
Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis

 , and
, and 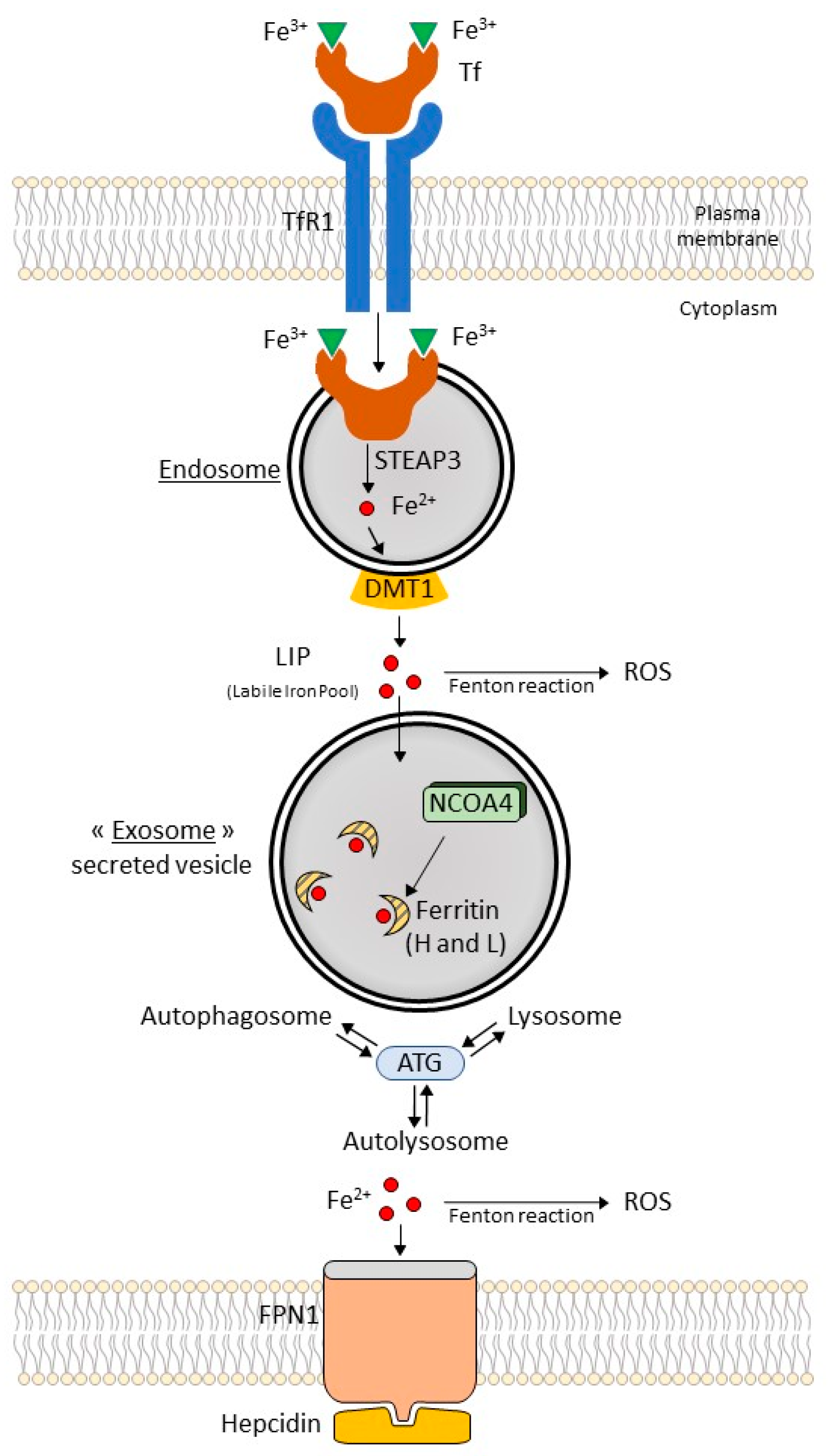{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mechanisms Governing Ferroptosis
2.1. The Process of Lipid Peroxidation
2.2. Steps of Lipid Peroxidation
2.3. Electrophilic Stress and Lipid-Derived Electrophiles
3. Iron Homeostasis
3.1. Iron Metabolism
3.2. Ferritinophagy: A Selective Autophagic Degradation of Ferritin (Figure 1)

4. Regulation of Ferroptosis
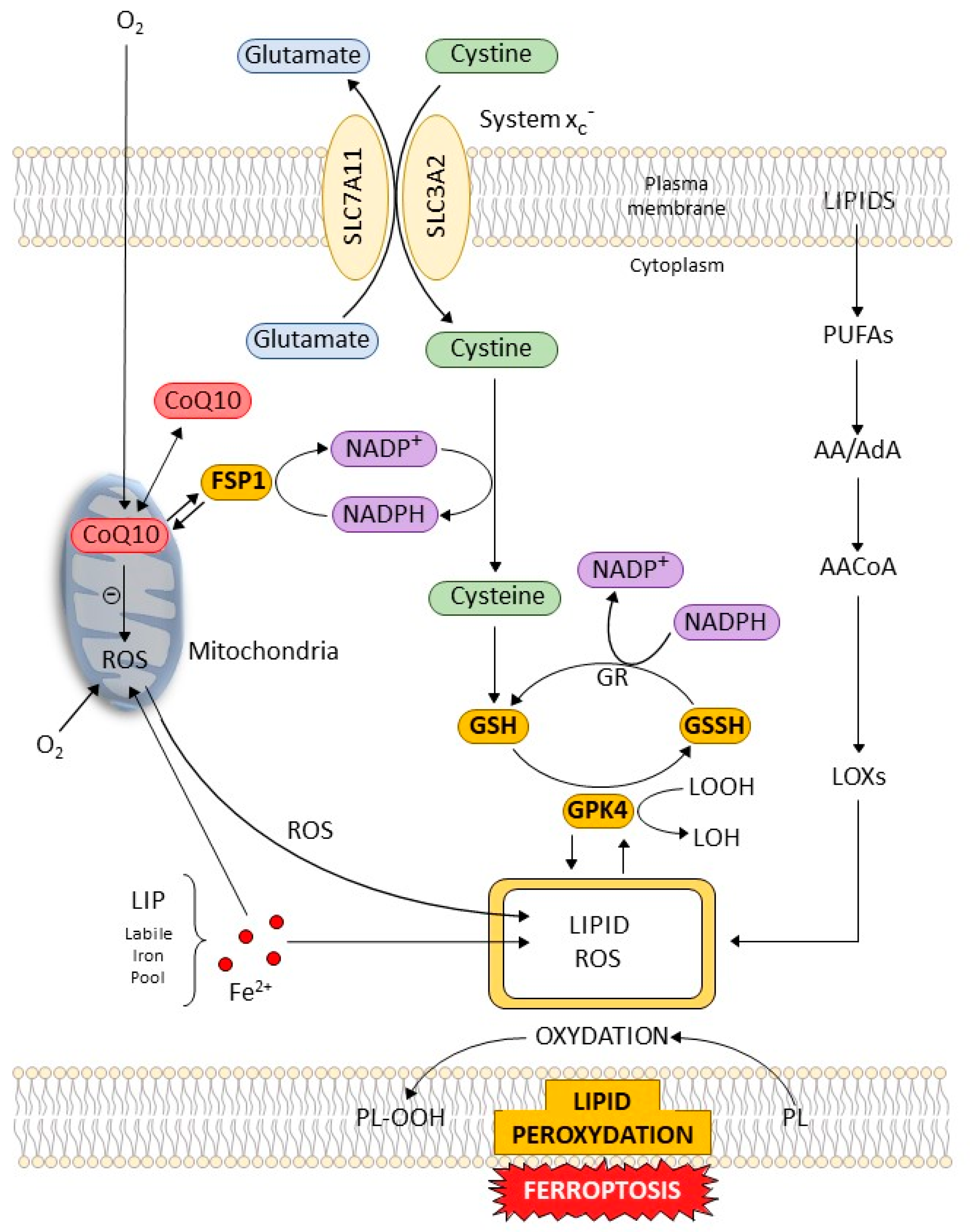
4.1. Cellular Pathways and Control of Ferroptosis (Figure 2)

4.2. Pharmacological Approach of Ferroptosis
4.2.1. Ferroptosis Inducers
4.2.2. Ferroptosis Inhibitors
5. Factors of Modulations in Ferroptosis Process
5.1. Key Role of Mitochondria in Relationship with Iron Homeostasis
5.2. Transforming Growth Factor-Beta (TGF-Beta) Modulations in Ferroptosis Process
5.3. Heme Oxygenase Modulations in Ferroptosis Process
6. Conclusions and Futures Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AA | arachidonic acid |
| ACSL | acyl-CoA synthetase long-chain |
| AdA | adrenic acid |
| AIFM | apoptosis inducing factor mitochondria |
| ARE | antioxidant-response element |
| ATG | autophagy-related proteins |
| CNS | central nervous system |
| CO | carbon monoxide |
| CoQ10 | coenzyme Q10 |
| COX | cyclooxygenase |
| CYP450 | cytochrome P450 |
| DAMP | damage-associated molecular pattern |
| FIN | ferroptosis inducer |
| FIO | free iron overload |
| FPN | ferroportin |
| FSP | ferroptosis suppressor protein |
| FtH | ferritin heavy chain |
| FtL | ferritin light chain |
| GDF | growth differentiation factor |
| GPX4 | glutathione peroxidase 4 |
| GR | glutathione reductase, |
| GSH | glutathione |
| GSSG | oxidized glutathione |
| HN | humanin |
| 4-HNE | 4-hydroxynon-2-enal |
| HO | heme oxygenase |
| HSF | heat shock transcription factor |
| HSP | heat shock protein |
| IMM | inner mitochondrial membrane |
| IRP | iron regulatory proteins |
| Keap Kelch | like ECH-associated protein |
| LDE | lipid-derived electrophile |
| LIP | labile iron pool |
| LOOH | lipid hydroperoxide |
| LOX | lipoxygenase |
| L-ROS | lipid reactive oxygen species |
| MDA | malondialdehyde |
| MDP | mitochondrial-derived peptide |
| Mfrn | mitoferrin |
| NCOA4 | nuclear receptor coactivator 4 |
| NOX | NADPH oxidase |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| PCBP1 | PCD programmed cell death |
| PL | phospholipid |
| PUFA | poly-unsaturated fatty acid |
| RBC | red blood cell |
| RCD | regulated cell death |
| RNS | nitrogen reactive species |
| ROS | reactive oxygen species |
| RSL RAS | selective lethal |
| SLC3A2 | solute carrier family 3 member 2 |
| SLC7A11 | subunit solute carrier family 7 member 11 |
| TBA | thiobarbituric acid |
| Tf | transferrin |
| TfR | transferrin receptor |
| TGF | transforming growth factor |
| YAP1 | Yes-associated protein |
References
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uyeda, A.; Muramatsu, R. Molecular Mechanisms of Central Nervous System Axonal Regeneration and Remyelination: A Review. Int. J. Mol. Sci. 2020, 21, 8116. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, H.N.; Hardman, M.J. Wound healing: Cellular mechanisms and pathological outcomes. Open Biol. 2020, 10, 200223. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 2020, 66, 89–100. [Google Scholar] [CrossRef]
- Hirschhorn, T.; Stockwell, B.R. The development of the concept of ferroptosis. Free Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Jiang, X. The Chemistry and Biology of Ferroptosis. Cell Chem. Biol. 2020, 27, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Vergely, C.; Maupoil, V.; Clermont, G.; Bril, A.; Rochette, L. Identification and quantification of free radicals during myocardial ischemia and reperfusion using electron paramagnetic resonance spectroscopy. Arch. Biochem. Biophys. 2003, 420, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Lorin, J.; Zeller, M.; Guilland, J.C.; Lorgis, L.; Cottin, Y.; Vergely, C. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: Possible therapeutic targets? Pharmacol. Ther. 2013, 140, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, V.N.; Oskolkova, O.V.; Birukov, K.G.; Levonen, A.L.; Binder, C.J.; Stockl, J. Generation and biological activities of oxidized phospholipids. Antioxid. Redox Signal. 2010, 12, 1009–1059. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef]
- Temple, M.D.; Perrone, G.G.; Dawes, I.W. Complex cellular responses to reactive oxygen species. Trends Cell Biol. 2005, 15, 319–326. [Google Scholar] [CrossRef]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Redox Functions of Heme Oxygenase-1 and Biliverdin Reductase in Diabetes. Trends Endocrinol. Metab. 2018, 29, 74–85. [Google Scholar] [CrossRef]
- Gutteridge, J.M.C.; Halliwell, B. Mini-Review: Oxidative stress, redox stress or redox success? Biochem. Biophys. Res. Commun. 2018, 502, 183–186. [Google Scholar] [CrossRef]
- Krysko, O.; Love Aaes, T.; Bachert, C.; Vandenabeele, P.; Krysko, D.V. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013, 4, e631. [Google Scholar] [CrossRef] [Green Version]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Muller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kossl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trostchansky, A.; Wood, I.; Rubbo, H. Regulation of arachidonic acid oxidation and metabolism by lipid electrophiles. Prostaglandins Other Lipid Mediat. 2021, 152, 106482. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Tatou, E.; Maupoil, V.; Zeller, M.; Cottin, Y.; Jazayeri, S.; Brenot, R.; Girard, C.; David, M.; Vergely, C. Atrial and vascular oxidative stress in patients with heart failure. Cell Physiol. Biochem. 2011, 27, 497–502. [Google Scholar] [CrossRef]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [Green Version]
- Milne, G.L.; Yin, H.; Morrow, J.D. Human biochemistry of the isoprostane pathway. J. Biol. Chem. 2008, 283, 15533–15537. [Google Scholar] [CrossRef] [Green Version]
- Richard, C.; Ghibu, S.; Delemasure-Chalumeau, S.; Guilland, J.C.; Des Rosiers, C.; Zeller, M.; Cottin, Y.; Rochette, L.; Vergely, C. Oxidative stress and myocardial gene alterations associated with Doxorubicin-induced cardiotoxicity in rats persist for 2 months after treatment cessation. J. Pharmacol. Exp. Ther. 2011, 339, 807–814. [Google Scholar] [CrossRef] [Green Version]
- Ghibu, S.; Delemasure, S.; Richard, C.; Guilland, J.C.; Martin, L.; Gambert, S.; Rochette, L.; Vergely, C. General oxidative stress during doxorubicin-induced cardiotoxicity in rats: Absence of cardioprotection and low antioxidant efficiency of alpha-lipoic acid. Biochimie 2012, 94, 932–939. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Li, J.; Zhang, Y.; Chang, Y.Z. Cellular Iron Metabolism and Regulation. Adv. Exp. Med. Biol. 2019, 1173, 21–32. [Google Scholar] [CrossRef]
- Rochette, L.; Gudjoncik, A.; Guenancia, C.; Zeller, M.; Cottin, Y.; Vergely, C. The iron-regulatory hormone hepcidin: A possible therapeutic target? Pharmacol. Ther. 2015, 146, 35–52. [Google Scholar] [CrossRef]
- Lill, R.; Hoffmann, B.; Molik, S.; Pierik, A.J.; Rietzschel, N.; Stehling, O.; Uzarska, M.A.; Webert, H.; Wilbrecht, C.; Muhlenhoff, U. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim. Biophys. Acta 2012, 1823, 1491–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudjoncik, A.; Guenancia, C.; Zeller, M.; Cottin, Y.; Vergely, C.; Rochette, L. Iron, oxidative stress, and redox signaling in the cardiovascular system. Mol. Nutr. Food Res. 2014, 58, 1721–1738. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Cai, Z.; Wang, H.; Han, D.; Cheng, Q.; Zhang, P.; Gao, F.; Yu, Y.; Song, Z.; Wu, Q.; et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ. Res. 2020, 127, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Arosio, P.; Elia, L.; Poli, M. Ferritin, cellular iron storage and regulation. IUBMB Life 2017, 69, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Tan, Y.; Qin, Q.; Yang, B.; Zhu, Q.; Xu, L.; Liu, Z.; Song, E.; Song, Y. Polybrominated Diphenyl Ethers Quinone Induces NCOA4-Mediated Ferritinophagy through Selectively Autophagic Degradation of Ferritin. Chem. Res. Toxicol. 2019, 32, 2509–2516. [Google Scholar] [CrossRef]
- Soares, M.P.; Hamza, I. Macrophages and Iron Metabolism. Immunity 2016, 44, 492–504. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, Y.; Guo, L.; Gao, W.; Tang, T.L.; Yan, M. Interaction between macrophages and ferroptosis. Cell Death Dis. 2022, 13, 355. [Google Scholar] [CrossRef]
- Fujimaki, M.; Furuya, N.; Saiki, S.; Amo, T.; Imamichi, Y.; Hattori, N. Iron Supply via NCOA4-Mediated Ferritin Degradation Maintains Mitochondrial Functions. Mol. Cell Biol. 2019, 39, e00010–e00019. [Google Scholar] [CrossRef] [Green Version]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Protchenko, O.; Baratz, E.; Jadhav, S.; Li, F.; Shakoury-Elizeh, M.; Gavrilova, O.; Ghosh, M.C.; Cox, J.E.; Maschek, J.A.; Tyurin, V.A.; et al. Iron Chaperone Poly rC Binding Protein 1 Protects Mouse Liver From Lipid Peroxidation and Steatosis. Hepatology 2021, 73, 1176–1193. [Google Scholar] [CrossRef]
- Belalcazar, A.D.; Ball, J.G.; Frost, L.M.; Valentovic, M.A.; Wilkinson, J.t. Transsulfuration Is a Significant Source of Sulfur for Glutathione Production in Human Mammary Epithelial Cells. ISRN Biochem. 2014, 2013, 637897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochette, L.; Vergely, C. Coronary artery disease: Can aminothiols be distinguished from reactive oxygen species? Nat. Rev. Cardiol. 2016, 13, 128–130. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Koppula, P.; Lei, G.; Zhang, Y.; Yan, Y.; Mao, C.; Kondiparthi, L.; Shi, J.; Liu, X.; Horbath, A.; Das, M.; et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat. Commun. 2022, 13, 2206. [Google Scholar] [CrossRef]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Sun, T.; Chi, J.T. Regulation of ferroptosis in cancer cells by YAP/TAZ and Hippo pathways: The therapeutic implications. Genes. Dis. 2021, 8, 241–249. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and cancer: Recent insights. Ann. N. Y. Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016, 23, 270–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- Levring, T.B.; Hansen, A.K.; Nielsen, B.L.; Kongsbak, M.; von Essen, M.R.; Woetmann, A.; Odum, N.; Bonefeld, C.M.; Geisler, C. Activated human CD4+ T cells express transporters for both cysteine and cystine. Sci. Rep. 2012, 2, 266. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Shchepinov, M.S. Polyunsaturated Fatty Acid Deuteration against Neurodegeneration. Trends Pharmacol. Sci. 2020, 41, 236–248. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chen, P.; Zhai, B.; Zhang, M.; Xiang, Y.; Fang, J.; Xu, S.; Gao, Y.; Chen, X.; Sui, X.; et al. The emerging role of ferroptosis in inflammation. Biomed. Pharmacother. 2020, 127, 110108. [Google Scholar] [CrossRef]
- Rochette, L.; Guenancia, C.; Gudjoncik, A.; Hachet, O.; Zeller, M.; Cottin, Y.; Vergely, C. Anthracyclines/trastuzumab: New aspects of cardiotoxicity and molecular mechanisms. Trends Pharmacol. Sci. 2015, 36, 326–348. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochette, L.; Rigal, E.; Dogon, G.; Malka, G.; Zeller, M.; Vergely, C.; Cottin, Y. Mitochondrial-derived peptides: New markers for cardiometabolic dysfunction. Arch. Cardiovasc. Dis. 2022, 115, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Gan, B. Mitochondrial regulation of ferroptosis. J. Cell Biol. 2021, 220, e202105043. [Google Scholar] [CrossRef] [PubMed]
- Drysdale, J.; Arosio, P.; Invernizzi, R.; Cazzola, M.; Volz, A.; Corsi, B.; Biasiotto, G.; Levi, S. Mitochondrial ferritin: A new player in iron metabolism. Blood Cells Mol. Dis. 2002, 29, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058. [Google Scholar] [CrossRef]
- Wang, P.; Cui, Y.; Ren, Q.; Yan, B.; Zhao, Y.; Yu, P.; Gao, G.; Shi, H.; Chang, S.; Chang, Y.Z. Mitochondrial ferritin attenuates cerebral ischaemia/reperfusion injury by inhibiting ferroptosis. Cell Death Dis. 2021, 12, 447. [Google Scholar] [CrossRef]
- Rizzollo, F.; More, S.; Vangheluwe, P.; Agostinis, P. The lysosome as a master regulator of iron metabolism. Trends Biochem. Sci. 2021, 46, 960–975. [Google Scholar] [CrossRef]
- Fujita, Y.; Ito, M.; Ohsawa, I. Mitochondrial stress and GDF15 in the pathophysiology of sepsis. Arch. Biochem. Biophys. 2020, 696, 108668. [Google Scholar] [CrossRef]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Insights Into Mechanisms of GDF15 and Receptor GFRAL: Therapeutic Targets. Trends Endocrinol. Metab. 2020, 31, 939–951. [Google Scholar] [CrossRef]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. GDF15: An emerging modulator of immunity and a strategy in COVID-19 in association with iron metabolism. Trends Endocrinol. Metab. 2021, 32, 875–889. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Qiao, L.; Bian, Y.; Sun, X. GDF15 knockdown promotes erastin-induced ferroptosis by decreasing SLC7A11 expression. Biochem. Biophys. Res. Commun. 2020, 526, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Zhang, Q.; Zhang, Y.; Li, R.; Zhao, T.; Chen, L.; Liu, Q.; Zheng, S.; Li, H.; Qian, Z.; et al. Growth Differentiation Factor 15 Regulates Oxidative Stress-Dependent Ferroptosis Post Spinal Cord Injury by Stabilizing the p62-Keap1-Nrf2 Signaling Pathway. Front. Aging Neurosci. 2022, 14, 905115. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Cottin, Y.; Zeller, M.; Vergely, C. Carbon monoxide: Mechanisms of action and potential clinical implications. Pharmacol. Ther. 2013, 137, 133–152. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W. Heme Oxgenase-1, a Cardinal Modulator of Regulated Cell Death and Inflammation. Cells 2021, 10, 515. [Google Scholar] [CrossRef]
- Lee, S.J.; Ryter, S.W.; Xu, J.F.; Nakahira, K.; Kim, H.P.; Choi, A.M.; Kim, Y.S. Carbon monoxide activates autophagy via mitochondrial reactive oxygen species formation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 867–873. [Google Scholar] [CrossRef] [Green Version]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Zhao, B.; Zhou, L.; Zhang, Z.; Shen, Y.; Lv, H.; AlQudsy, L.H.H.; Shang, P. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 2020, 483, 127–136. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef]
- Zhang, W.; Jiang, B.; Liu, Y.; Xu, L.; Wan, M. Bufotalin induces ferroptosis in non-small cell lung cancer cells by facilitating the ubiquitination and degradation of GPX4. Free Radic. Biol. Med. 2022, 180, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef]
- Yao, X.; Li, W.; Fang, D.; Xiao, C.; Wu, X.; Li, M.; Luo, Z. Emerging Roles of Energy Metabolism in Ferroptosis Regulation of Tumor Cells. Adv. Sci. (Weinh.) 2021, 8, e2100997. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Wu, S.; Li, T.; Liu, W.; Huang, Y. Ferroptosis and Cancer: Complex Relationship and Potential Application of Exosomes. Front. Cell Dev. Biol. 2021, 9, 733751. [Google Scholar] [CrossRef]
- Aydin, E.; Johansson, J.; Nazir, F.H.; Hellstrand, K.; Martner, A. Role of NOX2-Derived Reactive Oxygen Species in NK Cell-Mediated Control of Murine Melanoma Metastasis. Cancer Immunol. Res. 2017, 5, 804–811. [Google Scholar] [CrossRef] [Green Version]
- Turrell, F.K.; Kerr, E.M.; Gao, M.; Thorpe, H.; Doherty, G.J.; Cridge, J.; Shorthouse, D.; Speed, A.; Samarajiwa, S.; Hall, B.A.; et al. Lung tumors with distinct p53 mutations respond similarly to p53 targeted therapy but exhibit genotype-specific statin sensitivity. Genes. Dev. 2017, 31, 1339–1353. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rochette, L.; Dogon, G.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. Int. J. Mol. Sci. 2023, 24, 449. https://doi.org/10.3390/ijms24010449
Rochette L, Dogon G, Rigal E, Zeller M, Cottin Y, Vergely C. Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. International Journal of Molecular Sciences. 2023; 24(1):449. https://doi.org/10.3390/ijms24010449
Chicago/Turabian StyleRochette, Luc, Geoffrey Dogon, Eve Rigal, Marianne Zeller, Yves Cottin, and Catherine Vergely. 2023. "Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis" International Journal of Molecular Sciences 24, no. 1: 449. https://doi.org/10.3390/ijms24010449