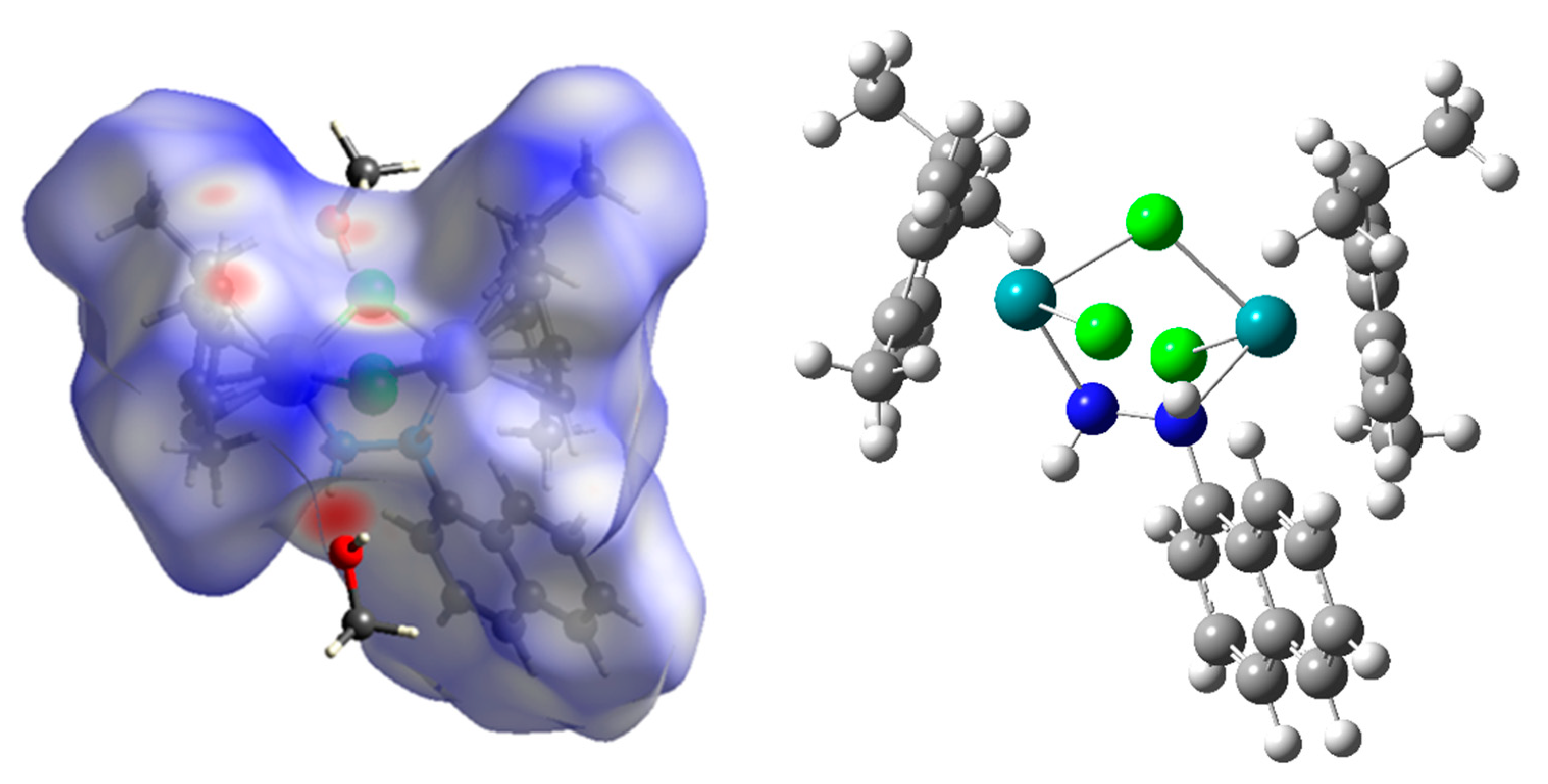
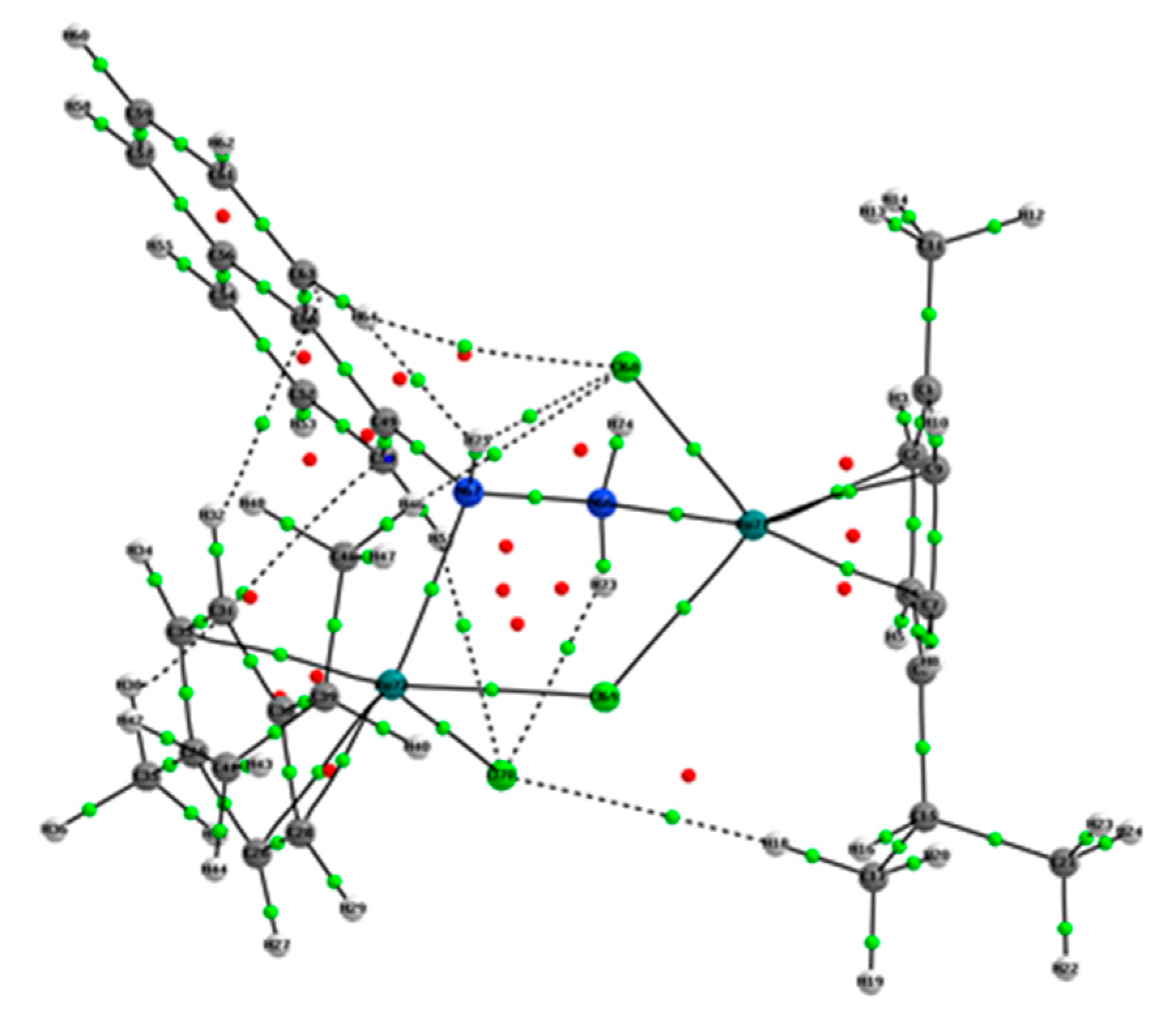
2.2. Hirshfeld Surface Analysis
2.3. Optimization of Structure, NBO, and QTAIM Analyses
2.4. Experimental and Theoretical IR and NMR Spectra
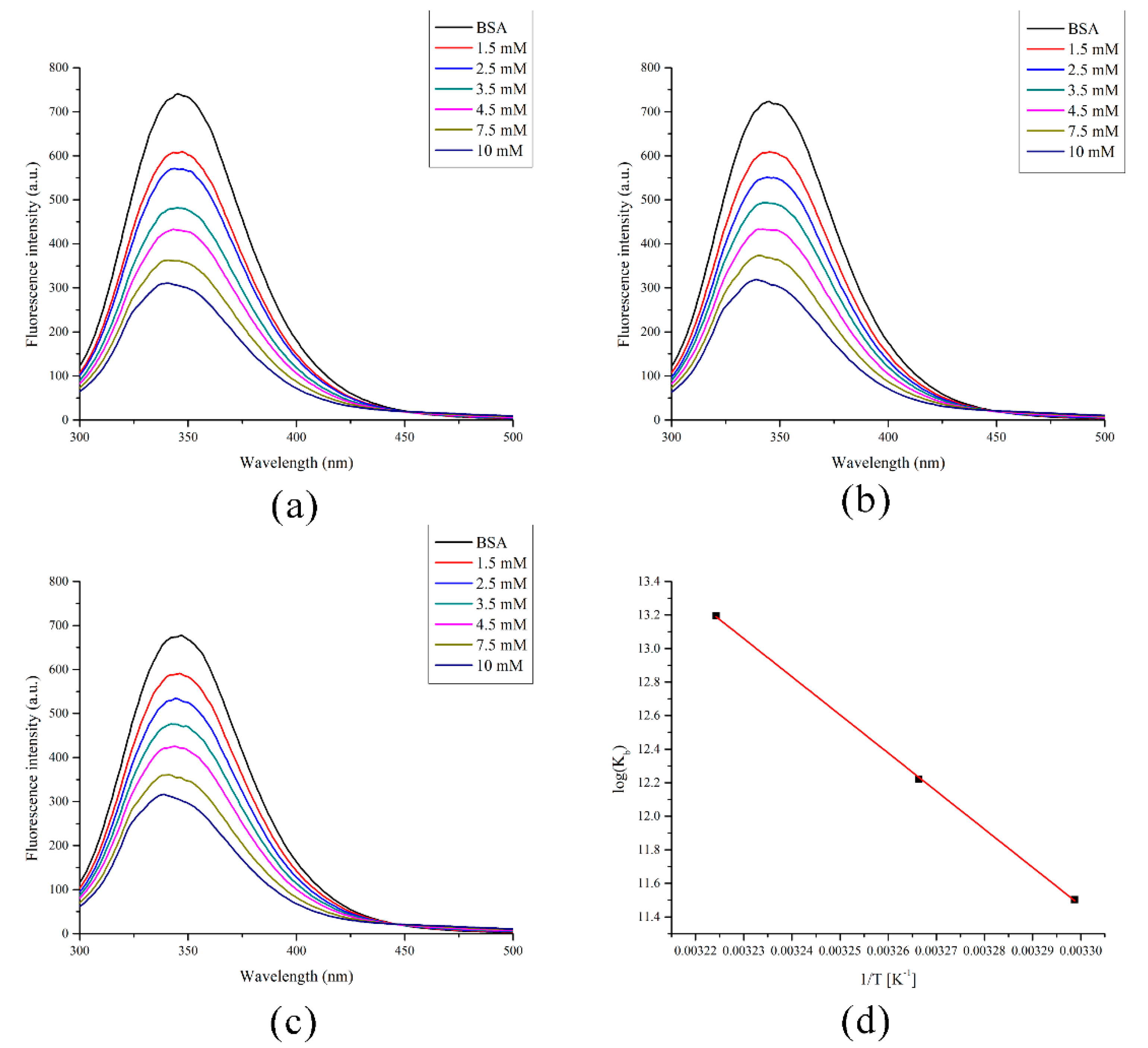
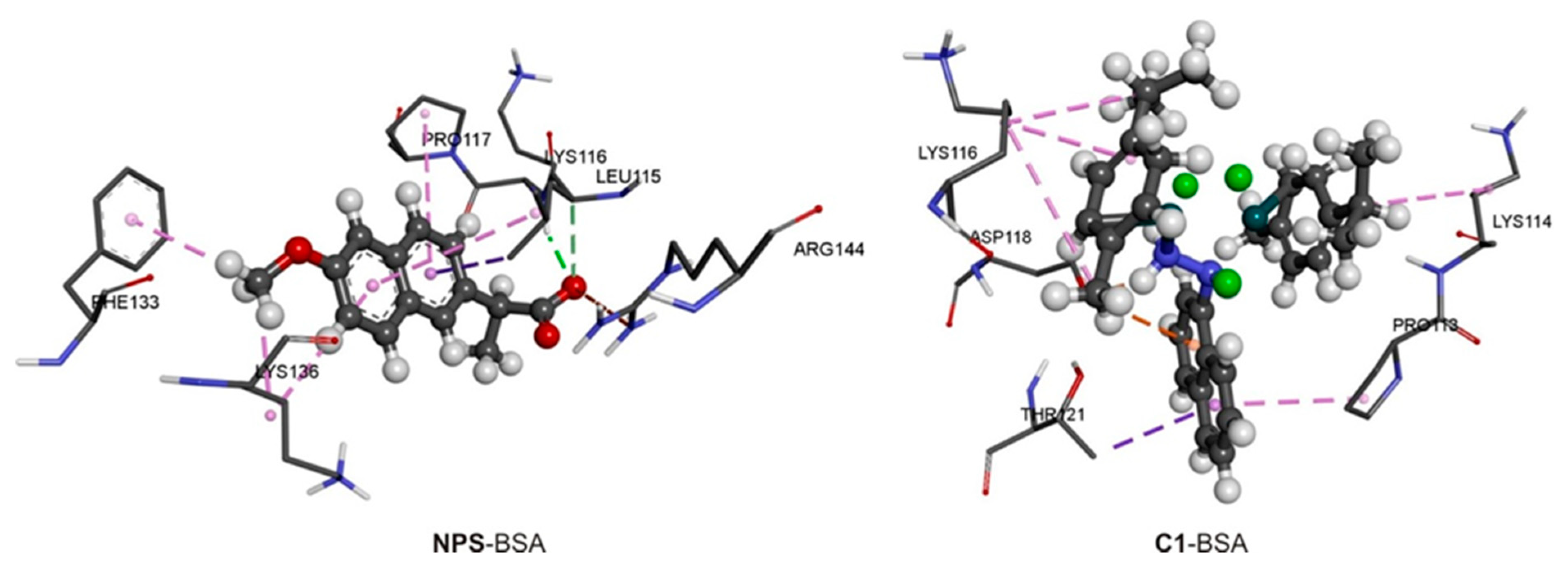
2.5. Spectrofluorometric Measurements and Molecular Docking with BSA
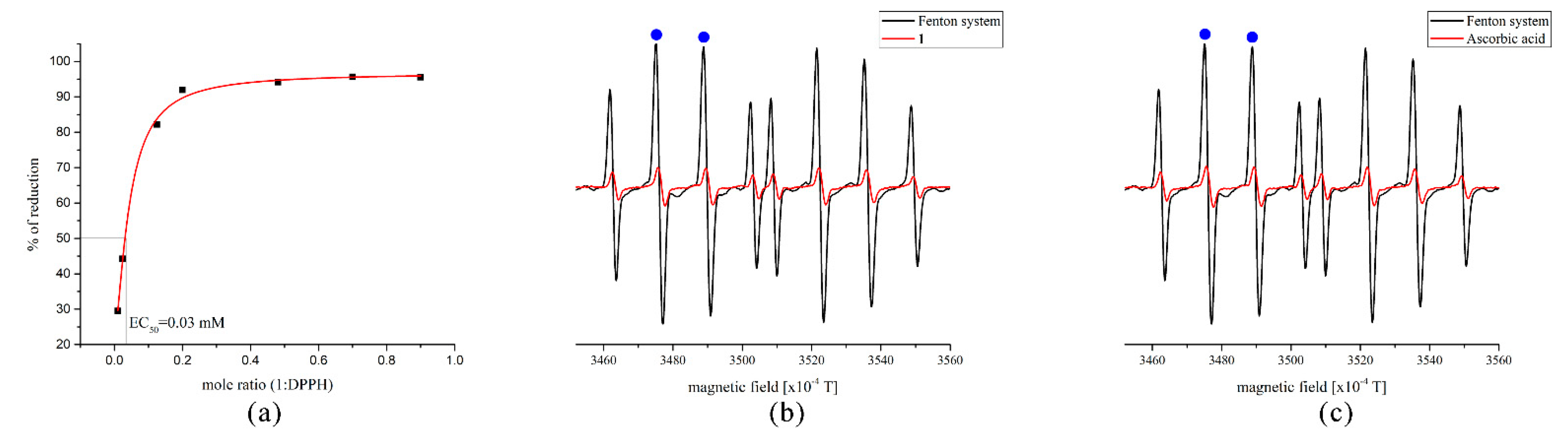
2.6. Radical Scavenging Activity
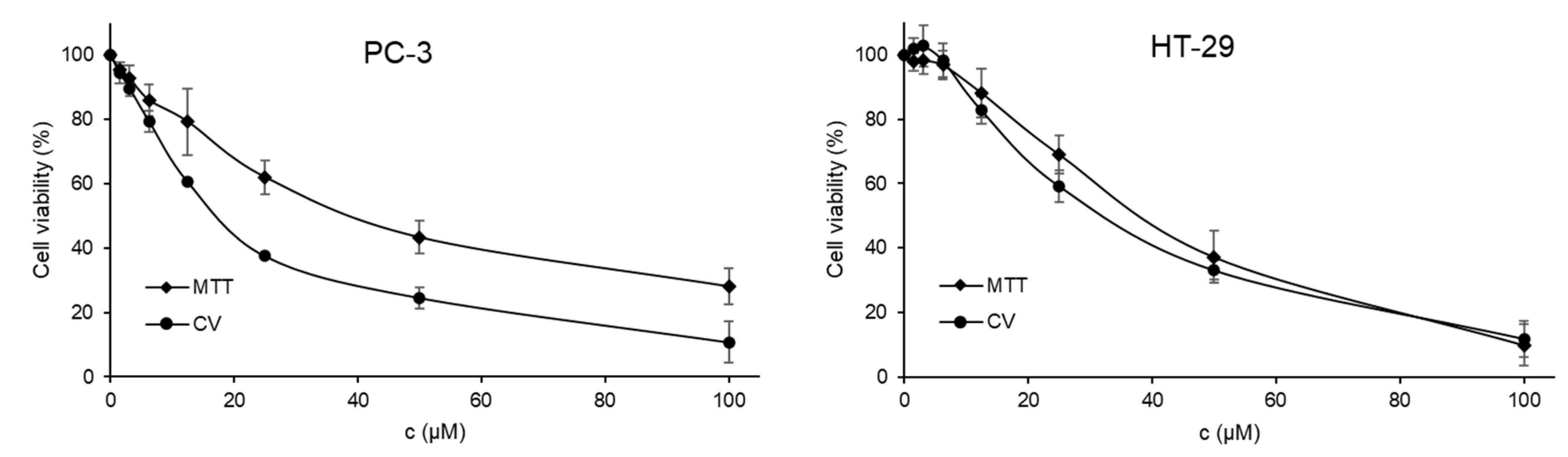
2.7. Cytotoxic Activity
3. Materials and Methods
3.1. Synthesis
3.1.1. Synthesis of [{RuCl(η6-p-Cymene)}2(μ-Cl)(μ-1-N,N′-Naphthyl)]Cl (1)
3.1.2. Synthesis of [{RuCl(η6-p-Cymene)}2(μ-Cl)(μ-1-N,N′-Naphthyl)]PF6 (2)
3.2. X-ray Analysis
3.3. Spectroscopic Analysis
3.4. Hirshfeld Surface Analysis
3.5. Theoretical Calculations
3.6. Spectofluorimetric Measurements
3.7. Antiradical Activity
3.8. Cell Culture Conditions and Viability Assays (MTT and CV)
3.9. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kenny, R.G.; Marmion, C.J. Toward Multi-Targeted Platinum and Ruthenium Drugs—A New Paradigm in Cancer Drug Treatment Regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [Green Version]
- Thota, S.; Rodrigues, D.A.; Crans, D.C.; Barreiro, E.J. Ru(II) Compounds: Next-Generation Anticancer Metallotherapeutics? J. Med. Chem. 2018, 61, 5805–5821. [Google Scholar] [CrossRef] [PubMed]
- Simović, A.R.; Masnikosa, R.; Bratsos, I.; Alessio, E. Chemistry and reactivity of ruthenium(II) complexes: DNA/protein binding mode and anticancer activity are related to the complex structure. Coord. Chem. Rev. 2019, 398, 113011. [Google Scholar] [CrossRef]
- Conti, L.; Macedi, E.; Giorgi, C.; Valtancoli, B.; Fusi, V. Combination of light and Ru(II) polypyridyl complexes: Recent advances in the development of new anticancer drugs. Coord. Chem. Rev. 2022, 469, 214656. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, C.Y.; Nam, T.-G. Ruthenium Complexes as Anticancer Agents: A Brief History and Perspectives. Drug Des. Dev. Ther. 2020, 14, 5375–5392. [Google Scholar] [CrossRef] [PubMed]
- Durig, J.R.; Danneman, J.; Behnke, W.D.; Mercer, E.E. The induction of filamentous growth in Escherichia coli by ruthenium and palladium complexes. Chem. Biol. Interact. 1976, 13, 287–294. [Google Scholar] [CrossRef]
- Therrien, B. Functionalised η6-arene ruthenium complexes. Coord. Chem. Rev. 2009, 253, 493–519. [Google Scholar] [CrossRef]
- Clarke, M.J. Ruthenium metallopharmaceuticals. Coord. Chem. Rev. 2002, 232, 69–93. [Google Scholar] [CrossRef]
- Smith, G.S.; Therrien, B. Targeted and multifunctional arene ruthenium chemotherapeutics. Dalton Trans. 2011, 40, 10793. [Google Scholar] [CrossRef]
- Kawano, M.; Hoshino, C.; Matsumoto, K. Synthesis, properties, and crystal structure of a novel μ-hydrazine-bridged mixed-valence ruthenium(II,III) complex stabilized by hydrazine hydrogen bonds, [RuCl(TMP)2]2(μ-Cl)(μ-N2H4)(μ-S2) (TMP=trimethylphosphite). Inorg. Chem. 1992, 31, 5158–5159. [Google Scholar] [CrossRef]
- Rozenel, S.S.; Arnold, J. Bimetallic Ruthenium PNP Pincer Complex as a Platform to Model Proposed Intermediates in Dinitrogen Reduction to Ammonia. Inorg. Chem. 2012, 51, 9730–9739. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Koyama, T.; Koide, Y. Oxidation of the Sulfide Ligands to SO42− in the Dinuclear Complex {RuCl[P(OMe)3]2}2(μ-S2)(μ-Cl)(μ-N2H4): Synthesis and Characterization of {RuCl[P(OMe)3]2}2(μ-S2)(μ-Cl)(μ-N2H4)+HSO4−, {RuCl2[P(OMe)3]2}2(μ-S)(μ-N2H4), and {RuCl2[P(OMe)3]2}2(μ-S2O5)(μ-N2H4). J. Am. Chem. Soc. 1999, 121, 10913–10923. [Google Scholar] [CrossRef]
- Mashima, K.; Kaneko, S.; Tani, K.; Kaneyoshi, H.; Nakamura, A. Synthesis and reactions of coordinatively unsaturated 16-electron chalcogenolate complexes, Ru(EAr)2(η6-arene) and cationic binuclear chalcogenolate complexes, [(η6-arene) Ru(μ-EPh)3Ru(η6-arene)]PF6. J. Organomet. Chem. 1997, 545–546, 345–356. [Google Scholar] [CrossRef]
- Ashworth, T.V.; Nolte, M.J.; Reimann, R.H.; Singleton, E. Dimeric ruthenium(II) complexes with bridging NN-dimethylhydrazine ligands: X-ray analysis of the structure of [{RuHCl(cyclo-octa-1,5-diene)}2NH2NMe2]. J. Chem. Soc. Chem. Commun. 1977, 21, 757–758. [Google Scholar] [CrossRef]
- Ashworth, T.V.; Singleton, E.; Hough, J.J. Cationic ruthenium(II) systems. Part 1. The preparation and reactivity of diene(hydrazine)ruthenium(II) cations, and the formation of aminobonded hydrazone complexes. J. Chem. Soc. Dalton Trans. 1977, 19, 1809–1815. [Google Scholar] [CrossRef]
- Albertin, G.; Antoniutti, S.; Botter, A.; Castro, J. Hydrazine complexes of ruthenium with cyclopentadienyl and indenyl ligands: Preparation and reactivity. J. Organomet. Chem. 2014, 774, 6–11. [Google Scholar] [CrossRef]
- Reindl, S.A.; Pöthig, A.; Drees, M.; Bechlars, B.; Herdtweck, E.; Herrmann, W.A.; Kühn, F.E. Pyrazolato-Bridged Dinuclear Complexes of Ruthenium(II) and Rhodium(III) with N-Heterocyclic Carbene Ligands: Synthesis, Characterization, and Electrochemical Properties. Organometallics 2013, 32, 4082–4091. [Google Scholar] [CrossRef]
- Rigby, W.; McCleverty, J.A.; Maitlis, P.M. Pentamethylcyclopentadienyl-rhodium and -iridium complexes. Part 20. Rhodium complexes of co-ordinated hydrazines. J. Chem. Soc. Dalton Trans. 1979, 2, 382–386. [Google Scholar] [CrossRef]
- Pal, S.; Pal, S. A Diruthenum(III) Complex Possessing a Diazine and Two Chloride Bridges: Synthesis, Structure, and Properties. Inorg. Chem. 2001, 40, 4807–4810. [Google Scholar] [CrossRef]
- Matsumoto, K.; Uemura, H.; Kawano, M. A Novel Diruthenium(II,III) Complex, [{Ru(AN)(TMP)2}2(μ-S2)(μ-NH2NH2)2](CF3SO3)3 ·Et2O, with Two Hydrazine Bridges (AN=acetonitrile, TMP=P(OMe)3). Chem. Lett. 1994, 23, 1215–1218. [Google Scholar] [CrossRef]
- Lalrempuia, R.; Kollipara, M.R.; Carroll, P.J. Syntheses and characterization of arene ruthenium (II) complexes containing N,N′-donor Schiff base ligands. Crystal and molecular structure of [(η6-C6Me6)Ru(C5H4N–2–CH=N–C6H4–p–NO2)]PF. Polyhedron 2003, 22, 605–609. [Google Scholar] [CrossRef]
- Al-Noaimi, M.; AlDamen, M.A. Ruthenium complexes incorporating azoimine and α-diamine based ligands: Synthesis, crystal structure, electrochemistry and DFT calculation. Inorg. Chim. Acta 2012, 387, 45–51. [Google Scholar] [CrossRef]
- Uahengo, V.; Cai, P.; Naimhwaka, J.; Rahman, A.; Daniel, L.S.; Bhakhoa, H.; Rhyman, L.; Ramasami, P. Photophysical, electrochemical, and DFT studies of the novel azacrown-bridged dinuclear ruthenium dye sensitizers for solar cells. Polyhedron 2019, 173, 114106. [Google Scholar] [CrossRef]
- Sikalov, A.A.; Pavlovskiy, V.V.; Kirilchuk, A.A. Ruthenium(II) p-cymene complexes bearing 2-(1,2,4-triazol-3-yl)pyridines: Linkage isomerism and related NMR/DFT studies. Inorg. Chem. Commun. 2019, 99, 156–159. [Google Scholar] [CrossRef]
- Mondal, A.; Sen, U.; Roy, N.; Muthukumar, V.; Sahoo, S.K.; Bose, B.; Paira, P. DNA targeting half sandwich Ru(II)-p-cymene-N^N complexes as cancer cell imaging and terminating agents: Influence of regioisomers in cytotoxicity. Dalton Trans. 2021, 50, 979–997. [Google Scholar] [CrossRef]
- Rupp, M.T.; Shevchenko, N.; Hanan, G.S.; Kurth, D.G. Enhancing the photophysical properties of Ru(II) complexes by specific design of tridentate ligands. Coord. Chem. Rev. 2021, 446, 214127. [Google Scholar] [CrossRef]
- Cabeza, J.A.; Van der Maelen, J.F.; García-Granda, S. Topological Analysis of the Electron Density in the N-Heterocyclic Carbene Triruthenium Cluster [Ru3(μ-H)2(μ3-MeImCH)(CO)9](Me2Im=1,3-dimethylimidazol-2-ylidene). Organometallics 2009, 28, 3666–3672. [Google Scholar] [CrossRef]
- Firme, C.L.; de Pontes, D.L.; Antunes, O.A.C. Topological study of bis(cyclopentadienyl) titanium and bent titanocenes. Chem. Phys. Lett. 2010, 499, 193–198. [Google Scholar] [CrossRef]
- Pocha, R.; Löhnert, C.; Johrendt, D. The metal-rich palladium chalcogenides Pd2MCh2 (M=Fe, Co, Ni; Ch=Se, Te): Crystal structure and topology of the electron density. J. Solid State Chem. 2007, 180, 191–197. [Google Scholar] [CrossRef]
- Gholivand, K.; Mahzouni, H.R.; Esrafili, M.D. Structure, bonding, electronic and energy aspects of a new family of early lanthanide (La, Ce and Nd) complexes with phosphoric triamides: Insights from experimental and DFT studies. Dalton Trans. 2012, 41, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Dindar, S.; Nemati Kharat, A.; Safarkoopayeh, B.; Abbasi, A. Ruthenium(II)β-diketimine as hydroamination catalyst, crystal structure and DFT computations. Transit. Met. Chem. 2021, 46, 403–413. [Google Scholar] [CrossRef]
- Zahirović, A.; Roca, S.; Kahrović, E.; Višnjevac, A. Low DNA and high BSA binding affinity of cationic ruthenium(II) organometallic featuring pyridine and 2′-hydroxychalcone ligands. J. Mol. Struct. 2021, 1236, 130326. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Ramon Spectra of Inorganic and Coordination Compounds; Wiley Interscience: New York, NY, USA, 1997. [Google Scholar]
- Turan, N.; Buldurun, K. Synthesis, characterization and antioxidant activity of Schiff base and its metal complexes with Fe(II), Mn(II), Zn(II), and Ru(II) ions: Catalytic activity of ruthenium(II) complex. Eur. J. Chem. 2018, 9, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Durig, J.R.; McAllister, W.A.; Mercer, E.E. On the I.R. spectra of the isomers of trichlorotriaquoruthenium(III). J. Inorg. Nucl. Chem. 1967, 29, 1441–1447. [Google Scholar] [CrossRef]
- Arlt, S.; Petković, V.; Ludwig, G.; Eichhorn, T.; Lang, H.; Rüffer, T.; Mijatović, S.; Maksimović-Ivanić, D.; Kaluđerović, G.N. Arene Ruthenium(II) Complexes Bearing the κ-P or κ-P, κ-S Ph2P(CH2)3SPh Ligand. Molecules 2021, 26, 1860. [Google Scholar] [CrossRef] [PubMed]
- Jeremias, L.; Novotný, J.; Repisky, M.; Komorovsky, S.; Marek, R. Interplay of Through-Bond Hyperfine and Substituent Effects on the NMR Chemical Shifts in Ru(III) Complexes. Inorg. Chem. 2018, 57, 8748–8759. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.D.; Truhlar, D.G. Performance of Density Functional Theory and Møller–Plesset Second-Order Perturbation Theory for Structural Parameters in Complexes of Ru. J. Chem. Theory Comput. 2011, 7, 2325–2332. [Google Scholar] [CrossRef]
- Novotný, J.; Přichystal, D.; Sojka, M.; Komorovsky, S.; Nečas, M.; Marek, R. Hyperfine Effects in Ligand NMR: Paramagnetic Ru(III) Complexes with 3-Substituted Pyridines. Inorg. Chem. 2018, 57, 641–652. [Google Scholar] [CrossRef]
- Novotný, J.; Sojka, M.; Komorovsky, S.; Nečas, M.; Marek, R. Interpreting the Paramagnetic NMR Spectra of Potential Ru(III) Metallodrugs: Synergy between Experiment and Relativistic DFT Calculations. J. Am. Chem. Soc. 2016, 138, 8432–8445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahirović, A.; Žilić, D.; Pavelić, S.K.; Hukić, M.; Muratović, S.; Harej, A.; Kahrović, E. Type of complex–BSA binding forces affected by different coordination modes of alliin in novel water-soluble ruthenium complexes. New J. Chem. 2019, 43, 5791–5804. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Huang, S.; Liu, Y.; Tian, F.; Zhu, J. Thermodynamics, Conformation and Active Sites of the Binding of Zn–Nd Hetero-bimetallic Schiff Base to Bovine Serum Albumin. J. Fluoresc. 2009, 19, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Devagi, G.; Dallemer, F.; Kalaivani, P.; Prabhakaran, R. Organometallic ruthenium(II) complexes containing NS donor Schiff bases: Synthesis, structure, electrochemistry, DNA/BSA binding, DNA cleavage, radical scavenging and antibacterial activities. J. Organomet. Chem. 2018, 854, 1–14. [Google Scholar] [CrossRef]
- Khan, T.A.; Bhar, K.; Thirumoorthi, R.; Roy, T.K.; Sharma, A.K. Design, synthesis, characterization and evaluation of the anticancer activity of water-soluble half-sandwich ruthenium(II) arene halido complexes. New J. Chem. 2020, 44, 239–257. [Google Scholar] [CrossRef]
- Kamatchi, T.S.; Kalaivani, P.; Poornima, P.; Padma, V.V.; Fronczek, F.R.; Natarajan, K. New organometallic ruthenium( II) complexes containing chelidonic acid (4-oxo-4H-pyran-2,6-dicarboxylic acid): Synthesis, structure and in vitro biological activity. RSC Adv. 2014, 4, 2004–2022. [Google Scholar] [CrossRef]
- Bujacz, A.; Zielinski, K.; Sekula, B. Structural studies of bovine, equine, and leporine serum albumin complexes with naproxen. Proteins Struct. Funct. Bioinform. 2014, 82, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- Foti, M.C. Use and Abuse of the DPPH• Radical. J. Agric. Food Chem. 2015, 63, 8765–8776. [Google Scholar] [CrossRef]
- Sârbu, C.; Casoni, D. Comprehensive evaluation of biogenic amines and related drugs’ antiradical activity using reactive 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical. Open Chem. 2013, 11, 679–688. [Google Scholar] [CrossRef]
- Foti, M.C.; Daquino, C.; Mackie, I.D.; DiLabio, G.A.; Ingold, K.U. Reaction of Phenols with the 2,2-Diphenyl-1-picrylhydrazyl Radical. Kinetics and DFT Calculations Applied To Determine ArO-H Bond Dissociation Enthalpies and Reaction Mechanism. J. Org. Chem. 2008, 73, 9270–9282. [Google Scholar] [CrossRef]
- Foti, M.C.; Daquino, C.; DiLabio, G.A.; Ingold, K.U. Kinetics of the Oxidation of Quercetin by 2,2-Diphenyl-1-picrylhydrazyl (dpph•). Org. Lett. 2011, 13, 4826–4829. [Google Scholar] [CrossRef] [Green Version]
- Foti, M.C.; Daquino, C.; Geraci, C. Electron-Transfer Reaction of Cinnamic Acids and Their Methyl Esters with the DPPH• Radical in Alcoholic Solutions. J. Org. Chem. 2004, 69, 2309–2314. [Google Scholar] [CrossRef] [PubMed]
- Dimić, D.; Milenković, D.; Marković, J.D.; Marković, Z. Antiradical activity of catecholamines and metabolites of dopamine: Theoretical and experimental study. Phys. Chem. Chem. Phys. 2017, 128, 16655–16663. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Mohanty, B.P.; Saini, G.S.S. Structure, spectra and antioxidant action of ascorbic acid studied by density functional theory, Raman spectroscopic and nuclear magnetic resonance techniques. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2016, 155, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.N.; Shukla, M.K.; Reed, D.; Leszczynski, J. Ab initio study of the structural properties of ascorbic acid (vitamin C). Int. J. Quantum Chem. 2006, 106, 2934–2943. [Google Scholar] [CrossRef]
- Milenković, D.A.; Dimić, D.S.; Avdović, E.H.; Amić, A.D.; Marković, J.M.D.; Marković, Z.S. Advanced oxidation process of coumarins by hydroxyl radical: Towards the new mechanism leading to less toxic products. Chem. Eng. J. 2020, 395, 124971. [Google Scholar] [CrossRef]
- Smolko, L.; Smolková, R.; Samoľová, E.; Morgan, I.; Saoud, M.; Kaluđerović, G.N. Two isostructural Co(II) flufenamato and niflumato complexes with bathocuproine: Analogues with a different cytotoxic activity. J. Inorg. Biochem. 2020, 210, 111160. [Google Scholar] [CrossRef]
- CrysAlisPRO; Oxford Diffraction/Agilent Technologies UK Ltd.: Yarnton, UK, 2017.
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; University of Western Australia: Perth, Australia, 2017. [Google Scholar]
- Shobana, D.; Sudha, S.; Ramarajan, D.; Dimić, D. Synthesis, crystal structure, spectral characterization and Hirshfeld surface analysis of (E)-N′-(3-ethoxy-4-hydroxybenzylidene)-4-fluorobenzohydrazide single-crystal—A novel NLO active material. J. Mol. Struct. 2022, 1250, 131856. [Google Scholar] [CrossRef]
- Spackman, M.A.; Byrom, P.G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 215–220. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitale. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Zieliński, R.; Szymusiak, H. Application of Dft B3Lyp/Giao and B3Lyp/Csgt Methods for Interpretation of Nmr Spectra of Flavonoids. Pol. J. Food Nutr. Sci. 2003, 12, 157–162. [Google Scholar]
- Bohmann, J.A.; Weinhold, F.; Farrar, T.C. Natural chemical shielding analysis of nuclear magnetic resonance shielding tensors from gauge-including atomic orbital calculations. J. Chem. Phys. 1997, 107, 1173. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Keith, T.A. TK Gristmill Software; (Version 19.10.12); AIMAll: Overland Park, KS, USA, 2019. [Google Scholar]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Dimić, D.; Petković, M. Control of a photoswitching chelator by metal ions: DFT, NBO, and QTAIM analysis. Int. J. Quantum Chem. 2016, 116, 27–34. [Google Scholar] [CrossRef]
- Nenadis, N.; Tsimidou, M. Observations on the estimation of scavenging activity of phenolic compounds using rapid 1, 1-diphenyl-2-picrylhydrazyl (DPPH•) tests. J. Am. Oil Chem. Soc. 2002, 79, 1191–1195. [Google Scholar] [CrossRef]
- Krajnović, T.; Pantelić, N.Đ.; Wolf, K.; Eichhorn, T.; Maksimović-Ivanić, D.; Mijatović, S.; Wessjohann, L.A.; Kaluđerović, G.N. Anticancer Potential of Xanthohumol and Isoxanthohumol Loaded into SBA-15 Mesoporous Silica Particles against B16F10 Melanoma Cells. Materials 2022, 15, 5028. [Google Scholar] [CrossRef] [PubMed]
- Morgan, I.; Wessjohann, L.A.; Kaluđerović, G.N. In Vitro Anticancer Screening and Preliminary Mechanistic Study of A-Ring Substituted Anthraquinone Derivatives. Cells 2022, 11, 168. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BIOVIA. Dassault Systèmes, Discovery Studio 2016; Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Rosselli, S.; Maggio, A.M.; Faraone, N.; Spadaro, V.; Morris-Natschke, S.L.; Bastow, K.F.; Lee, K.-H.; Bruno, M. The cytotoxic properties of natural coumarins isolated from roots of Ferulago campestris (Apiaceae) and of synthetic ester derivatives of aegelinol. Nat. Prod. Commun. 2009, 4, 1701–1706. [Google Scholar] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical formula | C32H46Cl3F6N2O2PRu |
| Formula weight [g mol−1] | 944.17 |
| Temperature [K] | 100 |
| Crystal System | monoclinic |
| Space group | I2/a |
| Radiation/Wavelength [Å] | Mo Ka/0.71073 |
| Unit cell dimension [Å, deg] | a = 21.3209(5), α = 90° b = 10.5848(2), β = 99.702(2)° c = 33.3109(8), γ = 90 |
| Volume | 7410.0(3) Å3 |
| Calculated density | 1.691 Mg m−3 |
| Z | 8 |
| Θ range [deg] | 3.116–29.015 |
| Reflections | |
| Measured: | 41,874 |
| Independent: | 8907 |
| Observed [I > 2s(I)]: | 7696 |
| Rint | 0.0411 |
| R1 [I > 2s(I)], R1 (all) | 0.0354, 0.0442 |
| wR2 [I > 2s(I)], wR2 (all) | 0.0827, 0.0883 |
| Goodness-of-fit on F2 | 1.037 |
| Largest diff. peak/hole (e Å−3) | 1.032/−0.720 |
| CCDC no. | 2217194 |
| D−H...Acceptor | D...A | D−H...A |
| Intramolecular Type | ||
| N2−H3N...Cl1 | 3.073 | 138 |
| N1−H1N...Cl3 | 3.144 | 126 |
| Intermolecular Type | ||
| N1−H1N...F5 | 3.322 | 167 |
| O2M−H2M...O1M | 2.801 | 156 |
| N1−H2N...O2M | 2.826 | 156 |
| T [K] | Kb [M−1] | ΔHb [kJ mol−1] | ΔSb [J mol−1 K−1] | ΔGb [kJ mol−1] | R |
|---|---|---|---|---|---|
| 303 | 9.90 × 104 | 189 | 719 | −29 | 0.998 |
| 306 | 2.03 × 105 | −31 | |||
| 310 | 5.37 × 105 | −34 |
| ΔGbind (kJ mol−1) | ΔGvdw+hbond+desolv (kJ mol−1) | ΔGelec (kJ mol−1) | ΔGtotal (kJ mol−1) | ΔGtor (kJ mol−1) | ΔGunb (kJ mol−1) | LE | |
|---|---|---|---|---|---|---|---|
| NPS-BSA | −21.3 | −24.5 | 0.3 | −1.5 | 2.8 | −1.5 | −0.3 |
| 1-BSA | −36.8 | −26.3 | −15.3 | −7.7 | 4.8 | −7.7 | −0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eichhorn, T.; Kolbe, F.; Mišić, S.; Dimić, D.; Morgan, I.; Saoud, M.; Milenković, D.; Marković, Z.; Rüffer, T.; Dimitrić Marković, J.; et al. Synthesis, Crystallographic Structure, Theoretical Analysis, Molecular Docking Studies, and Biological Activity Evaluation of Binuclear Ru(II)-1-Naphthylhydrazine Complex. Int. J. Mol. Sci. 2023, 24, 689. https://doi.org/10.3390/ijms24010689
Eichhorn T, Kolbe F, Mišić S, Dimić D, Morgan I, Saoud M, Milenković D, Marković Z, Rüffer T, Dimitrić Marković J, et al. Synthesis, Crystallographic Structure, Theoretical Analysis, Molecular Docking Studies, and Biological Activity Evaluation of Binuclear Ru(II)-1-Naphthylhydrazine Complex. International Journal of Molecular Sciences. 2023; 24(1):689. https://doi.org/10.3390/ijms24010689
Chicago/Turabian StyleEichhorn, Thomas, Franz Kolbe, Stefan Mišić, Dušan Dimić, Ibrahim Morgan, Mohamad Saoud, Dejan Milenković, Zoran Marković, Tobias Rüffer, Jasmina Dimitrić Marković, and et al. 2023. "Synthesis, Crystallographic Structure, Theoretical Analysis, Molecular Docking Studies, and Biological Activity Evaluation of Binuclear Ru(II)-1-Naphthylhydrazine Complex" International Journal of Molecular Sciences 24, no. 1: 689. https://doi.org/10.3390/ijms24010689