
Invadosome Formation by Lung Fibroblasts in Idiopathic Pulmonary Fibrosis
 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Several Key Genes of Invadosome Formation Are Upregulated in IPF Lung Tissue Samples and in Fibroblastic Foci Areas
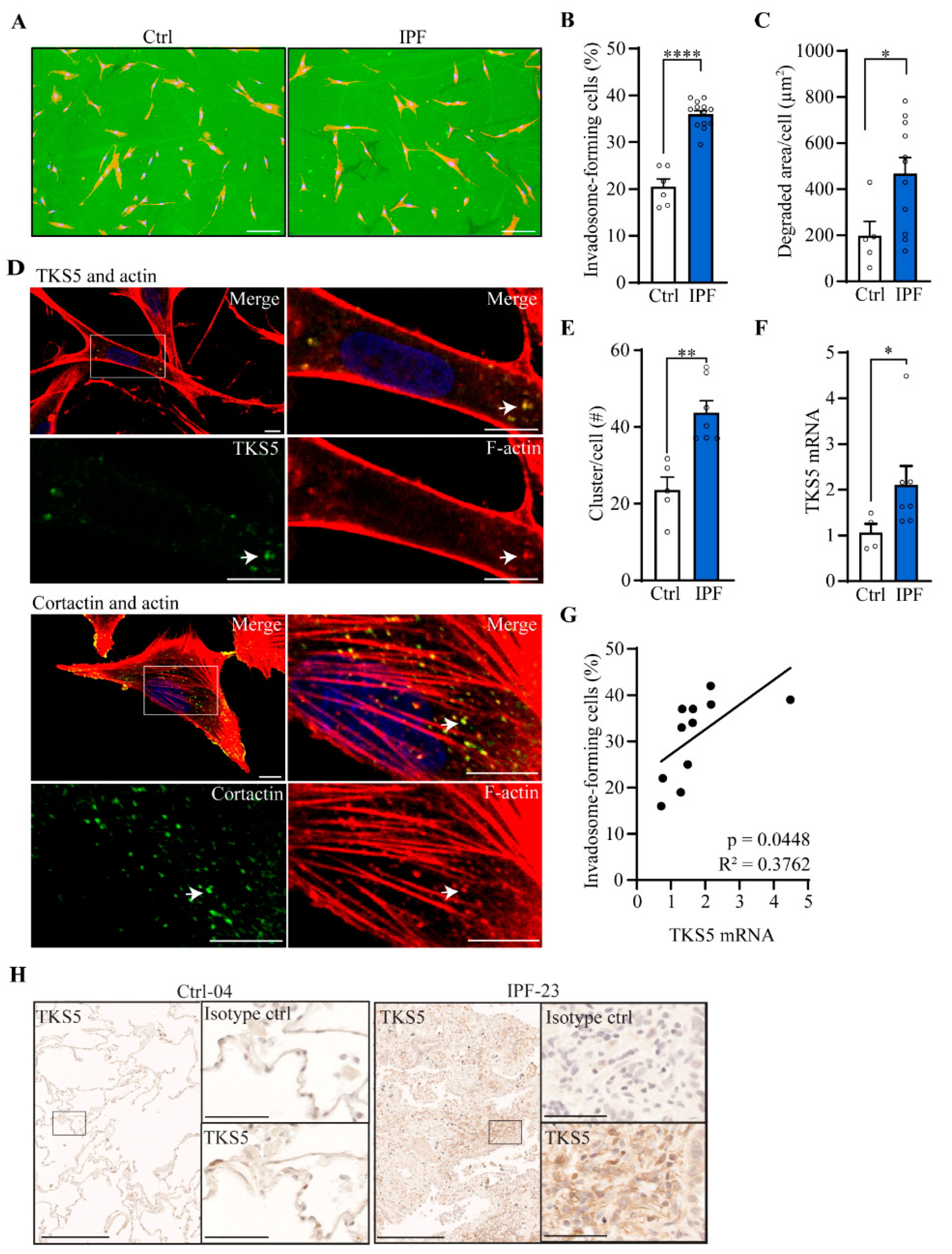
2.2. Invadosome Formation Is Increased in Lung Fibroblasts Isolated from IPF Patients and Fibrotic Mice
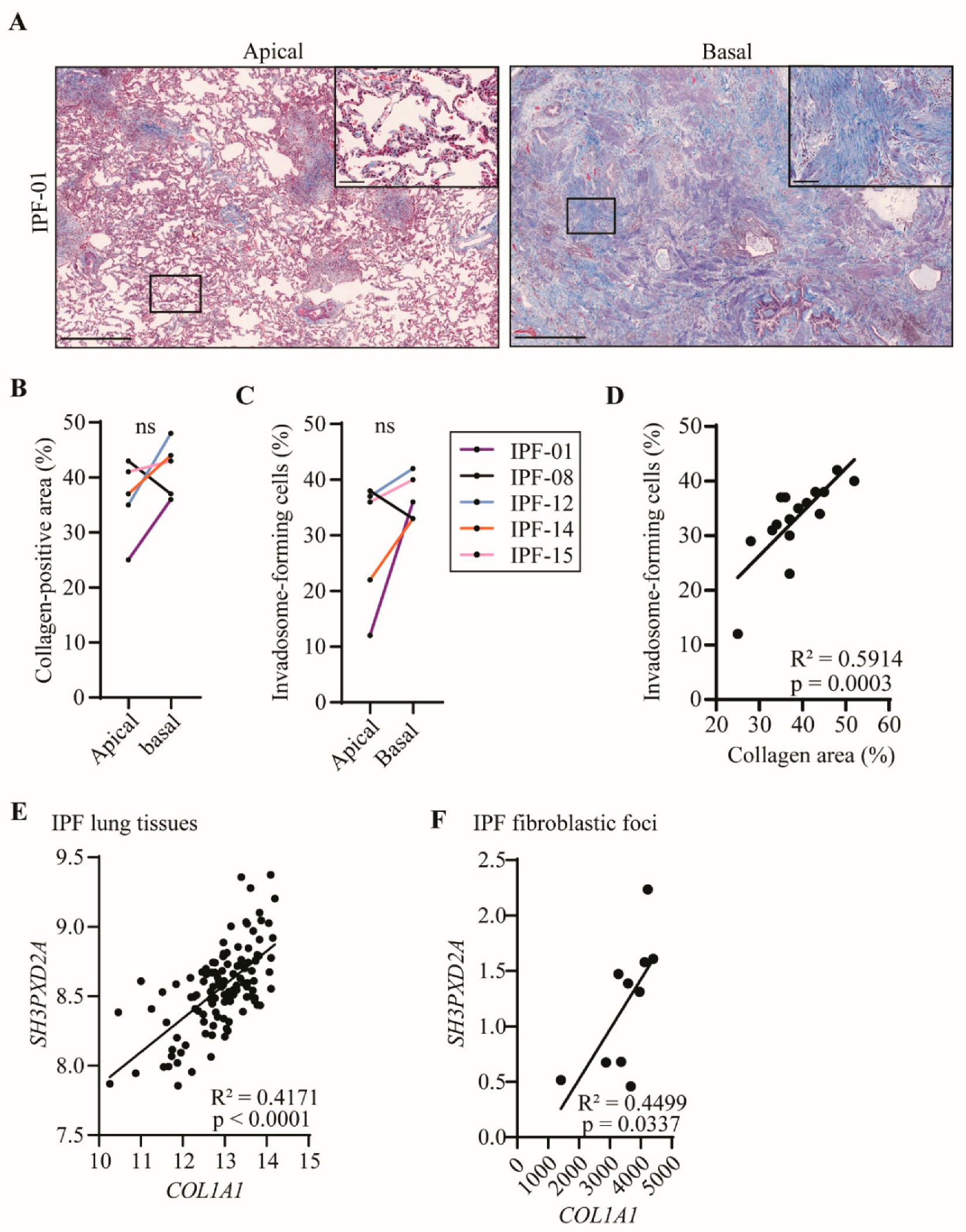
2.3. Fibroblast Invadosome Formation Correlates Positively with the Collagen Content of Neighbouring Tissue
2.4. Nintedanib and Pirfenidone Inhibit Invadosome Formation by Lung Fibroblasts from IPF Patients
2.5. Nintedanib Reduces the Expression of TKS5 and p-Akt in IPF Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Lung Fibroblast Isolation and Culture
4.2. Invadosome Formation Assessment
4.3. Lung Histology and Collagen Quantification
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple Angiokinase Inhibitor with Sustained Receptor Blockade and Good Antitumor Efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef] [Green Version]
- Wollin, L.; Maillet, I.; Quesniaux, V.; Holweg, A.; Ryffel, B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J. Pharmacol. Exp. Ther. 2014, 349, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Hough, K.P.; Curtiss, M.L.; Blain, T.J.; Liu, R.M.; Trevor, J.; Deshane, J.S.; Thannickal, V.J. Airway Remodeling in Asthma. Front. Med. 2020, 7, 191. [Google Scholar] [CrossRef]
- Selman, M.; Ruiz, V.; Cabrera, S.; Segura, L.; Ramirez, R.; Barrios, R.; Pardo, A. TIMP-1, -2, -3, and -4 in idiopathic pulmonary fibrosis. A prevailing nondegradative lung microenvironment? Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L562–L574. [Google Scholar] [CrossRef]
- Tjin, G.; White, E.S.; Faiz, A.; Sicard, D.; Tschumperlin, D.J.; Mahar, A.; Kable, E.P.W.; Burgess, J.K. Lysyl oxidases regulate fibrillar collagen remodelling in idiopathic pulmonary fibrosis. Dis. Model Mech. 2017, 10, 1301–1312. [Google Scholar] [CrossRef] [Green Version]
- Parker, M.W.; Rossi, D.; Peterson, M.; Smith, K.; Sikstrom, K.; White, E.S.; Connett, J.E.; Henke, C.A.; Larsson, O.; Bitterman, P.B. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J. Clin. Investig. 2014, 124, 1622–1635. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Qu, J.; Huang, X.; Kurundkar, A.; Zhu, L.; Yang, N.; Venado, A.; Ding, Q.; Liu, G.; Antony, V.B.; et al. Mechanosensing by the α6-integrin confers an invasive fibroblast phenotype and mediates lung fibrosis. Nat. Commun. 2016, 7, 12564. [Google Scholar] [CrossRef]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinkovic, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Pardo, A.; Cabrera, S.; Maldonado, M.; Selman, M. Role of matrix metalloproteinases in the pathogenesis of idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarone, G.; Cirillo, D.; Giancotti, F.G.; Comoglio, P.M.; Marchisio, P.C. Rous sarcoma virus-transformed fibroblasts adhere primarily at discrete protrusions of the ventral membrane called podosomes. Exp. Cell Res. 1985, 159, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Leong, H.S.; Robertson, A.E.; Stoletov, K.; Leith, S.J.; Chin, C.A.; Chien, A.E.; Hague, M.N.; Ablack, A.; Carmine-Simmen, K.; McPherson, V.A.; et al. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 2014, 8, 1558–1570. [Google Scholar] [CrossRef] [Green Version]
- Seals, D.F.; Azucena, E.F., Jr.; Pass, I.; Tesfay, L.; Gordon, R.; Woodrow, M.; Resau, J.H.; Courtneidge, S.A. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell 2005, 7, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Saini, P.; Courtneidge, S.A. Tks adaptor proteins at a glance. J. Cell Sci. 2018, 131, jcs203661. [Google Scholar] [CrossRef] [Green Version]
- Schoumacher, M.; Goldman, R.D.; Louvard, D.; Vignjevic, D.M. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J. Cell Biol. 2010, 189, 541–556. [Google Scholar] [CrossRef] [Green Version]
- Revach, O.Y.; Geiger, B. The interplay between the proteolytic, invasive, and adhesive domains of invadopodia and their roles in cancer invasion. Cell Adhes. Migr. 2014, 8, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Burns, S.; Thrasher, A.J.; Blundell, M.P.; Machesky, L.; Jones, G.E. Configuration of human dendritic cell cytoskeleton by Rho GTPases, the WAS protein, and differentiation. Blood 2001, 98, 1142–1149. [Google Scholar] [CrossRef]
- Lauzier, A.; Charbonneau, M.; Harper, K.; Jilaveanu-Pelmus, M.; Dubois, C.M. Formation of invadopodia-like structures by synovial cells promotes cartilage breakdown in collagen-induced arthritis: Involvement of the protein tyrosine kinase Src. Arthritis Rheum. 2011, 63, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Veillat, V.; Spuul, P.; Daubon, T.; Egana, I.; Kramer, I.; Genot, E. Podosomes: Multipurpose organelles? Int. J. Biochem. Cell Biol. 2015, 65, 52–60. [Google Scholar] [CrossRef]
- Paterson, E.K.; Courtneidge, S.A. Invadosomes are coming: New insights into function and disease relevance. FEBS J. 2018, 285, 8–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charbonneau, M.; Lavoie, R.R.; Lauzier, A.; Harper, K.; McDonald, P.P.; Dubois, C.M. Platelet-Derived Growth Factor Receptor Activation Promotes the Prodestructive Invadosome-Forming Phenotype of Synoviocytes from Patients with Rheumatoid Arthritis. J. Immunol. 2016, 196, 3264–3275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninio, L.; Nissani, A.; Meirson, T.; Domovitz, T.; Genna, A.; Twafra, S.; Srikanth, K.D.; Dabour, R.; Avraham, E.; Davidovich, A.; et al. Hepatitis C Virus Enhances the Invasiveness of Hepatocellular Carcinoma via EGFR-Mediated Invadopodia Formation and Activation. Cells 2019, 8, 1395. [Google Scholar] [CrossRef] [Green Version]
- Murphy, D.A.; Courtneidge, S.A. The ‘ins’ and ‘outs’ of podosomes and invadopodia: Characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 2011, 12, 413–426. [Google Scholar] [CrossRef] [Green Version]
- Martinet, Y.; Rom, W.N.; Grotendorst, G.R.; Martin, G.R.; Crystal, R.G. Exaggerated spontaneous release of platelet-derived growth factor by alveolar macrophages from patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 1987, 317, 202–209. [Google Scholar] [CrossRef]
- Kelley, L.C.; Ammer, A.G.; Hayes, K.E.; Martin, K.H.; Machida, K.; Jia, L.; Mayer, B.J.; Weed, S.A. Oncogenic Src requires a wild-type counterpart to regulate invadopodia maturation. J. Cell Sci. 2010, 123, 3923–3932. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, A.G.; Fulford, L.G.; Colby, T.V.; du Bois, R.M.; Hansell, D.M.; Wells, A.U. The Relationship between Individual Histologic Features and Disease Progression in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2002, 166, 173–177. [Google Scholar] [CrossRef]
- Moeller, A.; Gilpin, S.E.; Ask, K.; Cox, G.; Cook, D.; Gauldie, J.; Margetts, P.J.; Farkas, L.; Dobranowski, J.; Boylan, C.; et al. Circulating Fibrocytes Are an Indicator of Poor Prognosis in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 588–594. [Google Scholar] [CrossRef]
- Sava, P.; Ramanathan, A.; Dobronyi, A.; Peng, X.; Sun, H.; Ledesma-Mendoza, A.; Herzog, E.L.; Gonzalez, A.L. Human pericytes adopt myofibroblast properties in the microenvironment of the IPF lung. JCI Insight 2017, 2, e96352. [Google Scholar] [CrossRef] [PubMed]
- Habiel, D.M.; Hogaboam, C.M. Heterogeneity of Fibroblasts and Myofibroblasts in Pulmonary Fibrosis. Curr. Pathobiol. Rep. 2017, 5, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, N.; Grasberger, P.E.; Mugo, B.M.; Feghali-Bostwick, C.; Pardo, A.; Selman, M.; Lagares, D.; Tager, A.M. Fibrogenic Lung Injury Induces Non-Cell-Autonomous Fibroblast Invasion. Am. J. Respir. Cell Mol. Biol. 2016, 54, 831–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirillov, V.; Siler, J.T.; Ramadass, M.; Ge, L.; Davis, J.; Grant, G.; Nathan, S.D.; Jarai, G.; Trujillo, G. Sustained activation of toll-like receptor 9 induces an invasive phenotype in lung fibroblasts: Possible implications in idiopathic pulmonary fibrosis. Am. J. Pathol. 2015, 185, 943–957. [Google Scholar] [CrossRef]
- Chilosi, M.; Zamò, A.; Doglioni, C.; Reghellin, D.; Lestani, M.; Montagna, L.; Pedron, S.; Ennas, M.G.; Cancellieri, A.; Murer, B.; et al. Migratory marker expression in fibroblast foci of idiopathic pulmonary fibrosis. Respir. Res. 2006, 7, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moodley, Y.P.; Caterina, P.; Scaffidi, A.K.; Misso, N.L.; Papadimitriou, J.M.; McAnulty, R.J.; Laurent, G.J.; Thompson, P.J.; Knight, D.A. Comparison of the morphological and biochemical changes in normal human lung fibroblasts and fibroblasts derived from lungs of patients with idiopathic pulmonary fibrosis during FasL-induced apoptosis. J. Pathol. 2004, 202, 486–495. [Google Scholar] [CrossRef]
- Suga, M.; Iyonaga, K.; Okamoto, T.; Gushima, Y.; Miyakawa, H.; Akaike, T.; Ando, M. Characteristic elevation of matrix metalloproteinase activity in idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2000, 162, 1949–1956. [Google Scholar] [CrossRef] [Green Version]
- Garcia-de-Alba, C.; Becerril, C.; Ruiz, V.; Gonzalez, Y.; Reyes, S.; Garcia-Alvarez, J.; Selman, M.; Pardo, A. Expression of matrix metalloproteases by fibrocytes: Possible role in migration and homing. Am. J. Respir. Crit. Care Med. 2010, 182, 1144–1152. [Google Scholar] [CrossRef]
- Rowe, R.G.; Keena, D.; Sabeh, F.; Willis, A.L.; Weiss, S.J. Pulmonary fibroblasts mobilize the membrane-tethered matrix metalloprotease, MT1-MMP, to destructively remodel and invade interstitial type I collagen barriers. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L683–L692. [Google Scholar] [CrossRef] [Green Version]
- Courtneidge, S.A. Cell migration and invasion in human disease: The Tks adaptor proteins. Biochem. Soc. Trans. 2012, 40, 129–132. [Google Scholar] [CrossRef]
- Mrkonjic, S.; Destaing, O.; Albiges-Rizo, C. Mechanotransduction pulls the strings of matrix degradation at invadosome. Matrix Biol. 2017, 57–58, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Masi, I.; Caprara, V.; Bagnato, A.; Rosano, L. Tumor Cellular and Microenvironmental Cues Controlling Invadopodia Formation. Front. Cell Dev. Biol. 2020, 8, 584181. [Google Scholar] [CrossRef] [PubMed]
- Kolb, M.; Bonella, F.; Wollin, L. Therapeutic targets in idiopathic pulmonary fibrosis. Respir. Med. 2017, 131, 49–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiler, C.; Davuluri, G.; Abrams, J.; Byfield, F.J.; Janmey, P.A.; Pack, M. Smooth muscle tension induces invasive remodeling of the zebrafish intestine. PLoS Biol. 2012, 10, e1001386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artym, V.V.; Swatkoski, S.; Matsumoto, K.; Campbell, C.B.; Petrie, R.J.; Dimitriadis, E.K.; Li, X.; Mueller, S.C.; Bugge, T.H.; Gucek, M.; et al. Dense fibrillar collagen is a potent inducer of invadopodia via a specific signaling network. J. Cell Biol. 2015, 208, 331–350. [Google Scholar] [CrossRef]
- Asano, S.; Ito, S.; Takahashi, K.; Furuya, K.; Kondo, M.; Sokabe, M.; Hasegawa, Y. Matrix stiffness regulates migration of human lung fibroblasts. Physiol. Rep. 2017, 5, e13281. [Google Scholar] [CrossRef]
- Juin, A.; Planus, E.; Guillemot, F.; Horakova, P.; Albiges-Rizo, C.; Genot, E.; Rosenbaum, J.; Moreau, V.; Saltel, F. Extracellular matrix rigidity controls podosome induction in microvascular endothelial cells. Biol. Cell 2013, 105, 46–57. [Google Scholar] [CrossRef]
- Asmani, M.; Velumani, S.; Li, Y.; Wawrzyniak, N.; Hsia, I.; Chen, Z.; Hinz, B.; Zhao, R. Fibrotic microtissue array to predict anti-fibrosis drug efficacy. Nat. Commun. 2018, 9, 2066. [Google Scholar] [CrossRef] [Green Version]
- Ogura, T.; Taniguchi, H.; Azuma, A.; Inoue, Y.; Kondoh, Y.; Hasegawa, Y.; Bando, M.; Abe, S.; Mochizuki, Y.; Chida, K.; et al. Safety and pharmacokinetics of nintedanib and pirfenidone in idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1382–1392. [Google Scholar] [CrossRef] [Green Version]
- Kishi, M.; Aono, Y.; Sato, S.; Koyama, K.; Azuma, M.; Abe, S.; Kawano, H.; Kishi, J.; Toyoda, Y.; Okazaki, H.; et al. Blockade of platelet-derived growth factor receptor-β, not receptor-α ameliorates bleomycin-induced pulmonary fibrosis in mice. PLoS ONE 2018, 13, e0209786. [Google Scholar] [CrossRef]
- Chuang, Y.; Xu, X.; Kwiatkowska, A.; Tsapraillis, G.; Hwang, H.; Petritis, K.; Flynn, D.; Symons, M. Regulation of synaptojanin 2 5’-phosphatase activity by Src. Cell Adhes. Migr. 2012, 6, 518–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abram, C.L.; Seals, D.F.; Pass, I.; Salinsky, D.; Maurer, L.; Roth, T.M.; Courtneidge, S.A. The Adaptor Protein Fish Associates with Members of the ADAMs Family and Localizes to Podosomes of Src-transformed Cells. J. Biol. Chem. 2003, 278, 16844–16851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldassarre, M.; Pompeo, A.; Beznoussenko, G.; Castaldi, C.; Cortellino, S.; McNiven, M.A.; Luini, A.; Buccione, R. Dynamin participates in focal extracellular matrix degradation by invasive cells. Mol. Biol. Cell 2003, 14, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, Q.; Xu, C.-B. A convenient method for quantifying collagen fibers in atherosclerotic lesions by ImageJ software. Int. J. Clin. Exp. Med. 2017, 10, 14904–14910. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Criteria | Healthy Donors (n = 8) | IPF Patients (n = 16) |
|---|---|---|
| Age (mean years ± SD) | 56 ± 18 | 64 ± 8 |
| Sex | ||
| Male (%) | 75 | 81 |
| Female (%) | 25 | 19 |
| Smoking status | ||
| Ever smoker (%) | 37.5 | 56 |
| Never smoker (%) | 0 | 25 |
| Unknown (%) | 62.5 | 19 |
| Years with IPF disease (mean ± SD) | n/a * | 5.1 ± 3.1 |
| IPF treatment before transplantation | ||
| Nintedanib (%) | 0 | 0 |
| Pirfenidone (%) | 0 | 12.5 |
| FVC (% mean ± SD) | n/a * | 41 ± 12 |
| Target Gene | Primer Sequences |
|---|---|
| SH3PXD2A(TKS5) | Forward: 5′- TGC CAA GAA GGA GAT CAG CC-3′ Reverse: 5′-TGG AGG TCT TGT CCG TAG GT-3′ |
| RPL13 | Forward: 5′-CTC AAG GTC GTG CGT CTG- 3′ Reverse: 5′-TGG CTT TCT CTT TCC TCT TCT C-3′ |
| COL1A1 | Forward: 5′-AAG AGG AAG GCC AAG TCG AG-3′ Reverse: 5′-CAC ACG TCT CGG TCA TGG TA-3′ |
| CTGF | Forward: 5′-AAT GCT GCG AGG AGT GGG T -3′ Reverse: 5′- CGG CTC TAA TCA TAG TTG GGT CT-3′ |
| MMP2 | Forward: 5′-GGC ACC CAT TTA CAC CTA CA -3′ Reverse: 5′-CCA AGG TCA ATG TCA GGA GAG -3′ |
| ADAM12 | Forward: 5′- TCT CAT TGC CAG CAG TTT CAC -3′ Reverse: 5′- CGT GTA ATT TCG AGC GAG GG -3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebel, M.; Cliche, D.O.; Charbonneau, M.; Adam, D.; Brochiero, E.; Dubois, C.M.; Cantin, A.M. Invadosome Formation by Lung Fibroblasts in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2023, 24, 499. https://doi.org/10.3390/ijms24010499
Lebel M, Cliche DO, Charbonneau M, Adam D, Brochiero E, Dubois CM, Cantin AM. Invadosome Formation by Lung Fibroblasts in Idiopathic Pulmonary Fibrosis. International Journal of Molecular Sciences. 2023; 24(1):499. https://doi.org/10.3390/ijms24010499
Chicago/Turabian StyleLebel, Mégane, Dominic O. Cliche, Martine Charbonneau, Damien Adam, Emmanuelle Brochiero, Claire M. Dubois, and André M. Cantin. 2023. "Invadosome Formation by Lung Fibroblasts in Idiopathic Pulmonary Fibrosis" International Journal of Molecular Sciences 24, no. 1: 499. https://doi.org/10.3390/ijms24010499