
A Meta-Analysis Study of SOD1-Mutant Mouse Models of ALS to Analyse the Determinants of Disease Onset and Progression

Abstract
:1. Introduction
2. Results
2.1. Collection of Primary Studies
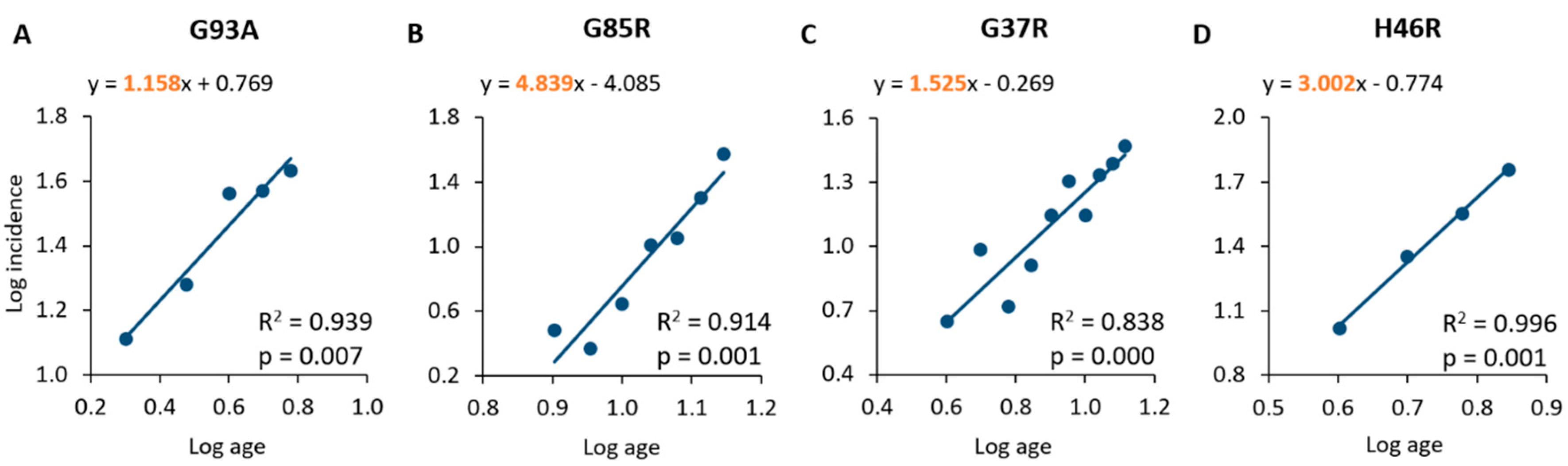
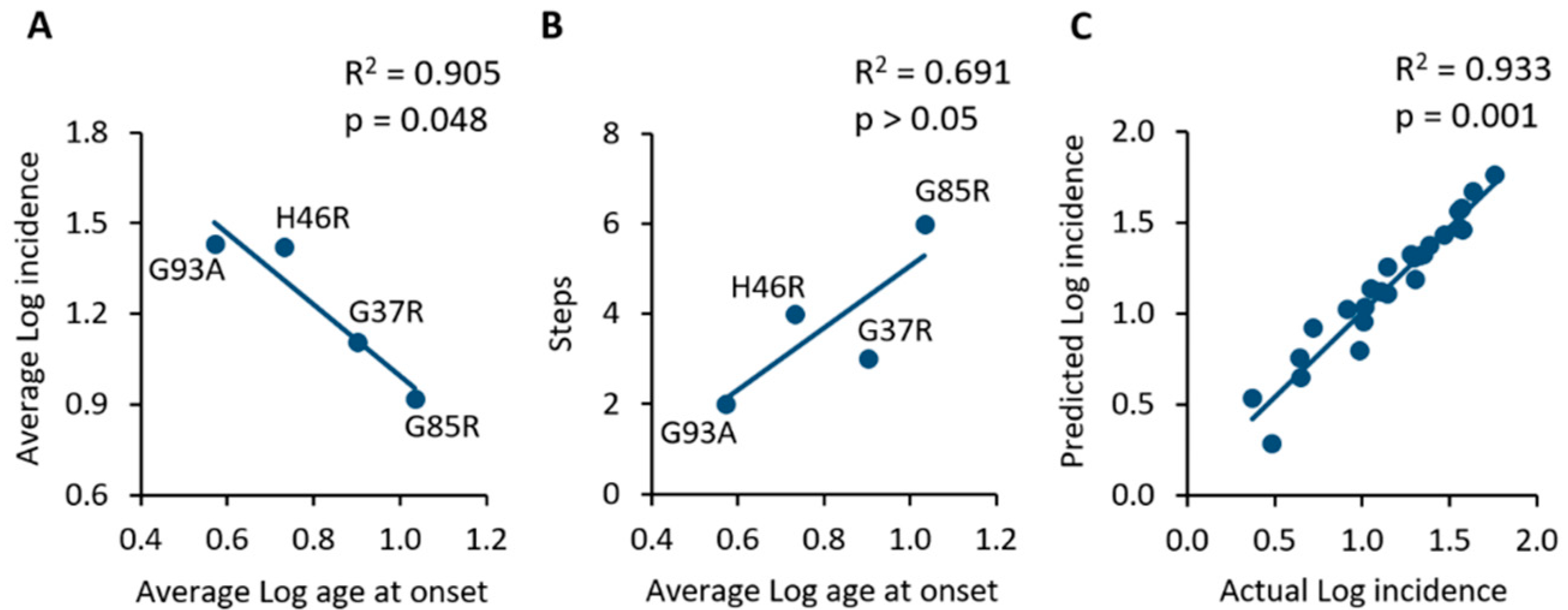
2.2. Analysis of Incidence Data Revealed a Multi-Step Disease Development
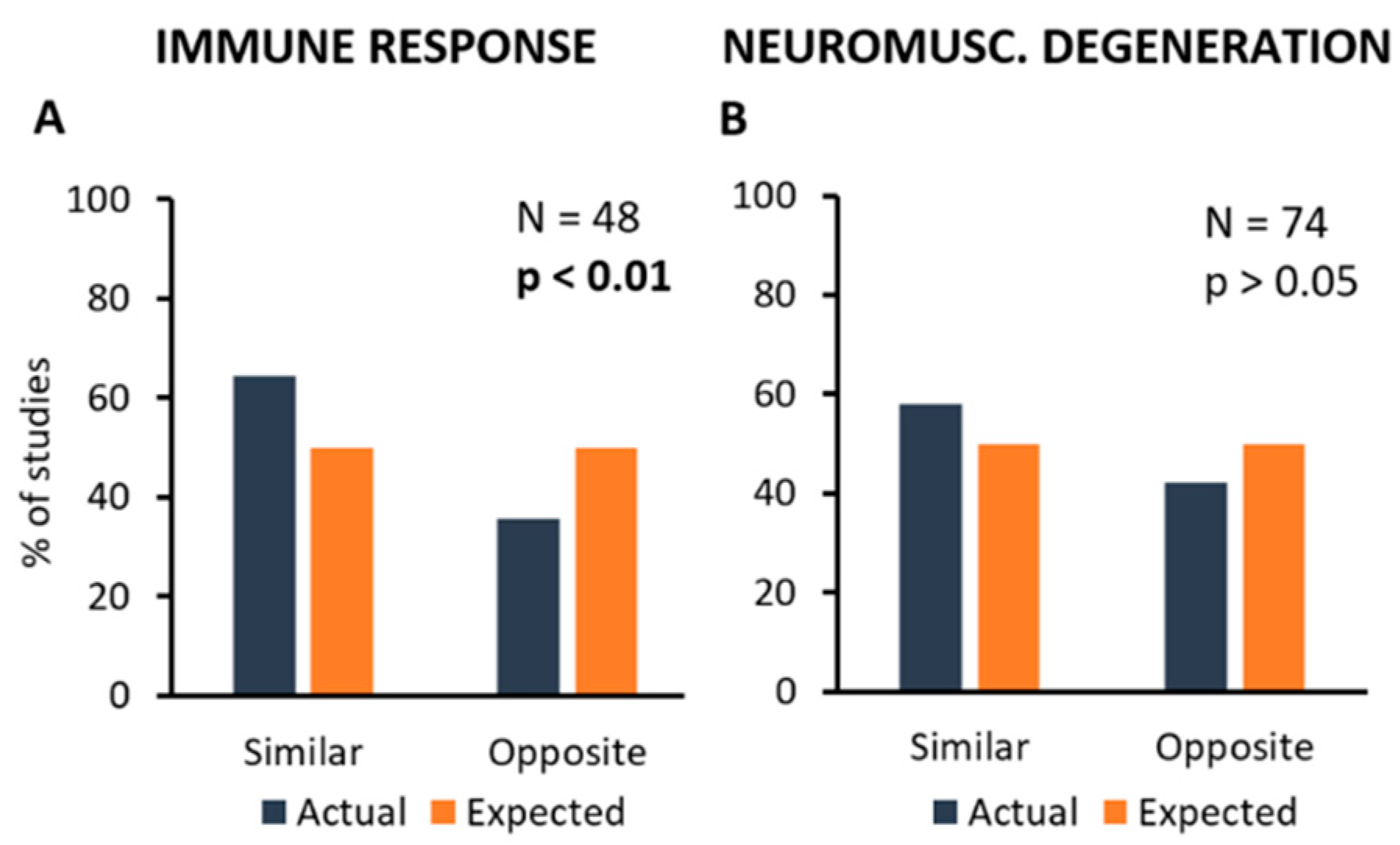
2.3. Extraction of Data concerning the Cellular and Molecular Mechanisms Associated with Disease Onset and Progression
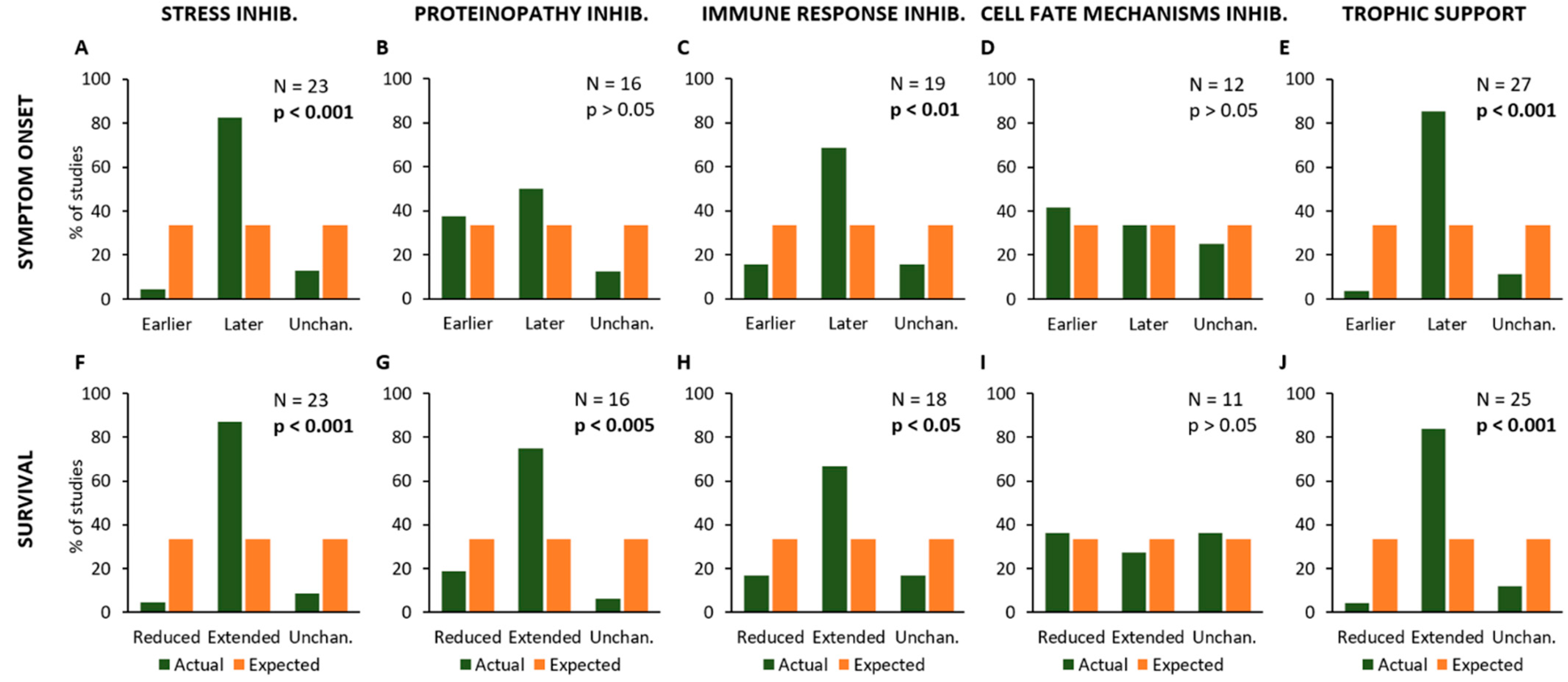
2.4. Effects of Experimental Procedures on Disease Onset and Survival
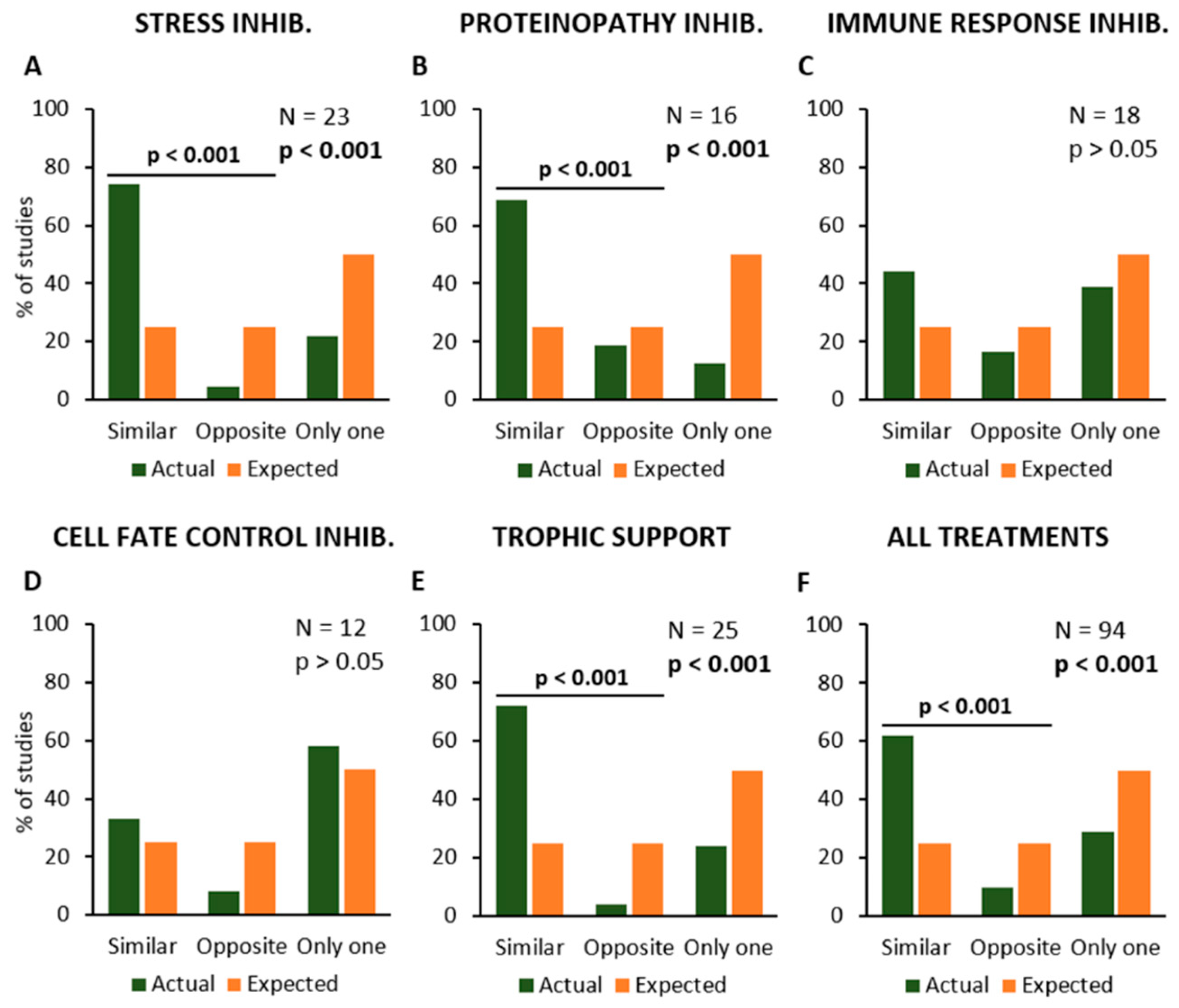
2.5. Effects of Experimental Procedures on the Pathological Mechanisms
3. Discussion
4. Materials and Methods
4.1. Study Selection and Eligibility Criteria
4.2. Calculation of Incidence Data
4.3. Multi-Step Model
4.4. Extraction of Data concerning Pathological Mechanisms and Effects of Treatments
4.5. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chiò, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G. Prognostic factors in ALS: A critical review. Amiotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef] [Green Version]
- Vucic, S.; Rothstein, J.D.; Kiernan, M.C. Advances in treating amyotrophic lateral sclerosis: Insights from pathophysiological studies. Trends Neurosci. 2014, 37, 433–442. [Google Scholar] [CrossRef]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Sever, B.; Ciftci, H.; DeMirci, H.; Sever, H.; Ocak, F.; Yulug, B.; Tateishi, H.; Tateishi, T.; Otsuka, M.; Fujita, M.; et al. Comprehensive research on past and future therapeutic strategies devoted to treatment of amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2022, 23, 2400. [Google Scholar] [CrossRef] [PubMed]
- Loscalzo, J.; Barabási, A.-L.; Silverman, E.K. Network Medicine: Complex Systems in Human Disease and Therapeutics; Harvard University Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Hardiman, O.; Al-Chalabi, A.; Chiò, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.; Freischmidt, A. Update on genetics of amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2022, 35, 672–677. [Google Scholar] [CrossRef]
- Dukkipati, S.S.; Garrett, T.; Elbasiouny, S.M. The vulnerability of spinal motoneurons and soma size plasticity in a mouse model of amyotrophic lateral sclerosis. J. Physiol. 2018, 596, 1723–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durand, J.; Amendola, J.; Bories, C.; Lamotte d’Incamps, B. Early abnormalities in transgenic mouse models of amyotrophic lateral sclerosis. J. Physiol. Paris 2006, 99, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Giusto, E.; Codrich, M.; de Leo, G.; Francardo, V.; Coradazzi, M.; Parenti, R.; Gulisano, M.; Vicario, N.; Gulino, R.; Leanza, G. Compensatory changes in degenerating spinal motoneurons sustain functional sparing in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. J. Comp. Neurol. 2020, 528, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Gulino, R.; Vicario, N.; Giunta, M.A.S.; Spoto, G.; Calabrese, G.; Vecchio, M.; Gulisano, M.; Leanza, G.; Parenti, R. Neuromuscular plasticity in a mouse model of spinal motoneuronal loss. Int. J. Mol. Sci. 2019, 20, 1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulino, R.; Forte, S.; Parenti, R.; Gulisano, M. TDP-43 as a modulator of synaptic plasticity in a mouse model of spinal motoneuron degeneration. CNS Neurol. Disord. Drug Targets 2015, 14, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Gulino, R.; Perciavalle, V.; Gulisano, M. Expression of cell-fate determinants and plastic changes after neurotoxic lesion of adult mice spinal cord by cholera toxin-B saporin. Eur. J. Neurosci. 2010, 31, 1423–1434. [Google Scholar] [CrossRef]
- Vicario, N.; Spitale, F.M.; Tibullo, D.; Giallongo, C.; Amorini, A.M.; Scandura, G.; Spoto, G.; Saab, M.W.; D’Aprile, S.; Alberghina, C.; et al. Clobetasol promotes neuromuscular plasticity in mice after motoneuronal loss via sonic hedgehog signaling, immunomodulation and metabolic rebalancing. Cell Death Dis. 2021, 12, 625. [Google Scholar] [CrossRef]
- Armitage, P.; Doll, R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br. J. Cancer 1954, 8, 1983–1989. [Google Scholar] [CrossRef] [Green Version]
- Al-Chalabi, A.; Calvo, A.; Chiò, A.; Colville, S.; Ellis, C.M.; Hardiman, O.; Heverin, M.; Howard, R.S.; Huisman, M.H.B.; Keren, N.; et al. Analysis of amyotrophic lateral sclerosis as a multistep process: A population-based modelling study. Lancet Neurol. 2014, 13, 1108–1113. [Google Scholar] [CrossRef] [Green Version]
- Chiò, A.; Mazzini, L.; D’Alfonso, S.; Corrado, L.; Canosa, A.; Moglia, C.; Manera, U.; Bersano, E.; Brunetti, M.; Barberis, M.; et al. The multistep hypothesis revisited: The role of genetic mutations. Neurology 2018, 91, e635–e642. [Google Scholar] [CrossRef] [Green Version]
- Bendotti, C.; Carrì, M.T. Lessons from models of SOD1-linked familial ALS. Trends Mol. Med. 2004, 10, 393–400. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- van Damme, P.; Robberecht, W.; van den Bosch, L. Modelling amyotrophic lateral sclerosis: Progress and possibilities. Dis. Model. Mech. 2017, 10, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philips, T.; Rothstein, J.D. Rodent models of amyotrophic lateral sclerosis. Curr. Protoc. Pharmacol. 2015, 69, 5–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, N. Transgenic mouse model for familial amyotrophic lateral sclerosis with superoxide dismutase-1 mutation. Neuropathology 2001, 21, 82–92. [Google Scholar] [CrossRef]
- Sasaki, S.; Nagai, M.; Aoki, M.; Komori, T.; Itoyama, Y.; Iwata, M. Motor neuron disease in transgenic mice with an H46R mutant SOD1 gene. J. Neuropathol. Exp. Neurol. 2007, 66, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burlando, B.; Milanese, M.; Giordano, G.; Bonifacino, T.; Ravera, S.; Blanchini, F.; Bonanno, G. A multistationary loop model of ALS unveils critical molecular interactions involving mitochondria and glucose metabolism. PLoS ONE 2020, 15, e0244234. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, F.; Iturria-Medina, Y.; Evans, A.C. Mathematical modeling of protein misfolding mechanisms in neurological diseases: A historical overview. Front. Neurol. 2018, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Lloret-Villas, A.; Varusai, T.M.; Juty, N.; Laibe, C.; Le Novère, N.; Hermjakob, H.; Chelliah, V. The impact of mathematical modeling in understanding the mechanisms underlying neurodegeneration: Evolving dimensions and future directions. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 73–86. [Google Scholar] [CrossRef]
- Shaabi, A. Modeling amyotrophic lateral sclerosis progression: Logic in the logit. Cureus 2022, 14, e24887. [Google Scholar] [CrossRef]
- Budini, M.; Buratti, E.; Morselli, E.; Criollo, A. Autophagy and its impact on neurodegenerative diseases: New roles for TDP-43 and C9orf72. Front. Mol. Neurosci. 2017, 10, 170. [Google Scholar] [CrossRef] [Green Version]
- Howland, D.S.; Liu, J.; She, Y.; Goad, B.; Maragakis, N.J.; Kim, B.; Erickson, J.; Kulik, J.; DeVito, L.; Psaltis, G.; et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. USA 2002, 99, 1604–1609. [Google Scholar] [CrossRef]
- Kabashi, E.; Agar, J.N.; Taylor, J.N.; Minotti, S.; Durham, H.D. Focal dysfunction of the proteasome: A pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2004, 89, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Körner, S.; Kollewe, K.; Fahlbusch, M.; Zapf, A.; Dengler, R.; Krampfl, K.; Petri, S. Onset and spreading patterns of upper and lower motor neuron symptoms in amyotrophic lateral sclerosis. Muscle Nerve 2011, 43, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Maniecka, Z.; Polymenidou, M. From nucleation to widespread propagation: A prion-like concept for ALS. Virus Res. 2015, 207, 94–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAlary, L.; Plotkin, S.S.; Yerbury, J.J.; Cashman, N.R. Prion-like propagation of protein misfolding and aggregation in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2019, 12, 262. [Google Scholar] [CrossRef] [Green Version]
- Münch, C.; O’Brien, J.; Bertolotti, A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3548–3553. [Google Scholar] [CrossRef] [Green Version]
- Heiman-Patterson, T.D.; Sher, R.B.; Blankenhorn, E.A.; Alexander, G.; Deitch, J.S.; Kunst, C.B.; Maragakis, N.; Cox, G. Effect of genetic background on phenotype variability in transgenic mouse models of amyotrophic lateral sclerosis: A window of opportunity in the search for genetic modifiers. Amyotroph. Lateral. Scler. 2011, 12, 79–86. [Google Scholar] [CrossRef]
- Marino, M.; Papa, S.; Crippa, V.; Nardo, G.; Peviani, M.; Cheroni, C.; Trolese, M.C.; Lauranzano, E.; Bonetto, V.; Poletti, A.; et al. Differences in protein quality control correlate with phenotype variability in 2 mouse models of familial amyotrophic lateral sclerosis. Neurobiol. Aging 2015, 36, 492–504. [Google Scholar] [CrossRef]
- Nardo, G.; Trolese, M.C.; Tortarolo, M.; Vallarola, A.; Freschi, M.; Pasetto, L.; Bonetto, V.; Bendotti, C. New insights on the mechanisms of disease course variability in ALS mutant SOD1 mouse models. Brain Pathol. 2016, 26, 237–247. [Google Scholar] [CrossRef]
- Pfohl, S.R.; Halicek, M.T.; Mitchell, C.S. Characterization of the contribution of genetic background and gender to disease progression in the SOD1 G93A mouse model of amyotrophic lateral sclerosis: A meta-analysis. J. Neuromuscul. Dis. 2015, 2, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Eisen, A.; Kiernan, M.; Mitsumoto, H.; Swash, M. Amyotrophic lateral sclerosis: A long preclinical period? J. Neurol. Neurosurg. Psychiatry 2014, 85, 1232–1238. [Google Scholar] [CrossRef] [Green Version]
- van Zundert, B.; Izaurieta, P.; Fritz, E.; Alvarez, F.J. Early pathogenesis in the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J. Cell. Biochem. 2012, 113, 3301–3312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klus, P.; Cirillo, D.; Botta Orfila, T.; Tartaglia, G.G. Neurodegeneration and cancer: Where the disorder prevails. Sci. Rep. 2015, 5, 15390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, B.E.; Patani, R. The microglial component of amyotrophic lateral sclerosis. Brain 2020, 143, 3526–3539. [Google Scholar] [CrossRef]
- Liao, B.; Zhao, W.; Beers, D.R.; Henkel, J.S.; Appel, S.H. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 2012, 237, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thonhoff, J.R.; Simpson, E.P.; Appel, S.H. Neuroinflammatory mechanisms in amyotrophic lateral sclerosis pathogenesis. Curr. Opin. Neurol. 2018, 31, 635–639. [Google Scholar] [CrossRef]
- KEGG: Kyoto Encyclopedia of Genes and Genomes/Amyotrophic Lateral Sclerosis—Reference Pathway. Available online: https://www.kegg.jp/pathway/map=map05014&keyword=ALS (accessed on 10 December 2022).
- Tann, J.Y.; Wong, L.-W.; Sajikumar, S.; Ibáñez, C.F. Abnormal TDP-43 function impairs activity-dependent BDNF secretion, synaptic plasticity, and cognitive behavior through altered Sortilin splicing. EMBO J. 2019, 38, e100989. [Google Scholar] [CrossRef]
- Gulino, R.; Parenti, R.; Gulisano, M. Novel mechanisms of spinal cord plasticity in a mouse model of motoneuron disease. Biomed. Res. Int. 2015, 2015, 654637. [Google Scholar] [CrossRef] [Green Version]
- Gulino, R.; Gulisano, M. Involvement of brain-derived neurotrophic factor and sonic hedgehog in the spinal cord plasticity after neurotoxic partial removal of lumbar motoneurons. Neurosci. Res. 2012, 73, 238–247. [Google Scholar] [CrossRef]
- Miranda-Laurenço, C.; Ribeiro-Rodrigues, L.; Fonseca-Gomes, J.; Tanqueiro, S.R.; Belo, R.F.; Ferreira, C.B.; Rei, N.; Ferreira-Manso, M.; de Almeida-Borlido, C.; Costa-Coelho, T.; et al. Challenges of BDNF-based therapies: From common to rare diseases. Pharmacol. Res. 2020, 162, 105281. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SOD1 Mutation | Total No. of Papers | No. of Excluded Papers | No. of Selected Papers | No. of Mice |
|---|---|---|---|---|
| G93A | 639 | 530 | 109 | 2073 |
| G85R | 29 | 18 | 11 | 479 |
| G37R | 38 | 26 | 12 | 314 |
| H46R | 19 | 14 | 5 | 172 |
| Totals | 725 | 588 | 137 | 3038 |
| SOD1 Mutation | Month | No. of Cases | No. of Susceptible Subjects | Monthly Incidence (%) | Log Age | Log Incidence |
|---|---|---|---|---|---|---|
| G93A | 2 | 35 | 272 | 12.9 | 0.30 | 1.11 |
| 3 | 476 | 2498 | 19.1 | 0.48 | 1.28 | |
| 4 | 1018 | 2798 | 36.4 | 0.60 | 1.56 | |
| 5 | 380 | 1025 | 37.1 | 0.70 | 1.57 | |
| 6 | 78 | 183 | 42.9 | 0.78 | 1.63 | |
| G85R | 8 | 6 | 198 | 3.0 | 0.90 | 0.48 |
| 9 | 31 | 1326 | 2.3 | 0.95 | 0.37 | |
| 10 | 62 | 1404 | 4.4 | 1.00 | 0.65 | |
| 11 | 129 | 1258 | 10.3 | 1.04 | 1.01 | |
| 12 | 106 | 943 | 11.2 | 1.08 | 1.05 | |
| 13 | 86 | 430 | 20.0 | 1.11 | 1.30 | |
| 14 | 47 | 125 | 37.6 | 1.15 | 1.58 | |
| G37R | 4 | 21 | 469 | 4.5 | 0.60 | 0.65 |
| 5 | 39 | 402 | 9.7 | 0.70 | 0.99 | |
| 6 | 19 | 362 | 5.2 | 0.78 | 0.72 | |
| 7 | 29 | 355 | 8.2 | 0.85 | 0.91 | |
| 8 | 56 | 401 | 14.0 | 0.90 | 1.15 | |
| 9 | 37 | 183 | 20.2 | 0.95 | 1.31 | |
| 10 | 16 | 114 | 14.0 | 1.00 | 1.15 | |
| 11 | 24 | 111 | 21.6 | 1.04 | 1.33 | |
| 12 | 32 | 131 | 24.4 | 1.08 | 1.39 | |
| 13 | 13 | 44 | 29.5 | 1.11 | 1.47 | |
| H46R | 4 | 10 | 96 | 10.4 | 0.60 | 1.02 |
| 5 | 65 | 288 | 22.6 | 0.70 | 1.35 | |
| 6 | 44 | 123 | 35.8 | 0.78 | 1.55 | |
| 7 | 8 | 14 | 57.6 | 0.85 | 1.76 |
| SOD1 Mutation | Average Age at Onset (Weeks) | Log Average Age at Onset | Average Incidence (%) | Log Average Incidence | Slope | Steps |
|---|---|---|---|---|---|---|
| G93A | 16.0 | 0.57 | 29.6 | 1.43 | 1.16 | 2 |
| G85R | 46.4 | 1.03 | 12.7 | 0.92 | 4.84 | 6 |
| G37R | 32.8 | 0.90 | 15.1 | 1.11 | 1.53 | 3 |
| H46R | 21.6 | 0.73 | 31.5 | 1.42 | 3.00 | 4 |
| Experimental Procedures | Main Pathological Mechanisms |
|---|---|
| Stress inhibition | Immune Response |
| Proteinopathy inhibition Immune Response inhibition | Neuromuscular Degeneration |
| Cell Fate Mechanisms inhibition | |
| Trophic Support |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciuro, M.; Sangiorgio, M.; Leanza, G.; Gulino, R. A Meta-Analysis Study of SOD1-Mutant Mouse Models of ALS to Analyse the Determinants of Disease Onset and Progression. Int. J. Mol. Sci. 2023, 24, 216. https://doi.org/10.3390/ijms24010216
Ciuro M, Sangiorgio M, Leanza G, Gulino R. A Meta-Analysis Study of SOD1-Mutant Mouse Models of ALS to Analyse the Determinants of Disease Onset and Progression. International Journal of Molecular Sciences. 2023; 24(1):216. https://doi.org/10.3390/ijms24010216
Chicago/Turabian StyleCiuro, Maria, Maria Sangiorgio, Giampiero Leanza, and Rosario Gulino. 2023. "A Meta-Analysis Study of SOD1-Mutant Mouse Models of ALS to Analyse the Determinants of Disease Onset and Progression" International Journal of Molecular Sciences 24, no. 1: 216. https://doi.org/10.3390/ijms24010216